
Abstract
The metal-free nitrogen-doped graphitic-carbon@graphene (Ng-C@G) is prepared from a composite of polyaniline and graphene by a facile polymerization following by pyrolysis for electrochemical oxygen reduction reaction (ORR). Pyrolysis creates a sponge-like with ant-cave-architecture in the polyaniline derived nitrogenous graphitic-carbon on graphene. The nitrogenous carbon is highly graphitized and most of the nitrogen atoms are in graphitic and pyridinic forms with less oxygenated is found when pyrolyzed at 800 °C. The electrocatalytic activity of Ng-C@G-800 is even better than the benchmarked Pt/C catalyst resulting in the higher half-wave potential (8 mV) and limiting current density (0.74 mA cm−2) for ORR in alkaline medium. Higher catalytic performance is originated from the special porous structure at microscale level and the abundant graphitic- and pyridinic-N active sites at the nanoscale level on carbon-graphene matrix which are beneficial to the high O2-mass transportation to those accessible sites. Also, it possesses a higher cycle stability resulting in the negligible potential shift and slight oxidation of pyridinic-N with better tolerance to the methanol.
Similar content being viewed by others


Introduction
To save the world from day-by-day increasing energy demands and environmental concerns, the clean, highly efficient and renewable energy technologies are immediately required to be implemented1. Among various renewable energy technologies, fuel cells (FCs) and metal-air batteries are regarded as the promising clean energy sources because of their high energy conversion efficiency and emission-free power generation2,3. However, the efficiency of those technologies is strongly depend on the reaction kinetics which are involving with4. For example, the sluggish kinetics of cathodic oxygen reduction reaction (ORR) hinders the overall performance of FCs5. Thus, FCs require a highly active and durable electrocatalysts for increasing ORR kinetics. Although, Pt-based materials have been used as the state-of-the-art electrocatalysts for ORR because of their higher current density with lower overpotential compared to other electrocatalysts6,7, but their poor durability and prohibitive cost are hampering widespread usage of FCs technology in practical life6,7.
In recent past, researchers are focusing on nitrogen-doped carbon materials (NCMs) with various extra-beneficial features as metal-free ORR catalysts which shown to be an efficient, durable and carbon monoxide (CO)-poisoning-free substitute to Pt-based catalysts4,8,9,10. Although, the previous research has shown that the significant improvement has done for ORR catalysis on NCMs, still the ORR performance needs to be improved11. There are two important approaches are strongly correlated to the improvement of the ORR electrocatalytic activity of NCMs. One is to enhance the intrinsic activity of doped nitrogen and the other one is to increase the number of active sites4. For enhancing the intrinsic activity of NCMs, they should be engineered to contain a high number of active sites with better exposure to O2 gas. Recent studies have confirmed that the graphitic- and pyridinic-N among all forms of nitrogen in NCMs are the most intrinsically active for ORR electrocatalysis12. Also, the porosity of NCMs leads to increase effective surface area and accessible active sites to electrolyte to accelerate mass transfer processes during electrochemical ORR13,14,15,16,17. For increasing active sites and porosity in NCMs, lots of preparation methods have been introduced such as chemical vapor deposition18,19, laser-induced pyrolysis20, arc-discharge21, and template-based growth22 which are involving with complicated and expensive instrumentation, critical atmosphere control and limited production. Also, they are suffering from unnecessary high cost and time-consuming approach23. Therefore, a facile and scalable method is necessary to obtain a highly active and porous NCMs for durable ORR electrocatalysis as the ideal replacement of Pt-catalysts in FCs.
The conducting polymers are widely used in various fields such as energy conversion, energy storage and sensors. Among various conducting polymers, polyaniline (PANI) is particularly interesting due to the low cost, superior stability in alkaline media, and conductivity24,25,26. Recently, PANI has also been used as the N-doping agent in metal-free electrochemical ORR catalysts preparation26,27,28,29. Although, the N-doped carbon derived from carbonized PANI upon heat treatment and has been applied as ORR catalyst26,27,28,29, but their ORR performances are not as good as other NCMs. This is probably due to the serious aggregation and less porous nature with lower amount of active sites in PANI derived carbon which lead to the low exposure to the active sites. There are only a few studies that focus on the development of N-doped carbon derived from PANI with graphene as a composite ORR catalyst30.
Graphene is a 2D sp2-carbon network which has well-known remarkable properties such as huge surface area, high mechanical strength and better electrical conductivity31,32,33,−34. Practically, the surface area of graphene-based materials is much lower than the theoretical value which is due to the self-stacking nature through π-π interaction. 3D structure engineering35,36 through composite making with polymer derived carbon37,38,39,40 is not only an advantageous strategy to prevent the self-stacking of graphene but also it facile to the electrochemical ORR37,38,41.
Considering the above mentioned observations, we have developed the scalable and facile fabrication of a composite electrocatalyst of PANI derived nitrogenous graphitic-carbon on graphene (Ng-C@G) which immobilized by polymerization followed by the pyrolysis at various temperatures for efficient ORR electrocatalysis (Scheme 1). The sponge-like with ant-cave-like carbon network are rendering sufficient exposure of graphitic- and pyridinic-N active sites in the robust and long term active catalyst. The Ng-C@G-800 demonstrates comparatively better ORR activity with more positively shifted half-wave potential (8 mV), higher limiting current density (0.74 mA cm−2) and better stability compared to commercial Pt/C (E-TEK) catalyst in alkaline electrolyte.

Schematic representation of the Ng-C@G-800 synthesis procedure.
Experimental
Catalyst synthesis
The 100 mg prepared graphene oxide (GO)42,43 was dispersed in 100 mL of 1 mol L−1 HCl ultrasonically until no visible clotted particles observed, followed by the addition of 100 µL of aniline monomer into the GO solution under ultrasonication for 3 h. Afterwards the 50 mL of ammonium peroxydisulfate [(NH4)2S2O8] (230 mg in 1 mol L−1 HCl) solution was slowly introduced (dropwise) to the mixed solution under vigorous stirring. The solution was then stirred at ~ 4 °C for 10 h to complete the polymerization reaction. Then the sample (denoted as GOPANI) was washed with deionized water till the pH reached at 7, followed by the drying in a vacuum oven for 24 h at 70 °C. Finally, the GOPANI was put into a crucible and was transferred to tube furnace for pyrolysis under N2-atmosphere at 800 °C for 3 h for producing Ng-C@G-800. For optimization of the best composition, four more Ng-C@G samples were prepared with the addition of 0, 25, 50, and 150 µL of aniline into the GO solution separately followed by the same protocol. Also, other Ng-C@G samples were prepared at various heating temperatures (200, 500, 700, and 900 °C). Then the instrumental and electrochemical characterizations were done systematically (see supporting information).
Results and discussions
Surface morphology
The field emission scanning electron microscopy (FESEM) was employed to understand the microstructure of best performing catalyst, as synthesized Ng-C@G-800. The FESEM image of Ng-C@G-800 shows that the PANI derived carbon matrix is nicely placed onto the surface of thin nanosheets-like pure RGO (prepared at 800 ºC, Figure S1a) with typical fibrillary morphology (Fig. 1a). The graphene nanosheets are embedded in the polymer network afterwards interconnected with carbon matrix through pyrolysis (Figure S1b). The magnified FESEM image (Fig. 1b) shows that the carbon matrix is in the sponge-like structure with ant-cave-like pore, although the wall thickness of sponge-wall is irregular. For better understanding, the transmission electron microscopy (TEM) was also employed and the TEM image shows as same as porous sponge-like structure (Fig. 1c). The magnified TEM image shows the layered graphitic structure of PANI derived carbon matrix (Fig. 1d) with the lattice d-spacing of 0.34 nm (Fig. 1d inset) which indicating better formation of graphitic carbon matrix44. The TEM image of before and after pyrolysis are also compared in Figure S2 which shown the thick polymer layers become porous carbon matrix upon pyrolysis. The energy-dispersive X-ray spectroscopy (EDS) spectrum shows that the Ng-C@G-800 sample mostly contains C (84.3 wt%) and O (8.8 wt%) with N (6.9 wt%) element (Fig. 1e). The elemental mapping is also showing well distribution of C, O, and N elements through the whole Ng-C@G-800 sample (Fig. 1f).

SEM (a,b), TEM (c,d) images, EDS spectrum (e) and elemental mapping (f) of Ng-C@G-800
The Ng-C@G-800 preparation with various amount of aniline addition is also observed by FESEM analysis in Fig. 2. Figure 2a shows most of the RGO surface is uncovered with visible PANI derived carbon matrix growth upon 25 µL of aniline addition. As aniline addition increases, PANI derived carbon is increasing onto the RGO surface upon 50 µL of aniline addition in Fig. 2b. In Fig. 2c, the RGO surface is fully covered by sponge-like architecture of carbon upon 100 µL of aniline addition. Further aniline addition is increased at 150 µL, the sponge-like architecture is increased with the thick wall onto RGO surface (Fig. 2d). For comparison, only PANI derived carbon surface shows thick and less-porous sponge-like agglomerates in Figure S3a. As a whole, the RGO helps to maintain the dispersion of PANI derived porous graphitic-carbon and the carbon prevents restacking of RGO by placing in between RGO sheets (Figure S3b), resulting in more accessible and utilization of active sites.

SEM images of Ng-C@G-800 at various amount of aniline addition 25 µL (a), 50 µL (b), 100 µL (c) and 150 µL (d).
Instrumental characterization
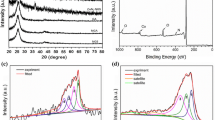
The structural variation of Ng-C@G upon various heat treatment was investigated by X-ray diffraction (XRD) in Fig. 3a. As prepared GO shows a sharp peak at 11.6° with respect to the 2θ which corresponding to the (002) plane of carbon is appeared with a lattice spacing of 0.78 nm due to presence of huge oxygen functionalities onto GO surface. Upon 200 °C heating on Ng-C@G-200, the typical peak at 2θ = 13.1° with a lattice spacing of 0.72 nm which indicating oxygen functionalities are still available in the composite45. However, upon 500 °C heating, the peak position of 002 plan for Ng-C@G-500 is shifted towards a higher angle 2θ = 23.6° with lower lattice spacing of 0.361 nm which indicating the significant number of oxygen functional groups are removed44. Upon further increasing the temperature at 700 and 800 °C, the peak of 002 plan becomes broad and shifted to higher angle at 24.2° and 25.6° on Ng-C@G-700 and Ng-C@G-800 samples, respectively, with significantly smaller lattice spacing (0.342 and 0.339 nm), indicating the residual oxygen functional groups such as epoxide and hydroxyl are removed and the intercalated structure of PANI polymer network is greatly deteriorated to make the better graphitic-carbon structure46. At 900 °C, the peak of 002 plan shifted to the lower degree (2θ = 25.1°) with increasing lattice spacing as 0.343 nm on Ng-C@G-900 sample due to the partial C−C bond breaking46.

XRD patterns at various heat treatment of Ng-C@G samples (a), Raman spectra of Ng-C@G samples which prepared at 500 to 900 °C (b) N2-adsorption desorption spectra of GO, GOPANI, RGO-800 and Ng-C@G-800 (c), and core level of N 1 s spectrum of Ng-C@G-500, Ng-C@G-700 and Ng-C@G-800 (d); insets: BJH pore size distribution (c) and various N-species percentage of corresponding samples (d).
The Raman spectra of Ng-C@G prepared from various heating treatment (500–900 °C) exhibit prominent D (at ~ 1,350 cm−1) and G (~ 1592 cm−1) bands in Fig. 3b. The intensity of D band corresponds to the sp3-carbon, whereas the G band corresponds to the E2g vibrational mode of the sp2-carbon47. Thus, the G band levelled Raman spectra shows that the D band gradually increasing up to 800 °C indicating defect density in the atomic structures of Ng-C@G-800 is increasing due to nitrogen heteroatom-doping9,15,18. However, at 900 °C, the D band is slightly lower than the 800 °C (Fig. 3b inset), indicative of less defect density in the atomic structure probably due to the removal of nitrogen-atom from graphitic hexagonal structure at high. This observation is consistent with the XRD analysis.
The porosity involves with the specific surface area (SSA) and catalytic properties of carbon-based materials48,49. The porous structure of Ng-C@G-800 can be revealed by the N2-adsorption/desorption test and compared with GO, GOPANI and RGO-800 (Fig. 3c). The hysteresis loop of all isotherms between relative pressures (P/P0) are confirming the presence of micro and mesopores in all tested samples50. The SSA (using Brunaue–Emmett–Teller method) of GOPANI (108 m2 g−1), which is significantly lower than the GO (224 m2 g−1). The lower surface area for GOPANI is attributed to the polymerization of PANI in between GO layers. This feature is also signified by comparing pore size distribution plots in the inset of corresponding figure. However, compared with all samples, the hysteresis loops in the isotherm of Ng-C@G-800 is considerably enlarged at high P/P0 region due to the significantly expanded pores upon hear treatment. As a result, the high pore distribution is observed (in corresponding inset) with higher SSA of 647 m2 g−1 which is about three times higher than the pure RGO-800 (205 m2 g−1). These results are due to the presence of sponge-like carbon matrix that substantially prevents the π-π restacking of RGO-800 nanosheets during pyrolysis. The interconnected porous network and high SSA are favorable for better mass transport at Ng-C@G-800 surface and providing more accessible active areas for electrochemical applications50,51.
Further, the X-ray photoelectron spectroscopy (XPS) was used to analyze nanoscale structure with elemental analysis of prepared samples. Mainly two peaks are appeared in XPS survey spectra of GO, GOPANI, RGO-800 and Ng-C@G samples at ~ 284 eV and ~ 532 eV which signifying the presence of C and O elements in those samples (Figure S4a)52. An additional peak at ~ 400 eV which signifying the N-element is present in GOPANI and Ng-C@G-800 samples due to the aniline addition. The atomic ratio of carbon and oxygen (C/O) increasing is an indication of oxygenated groups removal from carbon-based samples35. Thus, the value of C/O is increased from 2.1 to 3.2 for GO and GOPANI, respectively, due to addition of oxygen-free PANI. The C/O for RGO-800 (9.4) and Ng-C@G-800 (9.6) samples is significantly improved due to the substantial oxygen groups removal. The higher C/O ratio signifying the superior degree of GO reduction upon heat treatment4,53.
The core level of C1s XPS spectrum of GO is deconvoluted by three different peaks which representing the oxygen-free C=C with a peak at 284.8 eV, and oxygen-containing C–O at 287.1 and O–C=O at 288.9 eV carbon species in Figure S4b54,55. Compare to GO spectrum, the intensity of C–O and O–C=O species in GOPANI are slightly reduced, which indicating the C=C containing PANI is attached onto the GO surface with a partial condensation reaction in between –NH in PANI and –COOH/C–O in GO. The dramatic reduction of C–O and O–C=O species in RGO-800 and Ng-C@G-800 samples indicating the removal of oxygen groups due to heat treatment. At keen observation, the improved relative intensity of C–O peak in Ng-C@G-800 than the RGO-800 proves the presence of nitrogen-containing carbon (C–N) that appearing from condensation reaction and subsequent N-doping into graphene nanosheets through pyrolysis at 800 °C (Figure S4b)55,56. The high resolution N1s spectra of various heat-treated Ng-C@G are deconvoluted into three peaks corresponding to pyridinic-N at 398.5 eV, graphitic-N at 400.8 eV and oxygenated-N at ~ 402–403 eV (Fig. 3d)57. Among them, graphitic-N and pyridinic-N are critical in the formation of high-performance active sites for ORR12,58,59. As increasing the heating temperature up to 800 °C, the pyridinic-N is reduced with the increasing of graphitic-N (figure insets) while the graphitic-N is much stable than pyridinic-N and oxygenated-N at high temperatures13. However, the pyridinic-N is tremendously reduced with reducing of total N-content at Ng-C@G-900 (Figure S4c). The detail numerical analysis of elements from XPS is enlisted in Table 1 for all prepared Ng-C@G upon various heat treatments.
Electrochemical ORR
Nitrogen-active sites, 3D sponge-like structures and higher SSA containing PANI derived carbon materials exhibit an enhanced electrochemical performance27,59. Thus, various Ng-C@G samples with GOPANI and RGO-800 were tested for the ORR as metal-free electrocatalyst. The electrocatalytic activity of Ng-C@G for ORR is first evaluated by cyclic voltammetry (CV) in argon (Ar)- and O2-saturated 0.1 M KOH electrolyte as shown in Fig. 4a. In Ar-saturated electrolyte, no identical peak is observed for those samples (dotted lines) whereas a cathodic peak is appeared in a potential range of 0.9–1.0 V in O2-saturated electrolyte, implying electrocatalytic ORR activity of all samples (solid lines). As expected, the ORR onset potential (Eonset) is better at all Ng-C@G catalysts compared to GOPANI (0.9 V) and RGO-800 (0.91) signifying the presence of N-doped graphitic-carbon from PANI along with graphene enhances the ORR activity. Among all Ng-C@G catalysts, the Ng-C@G-800 has shown more positively shifted ORR peak at 0.96 V (0.94 and 0.92 V for Ng-C@G-700 and Ng-C@G-900, respectively) with highest intensity of current density. The abundant active sites of graphitic-N and pyridinic-N, and a high SSA with unique sponge-like with ant-cave-architecture play a crucial role in superior ORR performance.

The CVs for ORR at GOPANI, RGO-800 and Ng-C@G samples prepared at 700, 800 and 900 °C in Ar- and O2-saturated 0.1 M KOH electrolyte at a scan rate of 50 mV s−1 at a scale bar of 5 mA cm−2 (a), and the LSVs on RDE at same catalysts including with Pt/C catalyst in O2-saturated 0.1 M KOH electrolyte at a scan rate of 5 mV s−1 and at 1,600 rpm rotation speed (b).
The additional catalytic activity was evaluated by rotating disk electrode (RDE) for recording linear sweep voltammetry (LSV) curves conducted with the above mentioned samples in Fig. 4b. As can be seen in Fig. 4b, the Ng-C@G-800 sample shows the best ORR electrocatalytic activity, with higher positive half-wave potential (E1/2) and limiting current density (jL) among all tested catalysts including Pt/C (E-TEK). For instance, the Ng-C@G-800 catalyst exhibits 8 mV higher E1/2 and 0.74 mA cm−2 higher jL than the Pt/C catalyst (Figure S5a). The Ng-C@G-900 is poor ORR active because of lower N-content and lower graphitized sample as observed in XPS and XRD analysis. This observation suggests that the higher graphitic-N and pyridinic-N active sites with lower oxygenated-N and better graphitized-carbon is essential for better ORR catalysis60. Moreover, the interconnective ant-cave-architecture facilitates the penetration of the electrolyte inside the Ng-C@G-800, which allows an efficient mass transport at the interfacial area of electrode61. The ORR on prepared Ng-C@G-800 with various addition of aniline is also investigated by LSV (Figure S5b) which shows the Ng-C@G-800 upon 100 µL of aniline addition concomitant higher ORR in respect to both E1/2 and jL. The cyclic voltammetry of modified GCEs exhibits large redox peak current at Ng-C@G samples compared to the GOPANI and RGO-800 and highest large redox peak current at Ng-C@G-800 (Figure S6a), indicating that the Ng-C@G-800 samples have a larger electroactive surface area57. ORR result is chronological to the electroactive surface area which suggesting a good relationship with ORR to the electroactive surface area. More detailed results of ORR are presented in the Table 2 for all tested catalysts. Also, we have found the poor ORR performance at Ng-C@G-800 in 0.1 M HClO4 solution and the result is shown in Figure S6b.
Kinetics of ORR
To gain insight into the ORR on RGO-800 (Fig. 5a) and Ng-C@G-800 (Fig. 5b) catalysts which were prepared at identical conditions, the reaction kinetics are investigated by RDE at various rotation speeds. The LSV curves from both catalysts show that the jL of ORR is increased by the increasing rotating speed due to the increasing oxygen flux to the electrode surface. After addition of optimized amount of PANI derived carbon, the jL of ORR at the Ng-C@G-800 catalyst is always higher than the pure RGO-800 catalyst at any constant rpm, confirming an enhanced ORR process at Ng-C@G-800 catalyst27,62. The Koutecky–Levich (K–L) plots resultant from the corresponding LSV curves show the linear relationships between \(j_{L}^{ - 1}\) and angular velocity (ω–1/2, ω = 2π*rpm) under different potentials at 0.7–0.2 V (vs. RHE) (see K–L equation in the supporting information). Particularly, the K–L plots for Ng-C@G-800 catalyst maintained same slopes compared to the pure RGO-800 catalyst over the potential range is studied (in the corresponding Figure insets), indicating a consistent number of transferred electron (n) per O2 molecule during ORR and comparatively better first-order reaction kinetic of ORR possess on Ng-C@G-800 than RGO-800 catalyst63,64.

The RDE voltammograms on RGO-800 (a) and Ng-C@G-800 (b) catalysts at various rotation speed, the RRDE voltammograms on RGO-800, Ng-C@G-800 and Pt/C catalysts at 1,600 rpm rotation speed (c), the plots of transferred electron number and the corresponding peroxide synthesis at the cited potentials (d), LSV curves of Ng-C@G-800 and Pt/C catalysts at 1,600 rpm before and after 5,000 CV cycles (e) and the core level of N 1 s XPS spectrum of Ng-C@G-800 after 5,000 CV cycles (f); condition of voltammogram recording: in O2-saturated 0.1 M KOH electrolyte at a scan rate of 5 mV s−1, insets: the K–L plots of corresponding samples at 0.7 to 0.2 V vs. RHE (a,b), the kinetic current density comparison (c) and various N-species percentage of used Ng-C@G-800 for 5,000 cycles (f).
To further assess to the kinetics of ORR at Ng-C@G-800 and RGO-800 catalysts, rotating ring-disk electrode (RRDE) tests are performed at a rotation speed of 1,600 rpm in same electrolyte (Fig. 5c). The benchmark catalyst, 20 wt% Pt/C, is compared under identical conditions. As can be seen in Fig. 5c, all catalysts started to generate the ring current during ORR and the intensity of steady state current region indicated the degree of H2O2 generation65,66. Although, at Ng-C@G-800 catalyst, the H2O2 generation is little higher and significantly lower than that on Pt/C and pure RGO-800 catalysts, respectively, the steady state current region at disk electrode is much higher than that on both Pt/C and pure RGO-800 catalysts, which indicating the Ng-C@G-800 catalyst is comparatively better electrocatalyst than RGO-800 and Pt/C catalysts toward ORR13. This result is consistent with the relatively high kinetic current density (jk) at the Ng-C@G-800 when comparing pure RGO-800 and Pt/C (Fig. 5c inset) which calculated from an equation (see supporting information). Based on the RRDE data using Eq. (1)6,30, the n-values of Ng-C@G-800 catalyst are calculated as 3.96–4.0 over a potential range of 0.2–0.8 V, which is evidently superior to the n-values of the RGO-800 (< 3.8) and similar to the Pt/C (Fig. 5d), signifying a direct four-electron transfer ORR pathway on Ng-C@G-800 catalyst. The corresponding H2O2 synthesis (from Eq. (2)6,30) at those electrodes are also plotted along with the n-value. The average % of H2O2 synthesis is measured as 18.2, 5.3, and 5.1% for RGO-800, Ng-C@G-800 and Pt/C catalysts, respectively (Fig. 5d).
where id and ir are the disk and ring electrode currents, respectively, and N is the collection efficiency of the RRDE (0.37).
The ORR kinetics can also be investigated by the Tafel analysis (Figure S7). Typically, a Tafel plot has two slopes which are close to − 59 mV dec−1 at low and − 118 mV dec−1 at and high overpotential regions at room temperature27,33,67. The Tafel slope for Ng-C@G-800 catalyst can be obtained by plotting the logarithm of jk against respective potential and the values are calculated as 58 and 125 mV dec−1 which are close to those of Pt/C (61 and 121 mV dec−1) and much lower than RGO-800 (86 and 184 mV dec−1). This feature is consistent with other reported metal-free N-doped ORR electrocatalysts68,69, indicating the faster electron transfer at the rate-determining step on the surface of Ng-C@G-800 catalyst during ORR than those on RGO-800 and Pt/C catalysts. The overall ORR performance of Ng-C@G-800 catalyst is better over many other metal-free ORR catalysts (Table S1).
ORR stability
The Ng-C@G-800 and Pt/C catalysts are further subjected to investigate the stability and selectivity during ORR. The long term durability of Ng-C@G-800 and Pt/C for ORR is examined by cycling between the potentials cited in O2-saturated 0.1 M KOH for continuous 5,000 times as shown in Fig. 5e. After 5,000 cycles, the E1/2 of Ng-C@G-800 is slightly red shifted by 10 mV with no significant reduction of current density at steady state region which is much lower than the Pt/C, while the E1/2 of Pt/C is decreased by 25 mV with significant current density reduction under the same alkaline condition. Probably, the sponge-like with ant-cave-architecture allows the volume changes during the electrolyte penetration, which leads to the structural and the cycle stability of the Ng-C@G-80061.
The change upon cycle run in the ORR catalytic sites of Ng-C@G-800 is also investigated by core level of N 1 s XPS spectrum, which reflects the steady state ORR catalytic sites on the surface of the Ng-C@G-800 catalyst after stability test (Fig. 5f). As shown in Fig. 5f, no significant change in the total N-content by 6.6 at% which is only 4.3% of total N-content of fresh Ng-C@G-800 catalyst (6.9 at %). However, a prominent change in the pyridinic-N component which decreased from 2.35 to 2.2 at% and the graphitic-N component is remained nearly constant at 4.32 at%, whereas oxygenated-N increased from 0.18 to 0.61%. This result indicating the much stable graphitic-N sites and some of pyridinic-N sites become oxidized during stability test. This is fairly consistent with electrochemical stability test57. In addition, the fuel selectivity of the Ng-C@G-800 and Pt/C catalysts are evaluated by chronoamperometry (current vs. time) test (Figure S8). A quick decline in the relative current for the Pt/C catalyst is recorded upon 3 M methanol addition which is the inherent nature of CO-poisoning to the Pt-catalyst surface70 while methanol oxidation reaction leads to several reaction intermediates including CO. There is no notable change in the ORR current at Ng-C@G-800, implying the Ng-C@G-800 has higher fuel selectivity toward ORR than the Pt/C in presence of methanol.
Conclusion
A facile preparation method of metal-free N-doped graphitic-carbon@graphene through chemical polymerization followed by pyrolysis is developed. The PANI is used as nitrogenous graphitic-carbon source and a freestanding template to fabricate the sponge-like with ant-cave-architecture. The Ng-C@G-800 has a special porosity which is favorable to exposure of more active sites for ORR and a high flux mass transportation, resulting in a higher ORR catalytic activity with long term stability than the Pt/C catalyst. Considering the versatility of the preparation and the unique structure of graphitic-carbon, this work could be extended to prepare various low cost and superior active materials for many more electrochemical applications, such as supercapacitor, sensors and water splitting.
References
Whitesides, G. M. & Crabtree, G. W. Don’t forget long-term fundamental research in energy. Science 315, 796–798 (2007).
Steele, B. C. H. & Heinzel, A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001).
Begum, H., Ahmed, M. S., Lee, D. W. & Kim, Y. B. Carbon nanotubes-based PdM bimetallic catalysts through N4-system for efficient ethanol oxidation and hydrogen evolution reaction. Sci. Rep. 9, 11051 (2019).
Sun, J. et al. Ultrathin nitrogen-doped holey carbon@graphene bifunctional electrocatalyst for oxygen reduction and evolution reactions in alkaline and acidic media. Angew. Chem. Int. Ed. 57, 16511–16515 (2018).
Katsounaros, I., Cherevko, S., Zeradjanin, A. R. & Mayrhofer, K. J. Oxygen electrochemistry as a cornerstone for sustainable energy conversion. Angew. Chem. Int. Ed. 53, 102–121 (2014).
Ahmed, M. S., Choi, B. & Kim, Y.-B. Development of highly active bifunctional electrocatalyst using Co3O4 on carbon nanotubes for oxygen reduction and oxygen evolution. Sci. Rep. 8, 2543 (2018).
Ahmed, M. S., Begum, H. & Kim, Y.-B. Iron nanoparticles implanted metal-organic-frameworks based Fe–N–C catalysts for high-performance oxygen reduction reaction. J. Power Sources 451, 227733 (2020).
Qin, L. et al. Graphene-directed formation of a nitrogen-doped porous carbon sheet with high catalytic performance for the oxygen reduction reaction. J. Phys. Chem. C 122, 13508–13514 (2018).
Liu, R., Wu, D., Feng, X. & Müllen, K. Nitrogen-doped ordered mesoporous graphitic arrays with high electrocatalytic activity for oxygen reduction. Angew. Chem. Int. Ed. 49, 2565–2569 (2010).
Begum, H. & Kim, Y. B. Improvement of catalytic activity of platinum nanoparticles decorated carbon graphene composite on oxygen electroreduction for fuel cells. Processes 7, 586 (2019).
Dai, L. et al. Metal-free catalysts for oxygen reduction reaction. Chem. Rev. 115, 4823–4892 (2015).
Lu, Z. et al. N, B-codoped defect-rich graphitic carbon nanocages as high performance multifunctional electrocatalysts. Nano Energy 42, 334–340 (2017).
Lu, Z. et al. Isolated Zn-Co atomic pair for highly active and durable oxygen reduction. Angew. Chem. Int. Ed. 58, 2622–2626 (2019).
Zhao, Y. et al. Distinguished Zn, Co-Nx-C-Sy active sites confined in dentric carbon for highly efficient oxygen reduction reaction and flexible zn-air batteries. Nano Energy 58, 277–283 (2019).
Wu, X. et al. Mesoporous hollow nitrogen-doped carbon nanospheres with embedded MnFe2O4/Fe hybrid nanoparticles as efficient bifunctional oxygen electrocatalysts in alkaline media. ACS Appl. Mater. Interfaces 10, 20440–20447 (2018).
Ahmed, M. S. & Kim, Y. B. 3D graphene preparation via covalent amide functionalization for efficient metal-free electrocatalysis in oxygen reduction. Sci. Rep. 7, 43279 (2017).
Wang, H. et al. Hollow nitrogen-doped carbon spheres with Fe3O4 nanoparticles encapsulated as a highly active oxygen-reduction catalyst. ACS Appl. Mater. Interfaces 9, 10610–10617 (2017).
Wang, H. et al. Creating nitrogen-doped hollow multiyolk@shell carbon as high performance electrodes for flow-through deionization capacitors. ACS Sustain. Chem. Eng. 5, 3329–3338 (2017).
Shu, J., Qiu, Z., Lv, S., Zhang, K. & Tang, D. Cu2+-doped SnO2 nanograin/polypyrrole nanospheres with synergic enhanced properties for ultrasensitive room-temperature H2S gas sensing. Anal. Chem. 89, 11135–11142 (2017).
Zhang, Y., Yang, W., Luo, R. & Shang, H. Preparation of carbon nanospheres by non-catalytic chemical vapor deposition and their formation mechanism. New Carbon Mater. 31, 467–474 (2016).
Miao, J.-Y. et al. Synthesis and properties of carbon nanospheres grown by CVD using Kaolin supported transition metal catalysts. Carbon 42, 813–822 (2004).
Xiong, J. et al. Salt-templated synthesis of defect-rich MoN nano-sheet for boosted hydrogen evolution reaction. J. Mater. Chem. A 5, 24193–24198 (2017).
Dutta, S., Bhaumik, A. & Wu, K. C. W. Hierarchically porous carbon derived from polymers and biomass: effect of interconnected pores on energy applications. Energy Environ. Sci. 7, 3574–3592 (2014).
Gao, Z. et al. Chemically grafted graphene-polyaniline composite for application in supercapacitor. Electrochim. Acta 133, 325–334 (2014).
He, D. et al. Polyaniline-functionalized carbon nanotube supported platinum catalysts. Langmuir 27, 5582–5588 (2011).
Ye, B. et al. Polyaniline and perfluorosulfonic acid Co-stabilized metal catalysts for oxygen reduction reaction. Langmuir 33, 5353–5361 (2017).
Cao, S. et al. mesoporous hybrid shells of carbonized polyaniline/Mn2O3 as non-precious efficient oxygen reduction reaction catalyst. ACS Appl. Mater. Interfaces 8, 6040–6050 (2016).
Ding, W. et al. Shape fixing via salt recrystallization: a morphology-controlled approach to convert nanostructured polymer to carbon nanomaterial as a highly active catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 137, 5414–5420 (2015).
Wu, R. et al. Template-free synthesis of hollow nitrogen-doped carbon as efficient electrocatalysts for oxygen reduction reaction. J. Power Sources 274, 645–650 (2015).
Kumar, N. A. et al. Polyaniline-grafted reduced graphene oxide for efficient electrochemical supercapacitors. ACS Nano 6, 1715–1723 (2012).
Begum, H., Ahmed, M. S., Cho, S. & Jeon, S. Simultaneous reduction and nitrogen functionalization of graphene oxide using lemon for metal-free oxygen reduction reaction. J. Power Sources 372, 116–124 (2017).
Ahmed, M. S., Lee, D. W. & Kim, Y. B. Graphene supported silver nanocrystals preparation for efficient oxygen reduction in alkaline fuel cells. J. Electrochem. Soc. 163, F1169–F1176 (2016).
Ahmed, M. S., Han, H. S. & Jeon, S. One-step chemical reduction of graphene oxide with oligothiophene for improved electrocatalytic oxygen reduction reactions. Carbon 61, 164–172 (2013).
Begum, H., Ahmed, M. S. & Jeon, S. New approach for porous chitosan−graphene matrix preparation through enhanced amidation for synergic detection of dopamine and uric acid. ACS Omega 2, 3043–3054 (2017).
Ahmed, M. S. & Kim, Y. B. Amide-functionalized graphene with 1,4-diaminobutane as efficient metal-free and porous electrocatalyst for oxygen reduction. Carbon 111, 577–586 (2017).
Hu, C. et al. Scalable preparation of multifunctional fire-retardant ultralight graphene foams. ACS Nano 10, 1325 (2016).
Ramphal, I. A. & Hagerman, M. E. Water-processable laponite/polyaniline/graphene oxide nanocomposites for energy applications. Langmuir 31, 1505–1515 (2015).
Silva, R., Voiry, D., Chhowalla, M. & Asefa, T. Efficient metal-free electrocatalysts for oxygen reduction: polyaniline-derived N- and O-doped mesoporous carbons. J. Am. Chem. Soc. 135, 7823–7826 (2013).
Wang, Z. et al. Three-dimensional printing of polyaniline/reduced graphene oxide composite for high-performance planar supercapacitor. ACS Appl. Mater. Interfaces 10, 10437–10444 (2018).
Lin, Y.-H. et al. Facile synthesis of diamino-modified graphene/polyaniline semi-interpenetrating networks with practical high thermoelectric performance. ACS Appl. Mater. Interfaces 10, 4946–4952 (2018).
Han, T. H. et al. Ternary composite of polyaniline graphene and TiO2 as a bifunctional catalyst to enhance the performance of both the bioanode and cathode of a microbial fuel cell. Ind. Eng. Chem. Res. 57, 6705–6713 (2018).
Ahmed, M. S. & Jeon, S. Highly active graphene-supported NixPd100−x binary alloyed catalysts for electro-oxidation of ethanol in an alkaline media. ACS Catal. 4, 1830–1837 (2014).
Marcano, D. C. et al. Improved synthesis of graphene oxide. ACS Nano 4, 4806–4814 (2010).
Hu, Z. et al. Characterization of graphite oxide after heat treatment. New J. Chem. 36, 1373–1377 (2012).
Huang, H.-H., Silva, K. K. H. D., Kumara, G. R. A. & Yoshimura, M. Structural evolution of hydrothermally derived reduced graphene oxide. Sci. Rep. 8, 6849 (2018).
Ju, H.-M., Choi, S.-H. & Huh, S. H. X-ray diffraction patterns of thermally-reduced graphenes. J. Korean Phys. Soc. 57, 1649–1652 (2010).
Begum, H., Ahmed, M. S. & Jeon, S. A novel δ-MnO2 with carbon nanotubes nanocomposite as an enzyme-free sensor for hydrogen peroxide electrosensing. RSC Adv. 6, 50572–50580 (2016).
Begum, H., Ahmed, M. S. & Jeon, S. Highly efficient dual active palladium nanonetwork electrocatalyst for ethanol oxidation and hydrogen evolution. ACS Appl. Mater. Interfaces 9, 39303–39311 (2017).
Begum, H., Ahmed, M. S. & Jeon, S. δ-MnO2 nanoflowers on sulfonated graphene sheets for stable oxygen reduction and hydrogen evolution reaction. Electrochim. Acta 296, 235–242 (2019).
Yan, J. et al. Flexible mxene/graphene films for ultrafast supercapacitors with outstanding volumetric capacitance. Adv. Funct. Mater. 27, 1701264 (2017).
Wu, X., Wang, Z., Yu, M., Xiu, L. & Qiu, J. Stabilizing the mxenes by carbon nanoplating for developing hierarchical nanohybrids with efficient lithium storage and hydrogen evolution capability. Adv. Mater. 29, 1607017 (2017).
Ahmed, M. S., Kim, D. & Jeon, S. Covalently grafted platinum nanoparticles to multi walled carbon nanotubes for enhanced electrocatalytic oxygen reduction. Electrochim. Acta 92, 168–175 (2013).
Ahmed, M. S. & Jeon, S. New functionalized graphene sheets for enhanced oxygen reduction as metal-free cathode electrocatalysts. J. Power Sources 218, 168–173 (2012).
Ahmed, M. S. & Jeon, S. Electrochemical activity evaluation of chemically damaged carbon nanotube with palladium nanoparticles for ethanol oxidation. J. Power Sources 282, 479–488 (2015).
Xue, Q. et al. 3D nitrogen-doped graphene aerogels as efficient electrocatalyst for the oxygen reduction reaction. Carbon 139, 137–144 (2018).
Camisasca, A., Sacco, A., Brescia, R. & Giordani, S. Boron/nitrogen-codoped carbon nano-onion electrocatalysts for the oxygen reduction reaction. ACS Appl. Nano Mater. 1, 5763–5773 (2018).
Yan, Y. et al. Composites of thiol-grafted PEDOT with N-doped graphene or graphitic carbon nitride as an electrochemical sensor for the detection of paracetamol. J. Mater. Sci. https://doi.org/10.1007/s10853-020-04351-w (2020).
Lai, L. et al. Exploration of the active center structure of nitrogen-doped graphene-based catalysts for oxygen reduction reaction. Energy Environ. Sci. 5, 7936–7942 (2012).
Choe, J. E., Ahmed, M. S. & Jeon, S. Trouble free dopamine sensing by palladium nanoparticles fabricated poly(3,4-ethylenedioxythiophene) functionalized graphene. J. Electrochem. Soc. 163, B113–B118 (2016).
Lv, Q. et al. Nitrogen-doped porous graphdiyne: a highly efficient metal-free electrocatalyst for oxygen reduction reaction. ACS Appl. Mater. Interfaces 9, 29744–29752 (2017).
Ko, Y. N., Park, S. B., Jung, K. Y. & Kang, Y. C. One-pot facile synthesis of Ant-cave-structured metal oxide-carbon microballs by continuous process for use as anode materials in Li-ion batteries. Nano Lett. 13, 5462–5466 (2013).
Begum, H., Ahmed, M. S., Cho, S. & Jeon, S. Freestanding palladium nanonetworks electrocatalyst for oxygen reduction reaction in fuel cells. Int. J. Hydrogen Energy 43, 229–238 (2018).
Sohn, G. J. et al. Water-dispersible, sulfonated hyperbranched poly(ether-ketone) grafted multiwalled carbon nanotubes as oxygen reduction catalysts. ACS Nano 6, 6345–6355 (2012).
Park, D., Ahmed, M. S. & Jeon, S. Covalent functionalization of graphene with 1,5- diaminonaphthalene and ultrasmall palladium nanoparticles for electrocatalytic oxygen reduction. Int. J. Hydrogen Energy 42, 2061–2070 (2017).
Wang, C. H., Huang, H. C., Chang, S. T., Lin, Y. C. & Huang, M. F. Pyrolysis of melamine-treated vitamin B12 as a non-precious metal catalyst for oxygen reduction reaction. RSC Adv. 4, 4207–4211 (2014).
Ahmed, M. S., Jeong, H., You, J. M. & Jeon, S. Electrocatalytic reduction of dioxygen at a modified glassy carbon electrode based on Nafion-dispersed single-walled carbon nanotubes and cobalt–porphyrin with palladium nanoparticles in acidic media. Electrochim. Acta 56, 4924–4929 (2011).
Choe, J. E., Ahmed, M. S. & Jeon, S. 3,4-Ethylenedioxythiophene functionalized graphene with palladium nanoparticles for enhanced electrocatalytic oxygen reduction reaction. J. Power Sources 281, 211–218 (2015).
Panomsuwan, G., Saito, N. & Ishizaki, T. Nitrogen-doped carbon nanoparticle−carbon nanofiber composite as an efficient metal-free cathode catalyst for oxygen reduction reaction. ACS Appl. Mater. Interfaces 8, 6962–6971 (2016).
Lv, Y., Yang, L. & Cao, D. Nitrogen and fluorine-codoped porous carbons as efficient metal-free electrocatalysts for oxygen reduction reaction in fuel cells. ACS Appl. Mater. Interfaces 9, 32859–32867 (2017).
Wu, J. & Yang, H. Platinum-based oxygen reduction electrocatalysts. Acc. Chem. Res. 46, 1848–1857 (2013).
Acknowledgements
This work was supported by the National Research Foundation of Korea (15R1A4A1041746 and 18R1D1A1B07044005).
Author information
Authors and Affiliations
Contributions
H.B. has done all electrochemical experiments and written this manuscript with M.S.A. and analyzed all the results with due discussion with Y.-B.K. Also, H.B. and Y.-B.K. both are designed the experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Begum, H., Ahmed, M.S. & Kim, YB. Nitrogen-rich graphitic-carbon@graphene as a metal-free electrocatalyst for oxygen reduction reaction. Sci Rep 10, 12431 (2020). https://doi.org/10.1038/s41598-020-68260-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68260-3
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.