
Abstract
Genome-wide in-silico identification of microsatellites or simple sequence repeats (SSRs) in the Orf virus (ORFV), the causative agent of contagious ecthyma has been carried out to investigate the type, distribution and its potential role in the genome evolution. We have investigated eleven ORFV strains, which resulted in the presence of 1,036–1,181 microsatellites per strain. The further screening revealed the presence of 83–107 compound SSRs (cSSRs) per genome. Our analysis indicates the dinucleotide (76.9%) repeats to be the most abundant, followed by trinucleotide (17.7%), mononucleotide (4.9%), tetranucleotide (0.4%) and hexanucleotide (0.2%) repeats. The Relative Abundance (RA) and Relative Density (RD) of these SSRs varied between 7.6–8.4 and 53.0–59.5 bp/kb, respectively. While in the case of cSSRs, the RA and RD ranged from 0.6–0.8 and 12.1–17.0 bp/kb, respectively. Regression analysis of all parameters like the incident of SSRs, RA, and RD significantly correlated with the GC content. But in a case of genome size, except incident SSRs, all other parameters were non-significantly correlated. Nearly all cSSRs were composed of two microsatellites, which showed no biasedness to a particular motif. Motif duplication pattern, such as, (C)-x-(C), (TG)-x-(TG), (AT)-x-(AT), (TC)- x-(TC) and self-complementary motifs, such as (GC)-x-(CG), (TC)-x-(AG), (GT)-x-(CA) and (TC)-x-(AG) were observed in the cSSRs. Finally, in-silico polymorphism was assessed, followed by in-vitro validation using PCR analysis and sequencing. The thirteen polymorphic SSR markers developed in this study were further characterized by mapping with the sequence present in the database. The results of the present study indicate that these SSRs could be a useful tool for identification, analysis of genetic diversity, and understanding the evolutionary status of the virus.
Similar content being viewed by others

Introduction
Contagious ecthyma or Orf is a zoonotic viral disease of sheep, goats, and other small ruminants characterized by proliferative skin lesions in and around the oral cavity in the form of erythematous macule, papule, vesicle, pustule, and scabs. The causative agent is the Orf virus (ORFV), a member of the genus Parapoxvirus of the Poxviridae family. The virus is highly contagious, quite stable in the environment, and remains in the infectious form in wools or animal excreta for months to years1. The disease is manifested by proliferative lesions on the mouth and muzzle that usually get resolved in 1–2 months2. These facial and oral lesions in lambs may interfere with suckling, while lesions on the udder may interfere in feeding neonates. Similarly, foot lesions often cause transient lameness in infected animals, and together all these results in poor health and loss of body weight. Lesions progress through all clinical stages but are generally non-proliferative and usually resolve within 2–3 weeks. ORFV specific antibodies do not seem to confer protective immunity, although the IgG2 isotype is believed to provide some defense against ORFV infection3. As IgG2 is not secreted in the colostrum of ruminants, lamb and kids don’t get required protection4. Although Orf is normally non-fatal in adults, often comes with high morbidity (up to 100%). While in neonates, Orf can be life-threating as it interferes with suckling of milk from the infected udder or predisposing the animals to the secondary bacterial or fungal infections5. For these reasons, the mortality rate may reach up to 15%6. There is increasing evidence of ORFV to cross‐infect other species of animals such as camels, gazelles, reindeers, musk ox, and Japanese serows3.
The virus can infect humans, particularly those who are closely associated with animal handling. Zoonosis occurs most frequently during lambing, shearing, docking, drenching, or slaughtering of affected animals1,3. Orf infections in humans appear in hand7 but occasionally seen in the face8, nose9, axilla10, scalp11, genitals12,13, urethral8, and pericanthal eyelid skin and the wound heals spontaneously. However, in immunosuppressive individuals, large-sized poorly healing lesions could remain for an extended period up to a couple of months14. This possesses a significant health risk to animal-handlers and veterinarians who often get infected by direct contact and develop painful pustular lesions in the skins. Complications of Orf with secondary bacterial infections are potentially life-threatening and need urgent medical attention.
The ORFV is a classic epitheliotropic virus, having a double-stranded DNA genome with a higher (64%) GC content15. The genome consists of central conserved and terminal variable domains with size varying from 134 to 139 kbp having ~ 130 putative genes, 88 of which are conserved to Chordopoxviruses16,17. Having such a devastating character, this virus has got less attention in terms of genomic information, which is evident from the availability of only eleven complete genome sequences worldwide. Several conserved genomic regions such as envelope protein B2L (ORFV011), F1L (ORFV059), and A32L (ORFV108) were used for ORFV identification and phylogenetic tree construction18. Still, there is a lack of clarity regarding the real diversity of ORFV due to the absence of a reliable system for virus identification, which consists of hypermutable regions such as microsatellites rather than conventional conserved genes.
Simple sequence repeats (SSRs), also known as microsatellites, refer to mono-, di-, tri-, tetra-, penta- and hexanucleotide sequence units that are repeated in tandem in a genome19. Those short motifs of DNA are distributed ubiquitously in the genome of eukaryotes20, and prokaryotes21, and is regarded as the most variable type of DNA sequence within the viral genome22,23. The microsatellites may be classified as either simple or compound, depending on the constituent of nucleotide sequences. The interruptions present in the microsatellite will give rise to interrupted pure, compound, interrupted compound, complex and interrupted complex types. Two or more microsatellites resides directly adjacent to each other to form compound microsatellites by interruption of repeats24. Due to its unique characteristics, these SSRs play a major role in meiotic recombination25,26,27, the evolution of species28, genome mapping29, differentiation of viral strains30, studying population genetics31, and secondary structure formation32. Many studies have highlighted the presence of microsatellite repeats in viruses, such as menovirus33, vesicular stomatitis virus34, hepatitis C virus35, and human respiratory syncytial virus (RSV)36. Here, we report for the first time a comparative analysis of microsatellites with respect to the abundance, distribution, composition, and polymorphism of SSRs within ORFV through in-silico approach, followed by the development and characterization of thirteen microsatellites markers. Using these tools, we further tested its usefulness by screening the viral genome from an ORFV outbreak and constructing a concatenated phylogenetic tree, which elucidated that the investigated virus closely related to the Chinese isolate. These markers could be used as a tool for making multiplex PCR assays for virus identification, strain demarcation, and evolutionary analysis.
Materials and methods
Genome sequences
The publicly available eleven complete genome sequences of ORFV isolates obtained from the NCBI database (www.ncbi.nlm.nih.gov) were used for genome-wide in-silico microsatellites analysis. To compare genomic sequences of different lengths, we calculated the Relative Density (RD) and Relative Abundance (RA) values. RD is defined as the total length (bp) contributed by each microsatellite per kilobase (kb) of sequence analyzed whereas; RA is the number of microsatellites present per kb of the genome (kb). Among all the strains, we have chosen OV-SA00 (Acc. number: AY386264) as the reference to evaluate the polymorphism of microsatellites through in-silico approach as well as the development of SSRs for Indian origin ORFV (Table 1).
Microsatellites identification, investigation, and statistical analysis
For identification of perfect mono, di, tri, tetra, penta, hexa as well as compound microsatellites, IMEx software37 was utilized. Microsatellites from genomes were extracted using the ‘Advance-Mode’ of IMEx using the parameters previously used for RNA viruses38,39 and DNA viruses40. The parameters used were as follows: type of repeat: perfect; repeat size: all; minimum repeat number: 6, 3, 3, 3, 3, 3 for mono, di, tri, tetra, penta and hexanucleotide repeats, respectively. The maximum distance allowed between any two SSRs (dMAX) is 10 nucleotides. Other parameters were used as default. Compound microsatellites (cSSRs) were not standardized in order to determine real composition.
Multiple sequence alignment and identification of polymorphic SSRs
The microsatellites of OV-SA00 were considered for the identification of polymorphic microsatellites as well as consensus motifs. Sequences were first transferred to BioEdit version 7.2.5 software41 and aligned by CLUSTAL W42 module and checked manually for the presence of polymorphism. The Circos plot was generated using the Circos software to map the genome size, CDS, SSRs distribution, cSSRs distribution, and GC content in ORFV (OV-SA00) genome.
Disease outbreak and sample data collection
The study did not involve experiments on live vertebrates. Rather, samples were collected from the diseased goats (showing the symptoms of Orf) those reported for veterinary care where scab samples were collected by veterinary professionals as a routine practice. In October and November 2017, an outbreak of ORFV was noticed in Black Bengal goats in the Eastern-Indian state of Odisha with the geographical location (20.4625° N, 85.8830° E). Tissue samples in the form of scabs from four suspected goats were collected at both infective and recovery/convalescent phase and simultaneously treated for wounds with 2% boro glycerine and parenteral application of Enrofloxacin @ 5 mg/kg IM (Fig. 1).

ORFV infection in goat. Representative figure depicting clinical cases of ORFV infection in Black Bengal goat having proliferative lesions around the lip recorded in the study area.
About 5 g of tissue samples were collected from each animal and subsequently dissolved in phosphate-buffered saline (PBS, pH 7.2) added with antibiotics and antifungal supplements in a labeled sterile tube. The homogenized samples were then treated with tissue lysis buffer containing proteinase K, and the mixture was incubated at 56 °C overnight. Finally, the mixture was passed through a column, and DNA was purified from the column by using the standard phenol–chloroform method as described by Sambrook et al.43 and stored at − 20 °C until further use. The suspected samples collected during this outbreak produced the expected PCR-amplified fragment size of 140 bp using ORFV specific primers orf1 and orf244 having nucleotide sequences Orf1: 5′-CGCAGACGTGGCTGAGTACGT-3′ and Orf2: 5′-TGAGCTGGTTGGCGCTGTCCT-3′, which confirmed the presence of the virus.
Development of polymorphic SSRs
The polymorphic microsatellites identified through in-silico approach were further validated through in-vitro approach using ORFV positive clinical sample. Motifs located within defined flanking regions were PCR amplified using specially designed SSR-PCR primer pairs by Primer3Plus web tool (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/). The primer length was kept between 18 and 22 bp with product size in the range of 130–200 bp. For proper annealing to the template DNA, the annealing temperature was adjusted between 54 and 61 °C. The thermal cycling conditions for all genes were as follows: initial denaturation step at 95 °C for 5 min, with 35 cycles of denaturation at 95 °C for 50 s, with varying annealing temperature for each set of primers (55–61 °C) and extension step at 72 °C for 90 s with a final extension at 72 °C for 7 min. PCR amplification was performed in a Thermal Cycler system 2,720 (Applied Biosystems, USA) (Table 2).
The amplified products were resolved by electrophoresis in a 3% agarose gel. The PCR amplified products, stained with ethidium bromide, were visualized and photographed using a Gel Doc™ XR + System with Image Lab™ Software (Bio-Rad®). Subsequently, the amplified products were purified using QIAquick® purification kit (QIAGEN, USA) and the purified fragments were sent for sequencing using 3100 ABI sequencer (Applied Biosystems, USA) as described by Sanger et al.45. All sequences obtained were analyzed and verified twice in each direction.
Sequencing data analysis and phylogenetic tree construction
The sequencing results of the developed SSR markers were aligned by using discontiguous-MegaBLAST to identify specific regions among the reads (microsatellites) within the ORFV genome46. Next, the sequencing results were subjected to the BLASTx analysis, which compares translational products of the nucleotide query sequence to protein databases (https://www.ncbi. nlm.nih.gov). A concatenated phylogenetic tree was constructed using the bootstrap consensus tree building method of neighbor-joining with bootstrap value 500 through MEGA 5 to elucidate the genetic relationship of the outbreak sample with the global strains of ORFV.
Results
Distribution of SSRs and cSSRs in ORFV genome
Our study revealed a large number of SSRs scattered throughout the ORFV genomes varying from 1,036 to 1,181 in number with an average of 1,092 per genome. The RA and RD ranged from 7.6–8.4 and 53.0–59.5, respectively, in the analyzed ORFV genomes. However, previous reports in other DNA viruses such as human papillomaviruses (HPVs), the RA and RD ranged from 3.6–8.3 and 23.9–59.147. In the case of Herpesviruses, RA and RD occurred to be 4.1–13.3 and 26.9–102.948. On examining the SSR unit size classes, dinucleotide repeats were found to be most abundant (76.9%), followed by trinucleotide (17.7%) and mononucleotide repeats (4.9%) in all the genomes. Tetranucleotide and hexanucleotide repeats were least in number and represented 0.4% and 0.2% within the ORFV genome, respectively. There were no SSRs with pentanucleotide repeats observed in the ORFV genome. Approximately 90% and 10% of microsatellite motifs were distributed within coding and noncoding regions. Among the non-coding region, 4.8% are present in the UTR, while 5.4% in the intergenic regions, where functional protein and hypothetical protein occupied 68.8% and 21%, respectively. The genome-wide scan revealed the presence of 83–107 cSSRs, with an average of 93 occurrences per genome. In the case of compound microsatellites, the calculated RA and RD ranged from 0.6–0.8 and 12.1–17.0. However, in other DNA viruses such as HPVs, RA, and RD exhibited 0–1.2 and 0–27.3, whereas, in Herpesviruses, the RA and RD occurred 0.1–1.8 and 2.2–35.147,48. Approximately 89.5% and 10.5% of microsatellite motifs were distributed within coding or non-coding regions, respectively. Among the non-coding region, 5.0% were represented in the UTR while 5.5% in the intergenic region, where functional protein and hypothetical protein occupied 60.7% and 28.8%, respectively (Figure S2).
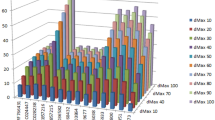
The percentage of individual microsatellites being part of compound microsatellite (cSSR%) ranged from 7.9 to 9.0 (Table 1). Based on dMAX value, the maximum distance between any two adjacent microsatellites and if the distance separating two microsatellites is less than or equivalent to dMAX, than microsatellites are classified as cSSRs49. To determine the impact of dMAX, all the studied genome sequences were chosen to determine the variability of cSSRs with increasing dMAX. The value of dMAX was set between 10 and 100 by Microsatellite Identification Search Analysis (MISA)50. Our analysis revealed an overall increase in the number of cSSRs with higher dMAX value and attained a plateau (Fig. 2).

Frequency of cSSRs in relation to varying dMAX (10–100) across eleven ORFV complete genomes represented on the right side of the graph. A higher cSSR incidence was observed with increasing dMAX in the genomes.
Genomic parameters influencing SSR and cSSR distribution
We tested for the correlation between genome size and GC content with the incidence, RA, RD of SSRs and cSSRs. Except incidence (R2 = 0.6162, p > 0.05), all other parameters such as RA and RD of SSRs had no correlation (R2 = 0.002374, p > 0.05; R2 = 0.18, p < 0.05) with the genome size and GC content (R2 = 0.09377, p < 0.05, R2 = 0.00126, p > 0.05; R2 = 0.08129, p < 0.05). The regression analysis of cSSRs showed significant correlation with the incidence (R2 = 0.6483, p > 0.05) and RA (R2 = 0.4823, p > 0.05) while displayed non-significant correlation with RD (R2 = 0.3759, p < 0.05). On the contrary, the GC content was weakly correlated with the number (R2 = 0.02903, p > 0.05), RD (R2 = 0.004839, p < 0.05) and RA (R2 = 0.03917, p < 0.05) of cSSRs.
The frequency of classified repeat types
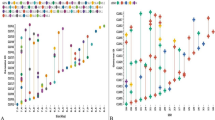
The overall frequency of mononucleotide repeats A/T (64.1%), dinucleotide repeat motif CG/GC (81.6%) were the most prevalent than poly G/C (35.9%), GA/TC (5.0%), AC/GT (4.5%), AG/CT (3.9%), CA/TG (3.6%) and AT/TA (1.4%), respectively. Analysis of the classified tri-repeat types revealed that the ORFV genome had 30 types of trinucleotide from which CGC/GCG, GCC/GGC, CAG/CTG, AGC/GCT, CCG/CGG were abundantly present exhibiting 18.2%, 14.5%, 6.3%, 6.2%, and 6.3%, respectively. The most common tetra and hexanucleotide repeats were CGAG/CTCG (34.9%), ACTC/GAGT (18.6%), GTGA/TCAC (9.3%) and AGTTAC/GTAACT (15.0%), ACACTC/GAGTGT (15.0%), respectively. However, the accession specific analysis illustrated that the frequency of mono, di, tri repeats varied from each other (Fig. 3a–c).

Types of SSRs distribution. (A) Distribution of different motifs of mononucleotide SSRs within ORFV genomes, (B) distribution of different motifs of dinucleotide SSRs within ORFV genomes, and (C) distribution of different motifs of trinucleotide SSRs within ORFV genomes.
Motif complexity of compound microsatellites
Compound microsatellites (cSSRs) are composed of two or more adjacent individual microsatellites. Generally, cSSR having the pattern like, m1-xn-m2, m1-xn-m2-xn-m3 are considered as ‘2-microsatellite’ and ‘3-microsatellite’, respectively49. Majority of cSSRs were composed of two motifs, followed by tri, tetra, and penta-motifs (Supplementary file 1). Interestingly, two long stretches of cSSR were composed of identical motifs repeated 12 times, which was exclusively found in the genome of AY386264. The CTG–CAG compound microsatellite composed of self-complementary motifs has been proposed to be created by recombination51. However, our study showed no such compound microsatellites which contained self-complementary motifs, suggesting that these compound microsatellites were not likely to be derived from recombination. Motifs exhibiting the form [m1]n-xn-[m2]n can be termed as SSR-couples and are represented the maximum time in the genome. In this study, SSR couples, such as (CG)-x-(GC), (GC)-x-(GC), (GC)-x-(CGC), (GT)-x-(GC), (GC)-x-(CG), (CT)-x-(C) were presented in all analyzed genome. A number of self-complementary motifs such as (CG)3-x1-(GC)3, (CG)4-x1-(GC)3, (CG)3-x7-(GC)3, (CG)3-x0-(GC)3, (GC)3-x8-(CG)3, (CG)3-x7-(CG)3, (GC)3-x0-(CG)3, (CG)3-x4-(GC)3 have been observed in ORFV, which played a pivotal role in secondary structure formation. Motif duplication is one of the phenomena in which a similar motif is located on both ends of the spacer sequence, for example (CA)n-(X)y-(CA)z. About 22.1% of the total cSSR were made up of duplicated sequences having the motif pattern (GC)-x-(GC), (CG)-x-(CG), (GA)-x-(GA), (CA)-x-(CA), (CT)-x-(CT), (TC)-x-(TC), (CA)-x-(CA)-x-(CA), (A)-x-(A), (AG)-x-(AG)-x-(AG)-x-(AG)-x-(AG)-x-(AG)3-x-(AG)-x1-(AG)-x-(AG)-x-(AG)-x-(AG)-x-(AG)-x-(AG), (AG)-x-(AG), (C)-x-(C), (CA)-x-(CA), and (CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT)-x-(CT) (Supplementary file 1).
Identification of polymorphic microsatellites through in silico approach
For a polymorphic microsatellite, the length of the repeat block should be non-identical with that of the other sequences in the database, and this length difference must be a multiple of the repeat unit19,30,52. For the identification of a polymorphic microsatellite, eleven strains of ORFV were used, where (AY386264) acted as the reference. A total thirteen number of polymorphic microsatellites were observed; among these, two were observed within the hypothetical protein, three in the intergenic regions, and rest eight in the protein-coding/genic regions. The polymorphic genic region containing the microsatellites encodes several important proteins such as Ankyrin repeat protein (ANK protein), DNA-binding phosphoprotein, virion core protein, Granulocyte–macrophage colony-stimulating factor (GM-CSF), Interleukin 10 protein (IL-10), Putative serine/threonine-protein kinase protein (Table 2, Figure S3). The Circos map provides a clear vision regarding the SSRs and cSSRs distribution and other related details in ORFV (OV-SA00) genome (Fig. 4).

Circos plot showing the Genome size, CDS, Distribution of SSRs, selected SSR markers, cSSRs and GC content in ORFV (OV-SA00) genome. From outer track to inner track: Genome size, CDS, SSRs, selected SSR markers (Black lines within the SSR), cSSRs and GC content.
Development and characterization of SSR markers
All clinical samples collected during the outbreak were found to be positive for ORFV tested by producing the desired PCR amplicon size of 140 bp (Fig. 5).

Clinical samples evaluation by universal OFRV primers. Electrophoresis gel showing the PCR amplicon of four suspected ORFV clinical samples collected from Black Bengal goats. M: 100 bp DNA ladder; -C: Negative control (PCR using nuclease-free water as DNA template); 1–4: Clinical samples.
We chose all thirteen polymorphic markers to validate in-vitro. Hence, PCR was set with each primer sets to amplify the DNA isolated from a positive clinical sample. The SSR name, primer sequences, expected size, targeted motif, functional region, protein motif position, gene, ORF number, and annealing temperature, were summarized in Table 2. All the SSR markers produced reliable and reproducible PCR products with the expected molecular size (Fig. 6).

Clinical sample validation using SSR markers. Electrophoresis gel showing the PCR amplicon of the developed SSR markers in ORFV. SSR markers from SSR1 to SSR13; M:50 bp DNA ladder; -C: Negative control (PCR using nuclease-free water as DNA template).
The amplified SSRs were further characterized by sequencing, mapping with the GenBank database through BLASTn and BLASTx. The results of BLASTn alignment revealed a 100% of query coverage and a high identity percentage (91–100%) between the respective sequencing product and their equivalent genes from the published OV-SA00 isolate genome sequence. The results of BLASTx alignment revealed various degrees of query coverage (38–96%) and a high identity percentage (91–100%) with their equivalent amino acid sequences (Table 3).
The concatenated phylogenetic tree showed the ORFV of our study closely related to Chinese isolate (MG712417) (Fig. 7). We observed the presence of 2–3 alleles within ORFV genomes.

The concatenated phylogenetic tree was constructed using the bootstrap consensus tree building method of neighbor-joining with bootstrap value 500 using MEGA 5. Black triangle represents the ORFV isolates of present investigation showing its relationship with eleven global strains.
Discussion
Microsatellites, otherwise known as short tandem repeats (STRs), or a variable number of tandem repeats (VNTRs) are being used to discriminate various viruses, such as human cytomegalovirus (hCMV)22,23, white spot syndrome virus (WSSV)53,54,55,56,57,58, Herpes Simplex virus type 130,59, Herpes Simplex virus type 260, Herpesvirus 361, Herpesvirus 662, Adenovirus63, Ostreid herpesvirus 164,65, Marek’s disease virus 166, and Spodoptera littoralis multiple nucleopolyhedrovirus (SpliMNPV)67 due to its polymorphic in nature. To get the insight into the microsatellite in ORFV, we have employed a comparative genomics approach for development and characterization through in-silico and in-vitro analysis and validated our findings using samples collected from the recent Orf outbreak for the first time.
The specific parameters, such as its incidence, RA and RD of SSR and cSSR in ORFV genomes, show abundance variation as compared to their genome size and GC content due to the heterogeneity of ORFVs. Until now, limited full-length ORF genomes exist in the database. Based on our analysis, we observed little variation in RA and RD in ORFV. However, in other viruses such as HPVs47 and Herpesviruses48, higher variation in RA and RD were reported. The large variation with the parameters was not observed in ORFV, probably due to the lack of enough size difference in the genome. However, a limited number of complete genome sequences are available for this virus, in comparison to HPV and herpesviruses, which act as a constraint to get the optimal range. Correlation analysis confirmed that incidence of both SSRs and cSSRs, RA of cSSRs were dependent on genome size, but independent of GC content, which was similar to that of HPV47, but opposite to HIV68, potexvirus, carlavirus, and tobamovirus69,70,71. The distribution of microsatellite in the viral genome is pathogen-specific rather than host-specific. The increase of cSSR is predominant when dMAX approaches 10–90 bp and further decreases with the increase of dmax (Fig. 2). This may be due to the occurrence of SSR in the overlapping regions of increasing dMAX. The ORFV genomes have more SSR within coding regions than non-coding regions in comparison with other DNA virus, such as herpes simplex virus. This might be due to higher relaxed selection pressure on coding regions in comparison to the non-coding region in the respective virus.
The cSSRs percentages of ORFV ranges from 7.9 to 9.0%, which is lower in comparison to HIV-1, 0–24.2%68, Geminivirus, 0–27.2%72, Herpesvirus, 8.1–33.3%48. Generally, the number of compound microsatellites decreases with an increase in complexity73. Moreover, the lack of sufficient genomic resources from diverse geographical locations may contribute to a stagnant range of cSSRs%. In ORFV, 22.1% of cSSRs were composed of similar motifs, probably contributed by genome duplication. Some study suggests that genome duplication may be helpful for the repeat tendency mechanism74, which promotes the expansion of genome size such as yeast75,76.
In ORFV genomes, the poly A/T repeats were significantly more prevalent than poly G/C repeats, similar to eukaryotic and prokaryotic genomes1,2. The presence of mononucleotide repeats in Mengovirus and Encephalomyocarditis virus affect virus growth in murine cell culture77. In the case of ORFV, its significance needs further validation. In this study, we also observed the microsatellites having polymorphism in poly A/T (ORF117), poly C/G (ORF121), within the important immune-regulatory genes, such as in GM-CSF and ANK protein, respectively (Supplementary file 2). GM-CSF secreted by a variety of cell types triggers neutrophil, monocyte, and eosinophil myelopoiesis and stimulate early events in immune responses, controlling the differentiation and function of antigen-presenting dendritic cells. IL-2 is a T-cell-derived lymphokine that stimulates T-cell and NK cell activation and proliferation and activated-B-cell proliferation78,79. ANK protein leads to the down-regulation of hypoxia-induced factor (HIF) activity and regulates energy metabolism, angiogenesis, the apoptotic cascade, the NF-kB signaling pathway, and cell cycle regulation80. The functional effects of this polymorphism in these regions require further investigations.
Dinucleotide CG/GC is more prevalent in most of the ORFV genomes, similar to that of DNA viruses such as HPVs47, Caulimoviruses, Geminiviruses52,81. CG/GC repeat could form Z‐conformation or other alternative secondary DNA to facilitate the recombination activity82. In our study, the polymorphism within dinucleotide (AC/CA)3 and (CG/CG)3 observed within the hypothetical protein. Dinucleotide repeats have the highest slippage rate as compared to any other type of repeats81. Among 257 viral genomes examined in a published study, the highest number of dinucleotide SSRs were found when compared to the other types83. Dinucleotide repeats are also speculated to be recombination hot spots84,85. In this study, the presence of higher di-nucleotide repeats over tri-nucleotide repeats suggests a possible role of hosts in the evolution of di-nucleotide repeats within poxvirus genomes. Inconsistency frequency of SSRs in different accession of the same virus may be attributed to instability because of a higher slippage rate86.
Trinucleotide motif ATA/TAA/AAT or ATT/TTA/TAT were most prevalent in most genomes of poxvirus whereas in other DNA virus GAG/AGA was most prevalent in HPVs and AAG/GAA in caulimoviruses. The higher density of trinucleotide repeats was observed compared to any other repeat type within coding regions of eukaryotic and prokaryotic genomes32. Interestingly, dynamic mutations within trinucleotide repeats responsible for the development of some diseases in humans87, as well as viral enzymes that interfere pathogenicity of Influenza virus88. Our study revealed the presence of trinucleotide CGC/GCG and GCC/GGC repeats to be most prevalent than others. The trinucleotide polymorphism was observed in some immunoregulatory genes such as ANK protein (GGC/GCC)3 (ORF008), IL-10 protein (AGT/ACT)3 (ORF127) and structural genes virion core protein (GAG/CTC)3, Putative serine/threonine-protein kinase (CGC/GCG)3, which needs further functional evaluation.
Three polymorphic SSRs such as (A/T)7, (T/A)6, (AGTTAC/ GTAACT)3 were observed within non-coding regions. The microsatellite present within the non-coding reasons was evolutionarily neutral and can be utilized as an excellent molecular marker30. Finally, we have characterized, those polymorphic markers present at non-genic as well as coding (genic) regions. These genic microsatellites, however, may provide adaptive variation important to viral evolution and genetic variability, perhaps similar to the functionally important mononucleotide runs found in VSV34 and RSV36 and virulence of avian influenza virus encephalo-myocarditis virus89,90. It is noteworthy to mention that, recently, the microsatellite present in HSV-1 glycoprotein coding region US4 was useful for strain differentiation30. The concatenated tree, which was constructed utilizing sequence information of characterized markers, confirmed that the ORFV of the present study closely related to Chinese isolate (MG712417). Our previous report, as well as several other studies, observed a similar pattern of relationship18,91. We speculate that trans-boundary and cross-species transfer of ORFV isolates could have resulted in this, as India is geographically adjacent to China. It is interesting to observe the presence of a number of the alleles (2–3) within ORFV genomes indicates the existence of polymorphism within microsatellites, which could act as a useful tool to estimate the diversity61. Using a single repeated mononucleotide was able to follow the dynamics of transmission of a human adenovirus during an epidemic63. Therefore, microsatellites constitute a potentially powerful tool for epidemiological studies of the transmission routes and evolution of ORFV and other related poxviruses. This study provides an important new type of molecular markers useful to investigate questions not only related to epidemiology but also for deciphering the diversity of the virus. However, the characterized microsatellites of the present study are not biased to the particular strain, which indicates the presence of recombinant strains circulating within the Indian subcontinent. This information is not concrete, which requires validation by several whole-genome sequence analysis of ORFV isolates from Indian origin. So far, our understanding of the functional and evolutionary role of microsatellites in ORFV biology is limited, which needs further in-depth evaluation and possible implementation.
In conclusion, the study of microsatellites in ORFV genome is a key step towards better understanding the nature, function, and evolutionary biology of the species. Our preliminary results can be considered as a useful tool for ORFV strain demarcation, diversity estimation, and evolutionary analysis. Our next plan is to characterize several ORFV strain complete genome from Indian origin through next-generation sequencing to get a better insight into genome organization, development of a suitable multiplex panel, which can be utilized as an effective tool for virus identification, genotyping and evolutionary analysis of the respective virus.
References
Haig, D. M. & Mercer, A. A. Ovine diseases. Orf. Vet. Res. 29, 311–326 (1998).
Gökce, H. I., Genc, O. & Gökce, G. Sero-prevalence of contagious ecthyma in lambs and humans in Kars, Turkey. Turk. J. Vet. Anim. Sci. 29, 95–101 (2005).
Hosamani, M., Scagliarini, A., Bhanuprakash, V., McInnes, C. J. & Singh, R. K. Orf: an update on current research and future perspectives. Expert Rev. Anti-infect. Therapy 7, 879–893 (2009).
Haig, D. M. et al. The orf virus OV20.0L gene product is involved in interferon resistance and inhibits an interferon-inducible, double-stranded RNA-dependent kinase. Immunology 93, 335–340 (1998).
Pugh, D. G. & Baird, N. N. Sheep & Goat Medicine-E-Book (Elsevier Health Sciences, Amsterdam, 2012).
Bora, D. P. et al. Identification and phylogenetic analysis of orf viruses isolated from outbreaks in goats of Assam, a northeastern state of India. Virus Genes 45, 98–104 (2012).
Uzel, M. et al. A viral infection of the hand commonly seen after the feast of sacrifice: human orf (orf of the hand). Epidemiol. Infect. 133, 653–657. https://doi.org/10.1017/S0950268805003778 (2005).
Duchateau, N. C., Aerts, O. & Lambert, J. Autoinoculation with Orf virus (ecthyma contagiosum). Int. J. Dermatol. 53, e60–e62. https://doi.org/10.1111/j.1365-4632.2012.05622.x (2013).
Maki, A. Jr., Hinsberg, A., Percheson, P. & Marshall, D. G. Orf: contagious pustular dermatitis. Can. Med. Assoc. J. 139, 971–972 (1988).
Weide, B. et al. Inflammatory nodules around the axilla: an uncommon localization of orf virus infection. Clin. Exp. Dermatol. 34, 240–242. https://doi.org/10.1111/j.1365-2230.2007.02567.x (2009).
Glass, J., Ghali, F. E., Sinkre, P., Ricotti, C. A. & Cockerell, C. J. Acute onset of erythematous scalp nodules in a child—quiz case. Arch. Dermatol. 145, 1053–1058. https://doi.org/10.1001/archdermatol.2009.184-a (2009).
Kandemir, H., Ciftcioglu, M. A. & Yilmaz, E. Genital ORF. Eur. J. Dermatol. 18, 460–461. https://doi.org/10.1684/ejd.2008.0444 (2008).
Vogel, T. A. & Schuttelaar, M.-L.A. Generalized orf superinfection in a child with atopic dermatitis. Eur. J. Dermatol. 23, 538–539 (2013).
Caravaglio, J. V. & Khachemoune, A. Orf virus infection in humans: a review with a focus on advances in diagnosis and treatment. J. Drugs Dermatol. 16, 684–689 (2017).
Cottone, R. et al. Analysis of genomic rearrangement and subsequent gene deletion of the attenuated Orf virus strain D1701. Virus Res. 56, 53–67. https://doi.org/10.1016/S0168-1702(98)00056-2 (1998).
Delhon, G. et al. Genomes of the parapoxviruses Orf virus and bovine papular stomatitis virus. J. Virol. 78, 168 (2004).
Mercer, A. A. et al. Comparative analysis of genome sequences of three isolates of Orf virus reveals unexpected sequence variation. Virus Res. 116, 146–158. https://doi.org/10.1016/j.virusres.2005.09.011 (2006).
Sahu, B. P., Majee, P., Sahoo, A. & Nayak, D. Molecular characterization, comparative and evolutionary analysis of the recent Orf outbreaks among goats in the Eastern part of India (Odisha). Agri Gene 12, 100088. https://doi.org/10.1016/j.aggene.2019.100088 (2019).
Chen, M. et al. Compound microsatellites in complete Escherichia coli genomes. FEBS Lett. 585, 1072–1076. https://doi.org/10.1016/j.febslet.2011.03.005 (2011).
Tóth, G., Gáspári, Z. & Jurka, J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 10, 967–981 (2000).
Mrázek, J., Guo, X. & Shah, A. Simple sequence repeats in prokaryotic genomes. Proc. Natl. Acad. Sci. 104, 8472 (2007).
Davis, C. L. et al. Numerous length polymorphisms at short tandem repeats in human cytomegalovirus. J. Virol. 73, 6265 (1999).
Walker, A. et al. Characterization of human cytomegalovirus strains by analysis of short tandem repeat polymorphisms. J. Clin. Microbiol. 39, 2219 (2001).
Chambers, G. K. & MacAvoy, E. S. Microsatellites: consensus and controversy. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 126, 455–476. https://doi.org/10.1016/S0305-0491(00)00233-9 (2000).
Schultes, N. P. Szostak JW A poly(dA.dT) tract is a component of the recombination initiation site at the ARG4 locus in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 322 (1991).
Gendrel, C. G., Boulet, A. & Dutreix, M. (CA/GT)(n) microsatellites affect homologous recombination during yeast meiosis. Genes Dev. 14, 1261–1268 (2000).
Kirkpatrick, D. T., Wang, Y.-H., Dominska, M., Griffith, J. D. & Petes, T. D. Control of meiotic recombination and gene expression in yeast by a simple repetitive DNA sequence that excludes nucleosomes. Mol. Cell. Biol. 19, 7661 (1999).
Bowcock, A. M. et al. High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368, 455. https://doi.org/10.1038/368455a0 (1994).
Sahoo, L. et al. Preliminary genetic linkage map of Indian major carp, Labeo rohita (Hamilton 1822) based on microsatellite markers. J. Genet. 94, 271–277. https://doi.org/10.1007/s12041-015-0528-7 (2015).
Deback, C. et al. Utilization of microsatellite polymorphism for differentiating herpes simplex virus type 1 strains. J. Clin. Microbiol. 47, 533–540. https://doi.org/10.1128/JCM.01565-08 (2009).
Sahoo, L. et al. Limited genetic differentiation in Labeo rohita (Hamilton 1822) populations as revealed by microsatellite markers. Biochem. Syst. Ecol. 57, 427–431. https://doi.org/10.1016/j.bse.2014.09.014 (2014).
Li, Y.-C., Korol, A. B., Fahima, T. & Nevo, E. Microsatellites within genes: structure, function, and evolution. Mol. Biol. Evol. 21, 991–1007. https://doi.org/10.1093/molbev/msh073 (2004).
Duke, G. M., Osorio, J. E. & Palmenberg, A. C. Attenuation of Mengo virus through genetic engineering of the 5′ noncoding poly(C) tract. Nature 343, 474. https://doi.org/10.1038/343474a0 (1990).
Barr, J. N., Whelan, S. P. & Wertz, G. W. cis-Acting signals involved in termination of vesicular stomatitis virus mRNA synthesis include the conserved AUAC and the U7 signal for polyadenylation. J. Virol. 71, 8718 (1997).
Yamada, N. et al. Genetic organization and diversity of the 3′ noncoding region of the hepatitis C virus genome. Virology 223, 255–261. https://doi.org/10.1006/viro.1996.0476 (1996).
García-Barreno, B., Delgado, T. & Melero, J. A. Oligo(A) sequences of human respiratory syncytial virus G protein gene: assessment of their genetic stability in frameshift mutants. J. Virol. 68, 5460 (1994).
Mudunuri, S. B. & Nagarajaram, H. A. IMEx: imperfect microsatellite extractor. Bioinformatics 23, 1181–1187. https://doi.org/10.1093/bioinformatics/btm097 (2007).
Chen, M. et al. Similar distribution of simple sequence repeats in diverse completed Human Immunodeficiency Virus Type 1 genomes. FEBS Lett. 583, 2959–2963. https://doi.org/10.1016/j.febslet.2009.08.004 (2009).
Alam, C. M., George, B., Sharfuddin, C., Jain, S. K. & Chakraborty, S. Occurrence and analysis of imperfect microsatellites in diverse potyvirus genomes. Gene 521, 238–244. https://doi.org/10.1016/j.gene.2013.02.045 (2013).
Wu, X., Zhou, L., Zhao, X. & Tan, Z. The analysis of microsatellites and compound microsatellites in 56 complete genomes of Herpesvirales. Gene 551, 103–109. https://doi.org/10.1016/j.gene.2014.08.054 (2014).
Hall, T. A. Nucleic Acids Symposium Series 95–98 (Information Retrieval Ltd., c1979-c2000, London, 1999).
Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protocols Bioinform. https://doi.org/10.1002/0471250953.bi0203s00 (2002).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular cloning: a laboratory manual. (Cold spring harbor laboratory press, 1989).
Torfason, E. G. & Guðnadóttir, S. Polymerase chain reaction for laboratory diagnosis of orf virus infections. J. Clin. Virol. 24, 79–84. https://doi.org/10.1016/S1386-6532(01)00232-3 (2002).
Sanger, F., Nicklen, S. & Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. 74, 5463 (1977).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Singh, A. K., Alam, C. M., Sharfuddin, C. & Ali, S. Frequency and distribution of simple and compound microsatellites in forty-eight Human papillomavirus (HPV) genomes. Infect. Genet. Evol. 24, 92–98. https://doi.org/10.1016/j.meegid.2014.03.010 (2014).
Ouyang, Q. et al. High GC content of simple sequence repeats in Herpes simplex virus type 1 genome. Gene 499, 37–40. https://doi.org/10.1016/j.gene.2012.02.049 (2012).
Kofler, R., Schlötterer, C., Luschützky, E. & Lelley, T. Survey of microsatellite clustering in eight fully sequenced species sheds light on the origin of compound microsatellites. BMC Genomics 9, 612. https://doi.org/10.1186/1471-2164-9-612 (2008).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. https://doi.org/10.1093/bioinformatics/btx198 (2017).
Jakupciak, J. P. & Wells, R. D. Genetic instabilities in (CTGCAG) repeats occur by recombination. J. Biol. Chem. 274, 23468–23479 (1999).
George, B., Gnanasekaran, P., Jain, S. K. & Chakraborty, S. Genome wide survey and analysis of small repetitive sequences in caulimoviruses. Infect. Genet. Evol. 27, 15–24. https://doi.org/10.1016/j.meegid.2014.06.018 (2014).
Hoa, T. T. et al. Genotypic variations in tandem repeat DNA segments between ribonucleotide reductase subunit genes of white spot syndrome virus (WSSV) isolates from Vietnam. Diseases in Asian Aquaculture, Asian Fisheries Society, Manila, 395–403 (2005).
Kiatpathomchai, W. et al. Target for standard Thai PCR assay identical in 12 white spot syndrome virus (WSSV) types that differ in DNA multiple repeat length. J. Virol. Methods 130, 79–82. https://doi.org/10.1016/j.jviromet.2005.06.006 (2005).
Pradeep, B., Shekar, M., Gudkovs, N., Karunasagar, I. & Karunasagar, I. Genotyping of white spot syndrome virus prevalent in shrimp farms of India. Dis. Aquat. Org. 78, 189–198. https://doi.org/10.3354/dao01878 (2008).
Pradeep, B., Shekar, M., Karunasagar, I. & Karunasagar, I. Characterization of variable genomic regions of Indian white spot syndrome virus. Virology 376, 24–30. https://doi.org/10.1016/j.virol.2008.02.037 (2008).
Tan, Y. et al. Molecular detection of three shrimp viruses and genetic variation of white spot syndrome virus in Hainan Province, China, in 2007. J. Fish Dis. 32, 777–784. https://doi.org/10.1111/j.1365-2761.2009.01055.x (2009).
Kang, H. H. & Lu, C. P. Comparison of variable region genes of shrimp White Spot Syndrome Virus (WSSV) in different areas in China. Chin. J. Virol. 23, 490–493 (2007).
Szpara, M. L. et al. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 88, 1209–1227. https://doi.org/10.1128/JVI.01987-13 (2014).
Burrel, S. et al. Molecular characterization of herpes simplex virus type 2 (HSV-2) strains by analysis of microsatellite polymorphism. J. Clin. Microbiol. 51, 3616–3623 (2013).
Avarre, J.-C. et al. Investigation of Cyprinid herpesvirus-3 genetic diversity by a multi-locus variable number of tandem repeats analysis. J. Virol. Methods 173, 320–327. https://doi.org/10.1016/j.jviromet.2011.03.002 (2011).
Achour, A. et al. Length variability of telomeric repeat sequences of human herpesvirus 6 DNA. J. Virol. Methods 159, 127–130. https://doi.org/10.1016/j.jviromet.2009.03.002 (2009).
Houng, H.-S.H. et al. Adenovirus microsatellite reveals dynamics of transmission during a recent epidemic of human adenovirus serotype 14 infection. J. Clin. Microbiol. 47, 2243 (2009).
Segarra, A. et al. Detection and description of a particular Ostreid herpesvirus 1 genotype associated with massive mortality outbreaks of Pacific oysters, Crassostrea gigas, in France in 2008. Virus Res. 153, 92–99. https://doi.org/10.1016/j.virusres.2010.07.011 (2010).
Renault, T. et al. Genotyping of a microsatellite locus to differentiate clinical Ostreid herpesvirus 1 specimens. Vet. Res. 45, 3. https://doi.org/10.1186/1297-9716-45-3 (2014).
Spatz, S. J. & Silva, R. F. Polymorphisms in the repeat long regions of oncogenic and attenuated pathotypes of Marek’s disease virus 1. Virus Genes 35, 41–53. https://doi.org/10.1007/s11262-006-0024-5 (2007).
Atia, M. A. M., Osman, G. H. & Elmenofy, W. H. Genome-wide in silico analysis, characterization and identification of microsatellites in Spodoptera littoralis Multiple nucleopolyhedrovirus (SpliMNPV). Sci. Rep. 6, 33741. https://doi.org/10.1038/srep33741 (2016).
Chen, M., Tan, Z., Zeng, G. & Zeng, Z. Differential distribution of compound microsatellites in various Human Immunodeficiency Virus Type 1 complete genomes. Infect. Genet. Evol. 12, 1452–1457. https://doi.org/10.1016/j.meegid.2012.05.006 (2012).
Alam, C. M., Singh, A. K., Sharfuddin, C. & Ali, S. Incidence, complexity and diversity of simple sequence repeats across potexvirus genomes. Gene 537, 189–196. https://doi.org/10.1016/j.gene.2014.01.007 (2014).
Alam, C. M., Singh, A. K., Sharfuddin, C. & Ali, S. Genome-wide scan for analysis of simple and imperfect microsatellites in diverse carlaviruses. Infect. Genet. Evol. 21, 287–294. https://doi.org/10.1016/j.meegid.2013.11.018 (2014).
Alam, C. M., Singh, A. K., Sharfuddin, C. & Ali, S. In-silico analysis of simple and imperfect microsatellites in diverse tobamovirus genomes. Gene 530, 193–200. https://doi.org/10.1016/j.gene.2013.08.046 (2013).
George, B., Mashhood Alam, C., Jain, S. K., Sharfuddin, C. & Chakraborty, S. Differential distribution and occurrence of simple sequence repeats in diverse geminivirus genomes. Virus Genes 45, 556–566. https://doi.org/10.1007/s11262-012-0802-1 (2012).
Kruglyak, S., Durrett, R., Schug, M. D. & Aquadro, C. F. Distribution and abundance of microsatellites in the yeast genome can be explained by a balance between slippage events and point mutations. Mol. Biol. Evol. 17, 1210–1219. https://doi.org/10.1093/oxfordjournals.molbev.a026404 (2000).
Fadda, Z., Daròs, J. A., Flores, R. & Duran-Vila, N. Identification in eggplant of a variant of citrus exocortis viroid (CEVd) with a 96 nucleotide duplication in the right terminal region of the rod-like secondary structure. Virus Res. 97, 145–149. https://doi.org/10.1016/j.virusres.2003.08.002 (2003).
Liti, G. & Louis, E. J. Yeast evolution and comparative genomics. Annu. Rev. Microbiol. 59, 135–153. https://doi.org/10.1146/annurev.micro.59.030804.121400 (2005).
Karaoglu, H., Lee, C. M. Y. & Meyer, W. Survey of simple sequence repeats in completed fungal genomes. Mol. Biol. Evol. 22, 639–649. https://doi.org/10.1093/molbev/msi057 (2005).
Martin, L. R., Neal, Z. C., McBride, M. S. & Palmenberg, A. C. Mengovirus and encephalomyocarditis virus poly(C) tract lengths can affect virus growth in murine cell culture. J. Virol. 74, 3074–3081. https://doi.org/10.1128/jvi.74.7.3074-3081.2000 (2000).
McNiece, I. Interleukin-3 and the colony-stimulating factors. In Cytokines in Health and Disease 2nd edn 41–53 (Marcel Dekker, Ann Arbor, 1997).
Farner, N. L. Interleukin-2: molecular and clinical aspects. In Cytokines Health and Diseases (Marcel Dekker, Ann Arbor, 1997).
Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721. https://doi.org/10.1038/nrc1187 (2003).
Di Rienzo, A. et al. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. U.S.A. 91, 3166–3170 (1994).
Karlin, S., Campbell, A. M. & Mrázek, J. Comparative DNA analysis across diverse genomes. Annu. Rev. Genet. 32, 185–225. https://doi.org/10.1146/annurev.genet.32.1.185 (1998).
Zhao, X. et al. Coevolution between simple sequence repeats (SSRs) and virus genome size. BMC Genomics 13, 435–435. https://doi.org/10.1186/1471-2164-13-435 (2012).
Treco, D. & Arnheim, N. The evolutionarily conserved repetitive sequence d(TG.AC)n promotes reciprocal exchange and generates unusual recombinant tetrads during yeast meiosis. Mol. Cell. Biol. 6, 3934–3947 (1986).
Wahls, W. P., Wallace, L. J. & Moore, P. D. The Z-DNA motif d(TG)30 promotes reception of information during gene conversion events while stimulating homologous recombination in human cells in culture. Mol. Cell. Biol. 10, 785–793 (1990).
Tachida, H. & Iizuka, M. Persistence of repeated sequences that evolve by replication slippage. Genetics 131, 471–478 (1992).
Usdin, K. The biological effects of simple tandem repeats: lessons from the repeat expansion diseases. Genome Res. 18, 1011–1019. https://doi.org/10.1101/gr.070409.107 (2008).
Steinhauer, D. A. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258, 1–20. https://doi.org/10.1006/viro.1999.9716 (1999).
Hahn, H. & Palmenberg, A. C. Encephalomyocarditis viruses with short poly(C) tracts are more virulent than their mengovirus counterparts. J. Virol. 69, 2697 (1995).
Perdue, M. L., Garcia, M., Senne, D. & Fraire, M. Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res. 49, 173–186. https://doi.org/10.1016/S0168-1702(97)01468-8 (1997).
Karki, M., Kumar, A., Arya, S., Ramakrishnan, M. A. & Venkatesan, G. Poxviral E3L ortholog (Viral Interferon resistance gene) of orf viruses of sheep and goats indicates species-specific clustering with heterogeneity among parapoxviruses. Cytokine 120, 15–21. https://doi.org/10.1016/j.cyto.2019.04.001 (2019).
Acknowledgments
BPS is thankful to the University Grant Commission (UGC, Govt India) for providing a Ph.D. fellowship as financial support.
Author information
Authors and Affiliations
Contributions
B.P.S. and A.S. have collected the samples. B.P.S. and P.M. have performed all the experiment and analysis. R.R.S. has analysed and created Ciros figure. D.N. managed the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sahu, B.P., Majee, P., Singh, R.R. et al. Comparative analysis, distribution, and characterization of microsatellites in Orf virus genome. Sci Rep 10, 13852 (2020). https://doi.org/10.1038/s41598-020-70634-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70634-6
- Springer Nature Limited
This article is cited by
-
Recent advances in diagnostic approaches for orf virus
Applied Microbiology and Biotechnology (2023)
-
Insights into genome plasticity and gene regulation in Orientia tsutsugamushi through genome-wide mining of microsatellite markers
3 Biotech (2023)