
Abstract
The lipidome of mammalian cells not only contain sphingomyelin (SM) but also, as a minor component, dihydrosphongomyelin (DHSM), in which the double bond at C4–C5 in the sphingosine base is reduced to a single-bond linkage. It has been indicated that DHSM forms ordered domains more effectively than SM due to its greater potential to induce intermolecular hydrogen bonds. However, direct information on partition and dynamic behaviors of DHSM in raft-like liquid-ordered (Lo) and non-raft-like liquid-disordered (Ld) phase-segregated membranes has been lacking. In the present study, we prepared fluorescent derivatives of DHSM and compared their behaviors to those of fluorescent SM and phosphatidylcholine (PC) derivatives. Fluorescence microscopy showed that DHSM is more preferentially localized to the Lo domains in the Lo/Ld phase-segregated giant unilamellar vesicles than SM and PC. Most importantly, diffusion coefficient measurements indicated that DHSM molecules form DHSM-condensed assembly inside the SM-rich Lo domain of the SM/dioleoylphosphatidylcholine/cholesterol system even when DHSM accounts for 1–3.3 mol% of total lipids. Such heterogeneous distribution of DHSM in the SM-rich Lo domains was further confirmed by inter-lipid FRET experiments. This study provides new insights into the biological functions and significance of minor component DHSM in lipid rafts.
Similar content being viewed by others

Introduction
In cell membranes, lipid rafts construct a platform for various important biological processes such as signal transduction and viral infections1,2. This finding has boosted interest in the heterogeneous distribution of sphingolipids (SLs), represented by sphingomyelin (SM, Fig. 1), in cell membranes, because distribution of SLs likely provides pivotal clues to understanding the mechanism underlying raft formation and raft-based biological events. The physicochemical properties of lipid rafts have been examined with artificial membrane systems consisting of SM, cholesterol (chol), and unsaturated phospholipids such as dioleoylphosphatidylcholine (DOPC) or palmitoyloleoylphosphatidylcholine (POPC), because these mixtures undergo macroscopic phase separation between raft-like liquid-ordered (Lo) and non-raft-like liquid-disordered (Ld) domains3,4,5,6,7,8. In order to understand dynamic and partition behaviors of SMs in artificial and biological membranes, fluorescent probes of SM have been essential and indispensable. However, direct information regarding diffusion and localization of SM has been difficult to obtain, owing to the lack of appropriate SM probes; the currently available fluorescent SM analogs, in which fluorophores are attached to the acyl chain, favor Ld domains rather than Lo domains in phase-separated artificial membranes9. Recently, we developed excellent fluorescent derivatives of SM: 488neg-SM and 594neg-SM (inclusively termed neg-SMs; Fig. 1)10, which reproduce partition and dynamic behaviors of native SM and thus enable the visualization of SM in artificial and biological membranes10,11,12.

Structures of dipalmitoylphosphatidylcholine (DPPC), palmitoylsphingomyelin (pSM), palmitoyldihydrosphingomielin (DHpSM), and their fluorescent derivatives, 488neg- and 594neg-DPPCs (neg-DPPCs), 488neg- and 594neg-pSMs (neg-pSMs), and 488neg- and 594neg-DHpSMs (neg-DHpSMs). While fluorescently labeled stearoyl-SMs (C18:0) were previously reported10, the lengths of the lipid acyl chains in this study were unified as palmitoylate (C16:0).
Dihydrosphingomyelin (DHSM, Fig. 1), in which the double bond between C4 and C5 of SM is reduced to a single bond, is a minor constituent of sphingolipids. In fact, it accounts for only 5–10% of all SMs in normal cells13,14. On the other hand, DHSM is the major phospholipid of the human lens membranes15, and a substantial amount of DHSM is contained in the HIV viral membrane16. Notably, several recent studies have indicated that DHSM has a higher potential to provide intermolecular hydrogen bonds than the usual type of SM17,18. For example, palmitoyl-SM (pSM) shows a main transition at 41.2 °C in its pure bilayer form19, while its dihydro-congener (DHpSM) shows this at 47.7 °C17,20,21. Concerning the domain formation of DHSM, fluorescent quenching measurements showed that DHpSM/chol bilayers form more ordered domains than pSM/chol bilayers22. In addition, DHSMs are more likely than SMs to undergo lateral phase separation from DOPC membranes23 and may thus, contribute to the formation of laterally condensed domains in biomembranes24. These results considered in combination suggest that DHSM forms more condensed and ordered domains than SM via effective hydrogen bond formation.
However, the underlying reason for cell membranes containing DHSM is currently not fully understood. Hence, to scrutinize the membrane properties of DHSM and its functions in lipid rafts, the development of new fluorescent derivatives that reproduce the behaviors of DHSM is desirable. In this study, applying the procedure used for the development of fluorescent SMs10, we prepared novel fluorescent derivatives of DHSM (488neg-DHpSM and 594neg-DHpSM, inclusively termed neg-DHpSMs; Fig. 1) and compared their properties with those of fluorescent SM and PC to discuss the functional role of DHSM in ordered membranes.
Results and discussion
Preparation of fluorescent lipid analogs
As mentioned above, we reported fluorescent SMs (Fig. 1) that show quite similar partition and dynamic behaviors of native SM10. Our strategy for their development was to connect hydrophilic fluorophores ATTO488 and ATTO594 to the polar head of SM via a hydrophilic nonaethylene glycol (neg) linker so as to place the fluorophores away from the membrane surface toward the bulk aqueous phase. Hence, based on this strategy, we first prepared DHpSM via catalytic hydrogenation of pSM and synthesized two kinds of fluorescent DHpSM, 488neg- and 594neg-DHpSM (Fig. 2). In this study, we adopted palmitoyl (C16:0) congeners of SM and DHSM (pSM and DHpSM, respectively) for preparation of fluorescent derivatives while stearoylated SM was used in our previous report10. For comparison, we also prepared 488neg- and 594neg-DPPCs in a similar manner.

Preparation of 488neg- and 594neg-DHpSM.
Partition and dynamic behavior of fluorescent lipids in ternary GUVs
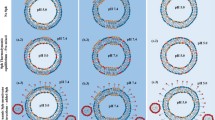
First, we examined the distribution of neg-DHpSMs, neg-pSMs, and neg-DPPCs in the Lo/Ld phase-separated giant unilamellar vesicles (GUVs) using fluorescence microscopy (Fig. 3). Here, we prepared three kinds of ternary GUVs consisting of DHpSM/DOPC/chol, pSM/DOPC/chol, and DPPC/DOPC/chol (1:1:1 molar ratio), which contain neg-DHSMs, neg-pSMs, and neg-DPPCs, respectively. The Ld phases were labeled by TexasRed-DPPE (TXred-DPPE) or 488neg-DOPC10,25,26.

Fluorescence micrographs of ternary-component GUVs consisting of (left) pSM/DOPC/chol, (center) DHpSM/DOPC/chol, and (right) DPPC/DOPC/chol at a molar ratio of 1:1:1. These samples contain 0.2 mol% neg-pSMs, neg-DHpSM, or neg-DPPC. The Ld phase was labeled by 0.2 mol% 488neg-DOPC or TXred-DPPE. All of these samples underwent phase separation between the Lo and Ld domains. A bar indicates 10 μm. The brightness and contrast have been enhanced for clarity.
As a result, all neg-pSMs, neg-DHpSMs, and neg-DPPCs showed contrasting partition behavior compared to the Ld markers (top and middle panels in Fig. 3), demonstrating that these fluorescent lipid analogs were favorably incorporated into the Lo domains. We also randomly selected 20–30 GUVs for each sample and calculated the average intensity ratios of the Lo to the Ld domains (Table 1). Notably, DHpSM is most preferentially localized in the Lo domains, followed by pSM. The lowest preference of DPPC for the Lo domains seems reasonable, since SMs bearing additional amide and hydroxy groups have higher ability to form intermolecular hydrogen bonds than PC27. To further explore the difference between DHpSM and pSM, we compared diffusion coefficients of 488neg-pSM and 488neg-DHpSM in the GUVs by fluorescence correlation spectroscopy (FCS) (Table 1). As expected, 488neg-DHpSM in DHpSM/DOPC/chol ternary GUVs showed a significantly slower diffusion in the Lo domain compared with 488neg-SM in pSM/DOPC/chol GUVs. These distribution and diffusion data can be accounted for by the notion that DHSM forms more ordered and condensed domains than SM due to its stronger intermolecular interaction, thus preferentially accumulating in the Lo domain.
Next, we further explored the distribution of 488neg-pSM and 488neg-DHpSM in inconsistent GUV systems: namely, 488neg-pSM (0.2 mol% of total lipids) in DHpSM/DOPC/chol GUVs, and 488neg-DHpSM (0.2 mol% of total lipids) in pSM/DOPC/chol GUVs (Table 1). Interestingly, the inter-phase distribution ratio of 488neg-DHpSM in pSM/DOPC/chol (4.2 ± 0.3) is significantly higher than that of 488neg-pSM in pSM/DOPC/chol (3.0 ± 0.1), and is similar to that of 488neg-DHpSM in DHpSM/DOPC/chol (4.4 ± 0.4; Table 1). These results clearly show that neg-DHpSM is effectively concentrated in the Lo domains, regardless of whether the main constituent of the Lo domain is pSM or DHpSM. To explain these results, we speculated that DHpSM molecules, even at such a low concentration, can form DHpSM-condensed assembly in the SM-rich Lo domain due to their higher propensity for intermolecular interaction and thus, they efficiently distribute in the Lo domains. If this speculation is valid, the diffusion coefficient of 488neg-DHpSM should be smaller than that of 488neg-pSM in pSM/DOPC/chol because diffusion coefficient is inversely related to the aggregation size28. However, in the Lo phase, the observed diffusion coefficient of 488neg-DHpSM (0.61 ± 0.07 μm2/s, Table 1) was not significantly different from that of 488neg-pSM in pSM/DOPC/chol (0.72 ± 0.06 μm2/s, Table 1). This is probably because the concentration of 488neg-DHpSM used for FCS measurements (0.002 mol%) is much lower than that for the determination of the Lo/Ld distribution ratio (0.2 mol%), which may reduce the clustering of 488neg-DHpSM under the FCS measurement conditions.
Formation of the DHpSM-condensed assembly in the Lo domains
To gain evidence for the formation of the DHpSM-condensed assembly inside the pSM-rich Lo domains, we prepared quaternary-component GUVs, which contain DHpSM as a minor constituent (1–3.3 mol% of total lipids), and measured diffusion coefficients of 488neg-pSM and 488neg-DHpSM (Table 2). In this experiment, we assumed two scenarios. One is that the minor component DHpSM is miscible in and dispersed into the pSM-rich Lo domains at a molecular level and therefore, both 488neg-pSM and 488neg-DHpSM have similar diffusion coefficients. The other is that the minor component DHpSM forms DHpSM-condensed assembly inside the pSM-rich Lo domains and thus, 488neg-DHpSM residing in the assembly has a decreased diffusion coefficient compared with 488neg-pSM.
The results showed that the diffusion coefficient of 488neg-DHpSM was significantly smaller than that of 488neg-pSM (Table 2), supporting the latter scenario. Importantly, 488neg-DHpSM diffusion decreased as the content of DHpSM increased, while 488neg-pSM diffusion was almost constant, irrespective of the content of DHpSM (Table 2). This means that the addition of DHpSM (< 3.3 mol% of all lipids) does not increase the overall order of the pSM-rich Lo domains because the diffusion coefficient of 488neg-pSM did not change significantly (Table 2). Therefore, it is most likely that even a small amount of DHpSM (1–3.3 mol% of total lipids) can form the DHpSM-condensed assembly in the pSM-rich Lo domains, thereby reducing the diffusion of only DHpSM without enhancing the overall order of the pSM-rich Lo domains. In other words, DHpSM molecules are heterogeneously distributed in the pSM-rich Lo domains at a molecular level.
We roughly estimated the aggregation sizes of pSM and DHpSM in the Lo phase from their diffusion coefficients using the Saffman–Delbrück formula (see “Materials and Methods” section)29,30. Consequently, the radius of the DHpSM-condensed assembly (ranging from 4 ± 2 nm to 18 ± 9 nm) is significantly larger than that of pSM (ranging from 0.9 ± 0.6 nm to 1.7 ± 1.4 nm), and the size the DHpSM-condensed assembly is getting larger with the increasing molar ratio of DHpSM (Table 2).
Figure 4 schematically summarizes the results of current diffusion experiments. DHpSM molecules are heterogeneously distributed and form DHpSM-condensed assembly in the pSM-rich Lo domains. As the mole ratio of DHpSM increases, the assembly size also increases, which reduces the diffusion of DHpSM. Meanwhile, the diffusion of pSM in the pSM-rich Lo domains is hardly affected by the presence of the DHpSM assembly.

Schematic drawing of the formation of DHpSM-condensed assembly in the Lo domain. The blue and red heads correspond to pSM and DHpSM, respectively. Orange membrane regions indicate the DHpSM-condensed assembly (see text for details).
The formation of the DHpSM-condensed assembly inside the SM-rich Lo domains was further confirmed by inter-lipid fluorescent energy transfer (inter-lipid FRET)11 using pSM/30 mol% chol binary membrane, which forms the homogeneous Lo phase31. Because fluorophores, ATTO488 and ATTO594, are an excellent donor–acceptor FRET pair, we included 488neg-pSM/594neg-pSM (neg-pSMs) pair or 488neg-DHpSM/594neg-DHpSM (neg-DHpSMs) pair in the pSM/chol binary membrane and, measured the fluorescence spectra. As shown in Fig. 5a, neg-DHpSMs FRET pair (red curve) showed a weaker emission intensity at 525 nm (namely a stronger FRET quenching) than neg-pSMs FRET pair (black curve). Note that 488neg-pSM and 594neg-pSM should be randomly distributed in this pSM/chol binary membrane because neg-SMs are reported to reproduce the behaviors of native SM in membranes10. The random distribution of neg-pSMs in pSM/chol membrane is also supported by Fig. 5a (see Fig. 5 legend for detail). Therefore, a stronger FRET quenching observed for neg-DHpSMs pair indicates the formation of DHpSM-condensed assembly in the pSM/chol Lo membrane, thereby supporting the above diffusion data.

Fluorescent spectra of (a) pSM/chol (70:30 by moles) and (b) pSM/DHpSM/chol (66.5:3.5:30 by moles) multi-lamellar vesicles. These samples contain 488neg-pSM/594neg-pSM FRET pair (neg-pSMs; black curve) or 488neg-DHpSM/594neg-DHpSM FRET pair (neg-DHpSMs; red curve). The content of each fluorescent SM is 0.4 mol%. Fluorescent measurements were repeated five and four times for the pSM/chol and pSM/DHpSM/chol samples, respectively, and averaged spectra are shown. Error bars show standard errors. The vertical axes indicate the normalized intensity, in which emission peak intensity at 525 nm of the acceptor-free sample was set to 1. Therefore, (1 − normalized peak intensity at 525 nm) corresponds to the FRET efficiency. Panel (a) shows that the FRET efficiency for 488neg-pSM/594neg-pSM pair in pSM/chol is ca. 50%. On the other hand, applying Refs.32,33 using the Förster-radius (5.7 nm) and acceptor molar ratio (0.4 mol%), the FRET efficiency between randomly distributed donor and acceptor is calculated to be ca. 60%. This is roughly consistent with the observed FRET efficiency for 488neg-pSM/594neg-pSM (ca, 50%), thus supporting their random distribution in pSM-chol membrane.
In addition, we measured the same spectra using pSM/DHpSM/chol (66.5:3.5:30) membrane (Fig. 5b). In this membrane system, the size of DHpSM assembly should be larger than that in pSM/chol membrane, and thus, the average distance between neg-DHpSMs FRET pair is expected to be elongated as compared in pSM/chol membrane. As expected, a significantly smaller FRET quenching (larger fluorescent intensity) was observed for pSM/DHpSM/chol system (Fig. 5b, red curve) compared with pSM-chol membrane (Fig. 5a, red curve), further confirming the formation of DHpSM cluster in the Lo membrane.
Preferential incorporation of DHpSM into detergent-resistant membrane domains compared to incorporation of pSM in horse erythrocyte ghost membranes
To contribute to the application of fluorescent DHpSM to biological membrane systems, we performed a partitioning assay using horse erythrocyte ghost membranes. In the assay, 488neg-DHpPC or 488neg-pSM was incorporated into the erythrocyte ghost membranes, which were fixed on a glass-bottom dish, and then the membranes were treated with detergent (Triton X-100) at 0 °C. Because the fluid membrane fractions are dissolved in the Triton X-100, only the detergent-resistant membrane (DRM) fractions, which are putative lipid rafts, are obtained on the glass-bottom dish10. In fact, erythrocyte ghosts showed rough membrane surfaces after detergent treatment owing to solubilization of the fluid membrane fractions (insertions in Fig. 6). Here, we randomly selected about 50 erythrocyte ghosts before and after detergent treatments, respectively, and compared their average fluorescent intensities (Fig. 6). This analysis shows mole fractions of fluorescent lipid derivatives incorporated into the DRMs. Consequently, although both fluorescent lipids were remained in the DRM fractions, 488neg-DHpPC showed a significantly higher distribution ratio than 488neg-pSM. Although it is reported that DRMs are not completely consistent with lipid rafts34,35, this result suggests that DHSM is favorably incorporated into lipid rafts in biomembranes, more than SM; this is consistent with the preceding GUV observation.

Fluorescent micrographs of (top) 488neg-pSM and (bottom) 488neg-DHpSM-labeled erythrocyte ghosts membranes (left) before and (right) after Triton X-100 treatment. The intensity ratio of after Triton X-100 treatment (I+TX) to before treatment (Icontrol) was described at the left-side of the corresponding micrographs. The insertion of each panel shows magnification of the region indicated by the dashed square.
Conclusion
Recently, it has been indicated that DHSM forms raft-like ordered domains more effectively than SM due to its greater potential to induce intermolecular hydrogen bonds36. Thus, we expected that DHSM would be a crucial molecule for raft formation and raft-based biological functions. However, owing to the lack of appropriate fluorescent probes, information regarding the partition and dynamic behaviors of DHSM has been difficult to obtain. In the present study, we synthesized promising fluorescent derivatives of DHpSMs based on our recently developed molecular design10 and directly observed the distribution and diffusion behavior of DHpSM in Lo/Ld phase-separated GUVs.
Fluorescence microscopy observation disclosed that in Lo/Ld phase-separated GUVs, DHpSM highly favors the Lo domains more than SM (Table 1). The FCS measurements (Table 2) further demonstrated that the diffusion coefficient of 488neg-DHpSM was significantly lower than that of 488neg-pSM in four-component GUVs consisting of SM, DOPC, chol, and a minor amount of DHSM (1–3.3 mol% of total lipids). This strongly suggests that DHpSM, even at a small molar ratio compared with pSM, is not completely miscible in pSM, forming DHpSM-rich aggregation in the pSM-rich Lo domain. The formation of the DHpSM-condensed assembly in the Lo domain was further confirmed by the inter-lipid FRET measurements, because FRET occurred more efficiently for neg-DHpSMs pair than for neg-pSMs pair in the pSM/chol binary membrane. Since the C4–C5 link in DHSM is a single bond, while it is a double bond in SM, DHSM may be able to adopt a conformation suitable for intermolecular hydrogen-bond formation.
While most previous raft-oriented studies have exclusively used SM as a representative sphingolipid, the disposition of DHSM to the domain formation is not sufficiently understood. This is because DHSM is a minor constituent of sphingolipids and accounts for only 5–10% of all SMs in normal cells13,14. However, our current data suggest that such a small amount of DHSM can form the DHSM-condensed assembly in SM-rich lipid rafts, which may have significant roles in membrane-related biological processes. For example, it may not be far-fetched to assume that some membrane proteins are preferentially localized to higher ordered DHSM-rich aggregation in lipid rafts and exert their functions37. In this context, this study will lead to a novel understanding of the biological functions and significance of minor-component DHSM in lipid rafts.
Materials and methods
Materials
Egg-yolk sphingomyelin and DOPC were purchased from Avanti Polar Lipids (Alabaster AL). Cholesterol was purchased from Sigma Aldrich (St. Louis MO). Palmitoyl-sphingomyelin (pSM) was purified from the egg-yolk SM using high-performance liquid chromatography (HPLC) and the purity was ascertained using thin-layer chromatography, which showed a single spot38. DHpSM was prepared from pSM by catalytic hydrogenation. These lipids were dissolved in chloroform/methanol (4:1, v/v) at a concentration of 1 mg/mL and stored at − 30 °C until use. NHS esters of ATTO488 and ATTO594 were purchased from ATTO-TEC GmbH (Siegen, Germany). Texas Red-DPPE (TXred-DPPE) was purchased from Invitrogen (Eugene, OR), and 488neg-DOPC was synthesized following previous report10. These dye compounds were dissolved in chloroform/methanol (4:1, v/v) at a concentration of 50 μg/mL or 25 μg/mL and, stored in the dark at − 30 °C until use.
Preparation of fluorescent lipids
Fluorescent labeled lipids, 488neg- and 594neg-labeled DPPC, pSM, and DHpSM, were prepared from DPPC, pSM, and DHpSM, respectively, as reported previously10. In brief, ATTO488- and ATTO594-neg-azides were conjugated to propargyl-DPPC, -pSM, and -DHpSM, all of which were prepared following the previously reported method39, using the Huisgen reaction, providing the objective compounds.
488neg-pSM red solid; Rf = 0.42 (CHCl3/MeOH/H2O = 6/4/1); MS(ESI-TOF) m/z [M + 3Na − H]2+ calc. for C88H145N9Na3O25PS22+, 945.9604; found 945.9661.
594neg-pSM blue solid; Rf = 0.76 (CHCl3/MeOH/H2O = 13/9/1); MS(ESI-TOF) m/z [M + 3Na − H]2+ calc. for C104H169N9Na3O25PS22+, 1,054.0545; found 1,054.0573.
488neg-DHpSM red solid; Rf = 0.49 (CHCl3/MeOH/H2O = 6:4:1); MS(ESI-TOF) m/z [M + 3Na − H]2+ calc. for C88H147N9Na3O25PS22+, 946.9685; found 946.9686.
594neg-DHpSM blue solid; Rf = 0.68 (CHCl3/MeOH/H2O = 8/1.9/0.1); MS(ESI-TOF) m/z [M + 3Na − H]2+ calc. for C104H171N9Na3O25PS22+, 1,055.0624; found 1,055.0694.
488neg-DPPC red solid; Rf = 0.48 (CHCl3/MeOH/H2O = 6/4/1); MS(ESI-TOF) m/z [M + 3Na − H]2+ calc. for C89H146N8Na3O27PS22+, 961.4579; found 961.4556.
594neg-DPPC blue solid; Rf = 0.48 (CHCl3/MeOH/H2O = 6/4/1); MS(ESI-TOF) m/z [M + 3Na − H]2+ calc. for C105H170N8Na3O27PS22+, 1,069.5518; found 1,069.5599.
Giant unilamellar vesicle (GUV) preparation and fluorescent observation
GUVs were prepared using an electroformation method40. Briefly, appropriate amounts of the lipid solutions were mixed in a glass vial (1 mg/mL), to which small amounts of fluorescent lipid analogs (0.2 mol% of total lipids) were added. The 10-μL aliquot of the lipid solution was spread on the surface of electrodes, platinum wires (100 μm diameter), and dried under vacuum for more than 12 h. Then parallel aligned electrodes were put into Milli-Q water sandwiched between two cover glasses (24 mm × 60 mm, 0.12–0.17 mm thickness) using an open-square-shaped rubber spacer. This chamber was fixed on a temperature-controlled aluminum block kept at 70 °C (Sahara 310, Rocker Scientific Co., Ltd., Taipei, Taiwan). The sample was incubated for 60 min, applying a low frequency alternating current (AC) field (sinusoidal wave function, 10 Vpp, 10 Hz), by a function generator (20 MHz function/arbitrary waveform function generator, Agilent, Santa Clara CA). The GUVs were then cooled to 25 °C. Fluorescent observation was carried out using the BZ-X700 (Keyence, Osaka, Japan) with an air objective lens (Plan Apoλ, 60 ×, N.A. 0.95, Nikon, Tokyo, Japan). The excitation (470 nm) and detection (525 nm) wavelengths were selected by dichroic mirrors, OP-87763 (Keyence, Osaka, Japan).
Inter-lipid fluorescent resonance energy transfer (FRET) measurements
Multi-lamellar vesicles (MLVs) of pSM/chol (70:30 by moles) and pSM/DHpSM/chol (66.5:3.5:30 by moles) containing 0.4 mol% FRET donor, 488neg-pSM or 488neg-DHpSM, and 0.4 mol% FRET acceptor, 594neg-pSM or 594neg-DHpSM, were prepared by a conventional method41. Briefly, appropriate amounts of SM, DHpSM and chol (total 0.100 μmol) were dissolved in chloroform/methanol (4:1 v/v) in the presence of the FRET donor and acceptor and dried under N2 flow. The remaining organic solvent was completely removed in vacuo for > 12 h. The dried lipid films were suspended in 1 mL of Milli-Q water at 80 °C with intermittent vortexing to form MLVs. The MLV suspensions were equilibrated for 30 min at room temperature for the subsequent FRET measurements. For preparation of the acceptor-free samples, the same procedure was adopted except getting rid of the FRET acceptor. Excitation was applied at 480 nm and the emission spectra were obtained from 450 to 700 nm at an interval step of 5 nm using a FP-8300 spectrofluorometer (JASCO Corp., Tokyo, Japan). The temperature was maintained at 25 °C with a CTU-100 water circulator (JASCO Corp., Tokyo, Japan).
Fluorescent correlation spectroscopy (FCS)
FCS measurements at GUV surfaces were performed at 23 °C with a confocal microscope (LSM880, Leica microsystems, Wetzlar, Germany), with a water immersion objective lens (C-Apochomat 40 × /1.2 W Korr M27), following the protocol published previously42,43. The diffusion coefficient, D, was obtained by fitting the autocorrelation function of the time-dependent changes of the signal intensities of fluorescent probe molecules in diffraction-limited spots, G(τ), with the following equation for two-dimensional simple Brownian diffusion:
where N is the average number of fluorescent particles in the detection area. The beam waist (radius) in the focal plane w0 (= 0.185 μm) was calibrated with Rhodamine 6G, for which the diffusion coefficient is widely known. D is the diffusion coefficient, and τ is the delay time. When the FCS measurements were performed at the Lo and Ld domains, 0.002 mol% and 0.004 mol% fluorescent lipids were added to the lipid mixture, respectively. The obtained FCS curves were analyzed by the software package ZEN 2.
Size estimation of the DHpSM and pSM clusters
Cluster sizes of pSM and DHpSM in the Lo phase were estimated by diffusion coefficients of 488neg-pSM and 488neg-DHpSM, respectively. Relationship between the radius R of cylindrical membrane inclusions, such as lipids and proteins, and their diffusion coefficients D was expressed by the Saffman–Delbrück formula29,30.
Here k, T and γ are Boltzmann’s constant (1.38 × 10−23 J/K), temperature (295 K) and Euler’s constant (0.5772), respectively. η (= 0.81 Pa s) and ηw (= 1 × 10−3 Pa s) are viscosities of the Lo membrane formed in brain-SM/DOPC/chol30 and water30, respectively. h (~ 4.5 nm) is membrane thickness of the Lo phase consisting of pSM/50 mol% chol44. In this calculation, we assumed that small amounts of DHpSM (< 3.3 mol% of total lipids) did not affect the viscosity and thickness of the Lo membrane.
Preparation of horse erythrocyte ghost membranes and incorporation of fluorescent lipid analogs
Horse erythrocyte ghost membranes were prepared essentially as previously reported45. Briefly, erythrocytes obtained from horses were pelleted by centrifugation, and after the removal of the buffy coat, 100 μL of the erythrocyte pellet was resuspended in 900 μL prechilled hypotonic 5 mM NaH2PO4-Na2HPO4 buffer (pH 8.0; 5P8) and incubated on ice for 20 min, to induce cell lysis. The cells were then washed four times with a 10 × volume of prechilled 5P8 buffer, by resuspension/centrifugation (12,000 × g, 10 min, 4 °C). Then, 5P8 buffer was replaced by phosphate-buffered saline (PBS; pH 7.6) buffer to form erythrocyte ghosts.
The erythrocyte ghost membranes were labeled with 488neg-pSM and 488neg-DHpSM as previously reported10. Briefly, erythrocyte ghosts were incubated with 1 μM fluorescent lipid analog in PBS buffer at 37 °C for 20 min in the dark. In order to remove the fluorescent lipid analog which was not incorporated into membranes, the samples were washed four times with a 10 × volume of PBS by resuspension/centrifugation (12,000 × g, 10 min, 4 °C). Finally, the sample was diluted in PBS at ~ 10% (v/v). For the subsequent microscopic observation, the erythrocyte ghosts were fixed on poly-L-lysine-coated glass-base dishes. For Triton extraction, the ghost membranes fixed on the glass surface were incubated with 1% cold Triton X-100 at 0 °C for 20 min and then, washed three times with prechilled PBS.
References
Lingwood, D. & Simons, K. Lipid rafts as a membrane-organizing principle. Science 327, 46–50. https://doi.org/10.1126/science.1174621 (2010).
Van Meer, G. & Hoetzl, S. Sphingolipid topology and the dynamic organization and function of membrane proteins. FEBS Lett. 584, 1800–1805. https://doi.org/10.1016/j.febslet.2009.10.020 (2010).
Veatch, S. L. & Keller, S. L. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85, 3074–3083. https://doi.org/10.1016/S0006-3495(03)74726-2 (2003).
Marsh, D. Cholesterol-induced fluid membrane domains: a compendium of lipid-raft ternary phase diagrams. Biochim. Biophys. Acta Biomembr. 1788, 2114–2123. https://doi.org/10.1016/j.bbamem.2009.08.004 (2009).
De Almeida, R. F. M., Fedorov, A. & Prieto, M. Sphingomyelin/phosphatidylcholine/cholesterol phase diagram: boundaries and composition of lipid rafts. Biophys. J. 85, 2406–2416. https://doi.org/10.1016/S0006-3495(03)74664-5 (2003).
Ariola, F. S., Li, Z., Cornejo, C., Bittman, R. & Heikal, A. Membrane fluidity and lipid order in ternary giant unilamellar vesicles using a new Bodipy-cholesterol derivative. Biophys. J. 96, 2696–2708. https://doi.org/10.1016/j.bpj.2008.12.3922 (2009).
Bunge, A., Müller, P., Stöckl, M., Herrmann, A. & Huster, D. Characterization of the ternary mixture of sphingomyelin, POPC, and cholesterol: support for an inhomogeneous lipid distribution at high temperatures. Biophys. J. 94, 2680–2690. https://doi.org/10.1529/biophysj.107.112904 (2008).
Yasuda, T., Tsuchikawa, H., Murata, M. & Matsumori, N. Deuterium NMR of raft model membranes reveals domain-specific order profiles and compositional distribution. Biophys. J. 108, 2502–2506. https://doi.org/10.1016/j.bpj.2015.04.008 (2015).
Klymchenko, A. S. & Kreder, R. Fluorescent probes for lipid rafts: from model membranes to living cells. Chem. Biol. 21, 97–113. https://doi.org/10.1016/j.chembiol.2013.11.009 (2014).
Kinoshita, M. et al. Raft-based sphingomyelin interactions revealed by new fluorescent sphingomyelin analogs. J. Cell Biol. 216, 1183–1204. https://doi.org/10.1083/jcb.201607086 (2017).
Kinoshita, M. et al. Emphatic visualization of sphingomyelin-rich domains by inter-lipid FRET imaging using fluorescent sphingomyelins. Sci. Rep. 7, 16801. https://doi.org/10.1038/s41598-017-16361-x (2017).
Kinoshita, M., Suzuki, K. G. N., Murata, M. & Matsumori, N. Evidence of lipid rafts based on the partition and dynamic behavior of sphingomyelins. Chem. Phys. Lipids 215, 84–95. https://doi.org/10.1016/j.chemphyslip.2018.07.002 (2018).
Nyholm, T. K. M., Nylund, M. & Slotte, J. P. A calorimetric study of binary mixtures of dihydrosphingomyelin and sterols, sphingomyelin or phosphatidylcholine. Biophys. J. 84, 3138–3146. https://doi.org/10.1016/S0006-3495(03)70038-1 (2003).
Ramstedt, B., Leppimaki, P., Axberg, M. & Slotte, J. P. Analysis of natural and synthetic sphingomyelins using high-performance thin-layer chromatography. Eur. J. Biochem. 266, 997–1002. https://doi.org/10.1046/j.1432-1327.1999.00938.x (1999).
Deeley, J. M. et al. Sphingolipid distribution changes with age in the human lens. J. Lipid Res. 51, 2473–2488. https://doi.org/10.1194/jlr.R004119 (2010).
Brügger, B. et al. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. USA 103, 2641–2646. https://doi.org/10.1073/pnas.0511136103 (2006).
Epand, R. M. Cholesterol in bilayers of sphingomyelin or dihydrosphingomyelin at concentrations found in ocular lens membranes. Biophys. J. 84, 3102–3110. https://doi.org/10.1016/S0006-3495(03)70035-6 (2003).
Yasuda, T., Sazzad, M. A. A., Jäntti, N. Z., Pentikäinen, O. T. & Slotte, J. P. The influence of hydrogen bonding on sphingomyelin/colipid interactions in bilayer membranes. Biophys. J. 110, 431–440. https://doi.org/10.1016/j.bpj.2015.11.3515 (2016).
Kuikka, M., Ramstedt, B., Ohvo-Rekilä, H., Tuuf, J. & Slotte, J. P. Membrane properties of D-erythro-N-acyl sphingomyelins and their corresponding dihydrospecies. Biophys. J. 80, 2327–2337. https://doi.org/10.1016/S0006-3495(01)76203-0 (2001).
Nyholm, T., Nylund, M., Söderholm, A. & Slotte, J. P. Properties of palmitoylphosphatidylcholine, sphingomyelin, and dihydrosphingomyelin bilayer membranes as reported by different fluorescent reporter molecules. Biophys. J. 84, 987–997. https://doi.org/10.1016/S0006-3495(03)74915-7 (2003).
Nyholm, T. K. M., Nylund, M. & Slotte, J. P. A calorimetric study of binary mixtures of dihydrosphingomyelin and sterols, sphingomyelin, or phosphatidylcholine. Biophys. J. 84, 3138–3146. https://doi.org/10.1016/S0006-3495(03)70038-1 (2003).
Kuikka, M., Ramstedt, B., Ohvo-Rekilä, H., Tuuf, J. & Slotte, J. P. Membrane properties of d-erythro-N-acyl sphingomyelins and their corresponding dihydro species. Biophys. J. 80, 2327–2337. https://doi.org/10.1016/S0006-3495(01)76203-0 (2001).
Kinoshita, M., Matsumori, N. & Murata, M. Coexistence of two liquid crystalline phases in dihydrosphingomyelin and dioleoylphosphatidylcholine binary mixtures. Biochim. Biophys. Acta Biomembr. 1838, 1372–1381. https://doi.org/10.1016/j.bbamem.2014.01.017 (2014).
Vieira, C. R. et al. Dihydrosphingomyelin impairs HIV-1 infection by rigidifying liquid-ordered membrane domains. Chem. Biol. 17, 766–775. https://doi.org/10.1016/j.chembiol.2010.05.023 (2010).
Kahya, N., Scherfeld, D. & Schwille, P. Differential lipid packing abilities and dynamics in giant unilamellar vesicles composed of short-chain saturated glycerol-phospholipids, sphingomyelin and cholesterol. Chem. Phys. Lipids 135, 169–180. https://doi.org/10.1016/j.chemphyslip.2005.02.013 (2005).
Kinoshita, M., Chitose, T. & Matsumori, N. Mechanism of local anesthetic-induced disruption of raft-like ordered membrane domains. Biochim. Biophys. Acta Gen. Subj. 1863, 1381–1389. https://doi.org/10.1016/j.bbagen.2019.06.008 (2019).
Slotte, J. P. & Ramstedt, B. Membrane properties of sphingomyelin. FEBS Lett. 531, 33–37. https://doi.org/10.1016/S0014-5793(02)03406-3 (2002).
Guigas, G. & Weiss, M. Size-dependent diffusion of membrane inclusions. Biophys. J. 91, 2393–2398. https://doi.org/10.1529/biophysj.106.087031 (2006).
Saffman, P. G. & Delbrück, M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA 72, 3111–3113. https://doi.org/10.1073/pnas.72.8.3111 (1975).
Wu, Y. et al. Molecular rheometry: direct determination of viscosity in Lo and Ld lipid phases via fluorescence lifetime imaging. Phys. Chem. Phys. Chem. 15, 14986–14993. https://doi.org/10.1039/c3cp51953h (2013).
Marsh, D. Liquid-ordered phases induced by cholesterol: A compendium of binary phase diagrams. Biochim. Biophys. Acta Biomembr. 1798, 688–699. https://doi.org/10.1016/j.bbamem.2009.12.027 (2010).
Loura, M. S., Fedorov, A. & Prieto, M. Resonance energy transfer in a model system of membranes: application to gel and Liquid crystalline phases. Biophys. J. 71, 1823–1836. https://doi.org/10.1016/S0006-3495(96)79383-9 (1996).
Wolber, P. K. & Hudson, B. S. An analytic solution to the Förster energy transfer problem in two dimensions. Biophys. J. 28, 197–210. https://doi.org/10.1016/S0006-3495(79)85171-1 (1979).
Munro, S. Lipid rafts: elusive or illusive?. Cell 115, 377–388. https://doi.org/10.1016/S00928674(03)00882-1 (2003).
Sezgin, E., Levental, I., Mayor, S. & Eggeling, C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 18, 361–374. https://doi.org/10.1038/nrm.2017.16 (2017).
Nyholm, T., Nylund, M., Sõderholm, A. & Slotte, J. P. Properties of palmitoyl phosphatidylcholine, sphingomyelin, and dihydrosphingomyelin bilayer membranes as reported by different fluorescent reporter molecules. Biophys. J. 84, 987–997. https://doi.org/10.1016/S0006-3495(03)74915-7 (2003).
Costello, M. J. et al. Ultrastructural analysis of damage to nuclear fiber cell membranes in advanced age-related cataracts from India. Exp. Eye Res. 87, 147–158. https://doi.org/10.1016/j.exer.2008.05.009 (2008).
Jungalwala, F. B., Hayssen, V., Pasquini, J. M. & McCluer, R. H. Separation of molecular species of sphingomyelin by reversed-phase high-performance liquid chromatography. J. Lipid Res. 20, 579–587 (1979).
Goretta, S. A. et al. Effects of chemical modification of sphingomyelin ammonium group on formation of liquid-ordered phase. Bioorg. Med. Chem. 20, 4012–4019. https://doi.org/10.1016/j.bmc.2012.05.015 (2012).
Angelova, M. I. & Dimitrov, D. S. Liposome electroformation. Faraday Discuss. Chem. Soc. 81, 303–311. https://doi.org/10.1039/DC9868100303 (1986).
Bangham, A. D., Standish, M. M. & Watkins, J. C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 13, 238–252. https://doi.org/10.1016/S0022-2836(65)80093-6 (1965).
Korlach, J., Schwille, P. & Webb, W. W. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 96, 8461–8466. https://doi.org/10.1073/pnas.96.15.8461 (1999).
Kahya, N., Scherfeld, D., Bacia, K., Poolman, B. & Schwille, P. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 278, 28109–28115. https://doi.org/10.1074/jbc.M302969200 (2003).
Maulik, P. R. & Shipley, G. G. N-palmitoyl sphingomyelin bilayers: structure and interactions with cholesterol and dipalmitoylphosphatidylcholine. Biochemistry 35, 8025–8034. https://doi.org/10.1021/bi9528356 (1996).
Dodge, J. T., Mitchell, C. & Hanahan, D. J. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 100, 119–130. https://doi.org/10.1016/0003-9861(63)90042-0 (1963).
Acknowledgements
We thank Profs. Masahiko Annaka and Tohru Oishi (Kyushu University, Fukuoka, Japan) for their approval of use of confocal microscope and ESI–MS. This research was supported by JSPS KAKENHI (JP18K19149, JP20H04781, JP20H00405, and JP20K06590).
Author information
Authors and Affiliations
Contributions
T.K. synthesized new fluorescent lipid analogs and performed a large majority of observations using GUVs and erythrocyte ghost. M.K. conducted FCS measurements. N.M. conceived and formulated this project, and evaluated and discussed data. M.K. and N.M. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kinoshita, M., Kyo, T. & Matsumori, N. Assembly formation of minor dihydrosphingomyelin in sphingomyelin-rich ordered membrane domains. Sci Rep 10, 11794 (2020). https://doi.org/10.1038/s41598-020-68688-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68688-7
- Springer Nature Limited