
Abstract
The European honeybee (Apis mellifera) is a key pollinator and has in the last decades suffered significant population decline. A combination of factors, including decrease in genetic diversity and introduction of Varroa mites, have been suggested to be responsible for these losses, but no definitive cause has yet been appointed. In Europe not only have wild colonies been severely affected, but managed hives have had a massive decline in numbers. To test the hypothesis that honeybees’ genetic diversity has decreased in the recent past, we used reduced representation genome sequencing of 40 historical honeybee specimens collected in Natural History collections across Europe and compared them to genomic data from 40 individuals from extant populations (collected post 2006). Our results are consistent with the existence of five evolutionary lineages as previously described, and show a decrease in genetic diversity between historical and extant individuals of the same lineage, as well as high levels of admixture in historical specimens. Our data confirm that a loss of genetic diversity has occurred during the last century, potentially increasing honeybees’ vulnerability to contemporary ecological and anthropogenic stressors.
Similar content being viewed by others

Introduction
Honeybees are one of the most important pollinator species and the most widely used insect, managed for its pollination services and production of honey. The number of managed honeybee colonies in Europe has generally decreased since the 1960′s, at least in Central Europe1. The number of wild or feral honey bees is less known, with many believing or assuming that they no longer exist. Recent work however suggests that feral honey bees still colonize beech forests in Germany, and probably much of Central Europe2.
Even though bees have been used to produce honey and for pollination purposes for over 7,000 years, since at least Ancient Egypt civilizations3,4,5, it was only when beekeeping techniques were perfected in the seventeenth and eighteenth centuries that it became possible to maintain large bee colonies giving rise to modern apiculture6. More recent practices, such as the commercial mass rearing of queens, artificial selection of behaviours favouring honey production, and the presence of thousands of bees in limited spaces, may have altered the natural processes and affected the genetic diversity of domestic and wild (or feral) hives, increasing their susceptibility and the transmission rate of diseases between bees7. There is an ongoing debate about whether European honeybees are domesticated (in the sense that selective breeding over generations has led to artificial selection) or not8,9,10. Traits favourable to beekeepers, such as docility, lack of propensity to swarming, honey yield, and others may be selected for, but as it is difficult to have controlled mating, this is usually done through the import of stock from other areas, where these traits are more frequent. This has consequences for wild populations, as due to the wide freedom honeybees have even when in artificial hives9, factors influencing one of them will have a similar effect on the other.
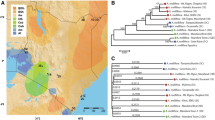
There are at least 28 subspecies of Apis mellifera11,12 described based on geography and morphological variation; morphometric and genetic studies have consistently showed that there are only four or five major evolutionary lineages, with one or two of them only occurring in Africa. Honeybees were first aggregated into four major lineages (A, C, M, and O) based on morphometry and biogeography13. Lineage A is present in Africa and in the Iberian Peninsula, O in the Middle East, M in Northern and Western Europe, and C in South Eastern Europe. The existence of a fifth lineage from north-eastern Africa named Y was proposed and supported using mitochondrial DNA14. The approximate distribution of the lineages in Africa, Europe, and Western Asia is shown in Fig. 1. The main hypothesis for the origin of Apis mellifera is of an Asian origin, as all other Apis species are endemic to Asia. However, genetic diversity is higher in Africa, which has led some authors to speculate on an African origin for the species14.

Map showing approximate limits of the A, C, M, O, and Y evolutionary lineages of Apis mellifera, and original sampling locations of historic (red) and contemporary (blue) specimens used in this study.
Early genomic work on honeybees focused on molecular determinants in behaviour15,16. After the release of the honeybee genome17, several specific pathways involved in immunity18, and olfaction, and gustation19 have been identified.
Considering the important role of honeybees for the pollination of monocultures, that are essential for human food security and variety, and other ecological services performed by honeybees, there is dire need for a better understanding of changes in genetic variability of (semi)natural populations and how these may be related to domestication and artificial selection.
Museum collections are ideal for this purpose, as they provide a temporal series of honeybees from different areas of their natural range, from which trends can be derived, and they are invaluable sources of chitinous material from which DNA can be retrieved without conferring any external damage to the specimen20,21,22. Museum specimens have been widely used to provide insight into past genetic diversity, domestication, taxonomic placement, and migration of several species23,24 . However, specimens were not always collected considering the preservation of genetic material, and DNA degrades as a function of temperature and time. For this reason, it is necessary to use ancient DNA (aDNA) techniques to retrieve usable DNA from these specimens. We therefore use the term ancient DNA not as a reference to the age of our specimens, but to the likely state of degradation of the molecules. The low percentage of endogenous DNA (that belonging to the organism, and not contaminating fungi or bacteria) in museum samples still poses limitations to aDNA shotgun sequencing experiments, as most sequences yielded would not be from the specimen. DNA capture-enrichment methods, in contrast, allow targeted sequencing by selective enrichment of sequences of interest prior to sequencing, hence increasing the depth of sequencing over target regions and lowering costs per target25.
As honeybees are thought of being at least partially domesticated, they were not usually collected with purpose by naturalists, but rather as incidental bycatches when collecting other insect species thought to have greater natural history value. Curation and annotations were therefore sparse and incomplete. Despite this, several historical collections exist. We took advantage of this, and generated genomic data on historical European honeybee specimens, allowing us to explore the genetic diversity of the species over the last 150 years. In this study, we assess the genetic diversity of honeybees across Europe from different time periods using historical museum collections and ancient DNA (aDNA) techniques.
Material and methods
Data collection
Pin-dried specimens of Apis mellifera covering most of the natural range of honeybees in Europe were obtained from museum collections. As collections were not digitized at the time of sampling, all information from labels were recorded manually, including when available: date of sampling, location name, geographical coordinates, sex, and name of collector. None of the collections had unique identifiers (voucher numbers) for these specimens, so internal identifiers were used consisting of two letter country code, four-digit year of sampling, and, if more than one specimen matched the country/year combination, one letter (a through e). This is the only information available for these specimens, and we therefore do not know if their origin is from feral, wild or managed colonies. As bees in managed hives can freely move around their area, gene flow between managed and feral hives is most likely unrestricted, so we believe this does not affect the interpretation of our results. Subspecies/lineage information was not known a priori when selecting specimens, so sampling attempted to cover a wide geographic and temporal span.
The time span of the specimens is from 1850 to 2002 (See Fig. 1 and Supplementary Table S1). DNA extraction, Illumina library preparations, and PCR setups were performed in a dedicated ancient DNA laboratory. Total genomic DNA was extracted using the non-destructive method described in Gilbert et al.20 and detailed in Campos and Gilbert21. Probes for in-solution, hybridization capture enrichment kits (MYcroarray) were designed for randomly selected gene locations present in gene set AmelOGSV3.226, but also for immune27, sensory19, and behavioural15 biochemical pathways (SI Appendix and Supplementary Table S2). Libraries were pooled with other indexed DNA libraries and sequenced on Illumina HiSeq platforms at the Danish National High-Throughput DNA Sequencing Centre. Whole genome sequences of 40 honeybees collected after 2006 from lineages A, C, M, and Y and one Apis cerana were retrieved from NCBI’s short read archive28.
Analysis
Sequencing data were analysed using a set of custom scripts and software (SI Appendix). Adapter sequences were trimmed and filtered for N’s and reads shorter than 30 bp were removed using AdapterRemoval29. Trimmed reads were initially mapped to Amel 4.517, 26 using bwa-0.7.5a-r405, with seed length disabled to improve mapping efficiency in ancient DNA datasets30. The alignments were sorted using Samtools31 and filtered for PCR duplicates using Picard MarkDuplicates-1.88 (https://picard.sourceforge.net), and for paralogs using BWA. We used ANGSD (Analysis of Next Generation Sequencing Data)32 for quality filtering and data processing. In all ANGSD analyses, we required a minimum mapping quality of 30 and minimum base quality score of 20. We calculated error rates using an outgroup individual and an error free individual. We randomly selected a modern sample from lineage M from Poland (SRR957058) as the error-free individual, and the outgroup individual used was a modern Apis cerana (SRR957079). We used mapDamage33 to display nucleotide misincorporation patterns and rescale the quality scores in the bam files. After rescaling we recalculated error rates and compared them with the previous estimates. The rescaled sequences were used in subsequent analyses. We calculated genome-wide coverage in the modern individuals and depth of coverage within the capture regions for both modern and historical individuals. Five historical samples were excluded from further processing, as they had an average coverage below 0.5 ×. Genotype likelihoods were estimated based on the aligned reads and associated mapping, and sequencing quality scores for all individuals.
Population structure
We used NGSadmix version 32 to test the number of genetically distinguishable populations in our data34. As the presumptive number of evolutionary lineages in Apis mellifera is five, we ran NGSadmix for K between two and nine. The evolutionary history of the individuals was inferred using Neighbor-Joining35. Haploid genotypes from ancient and modern samples were obtained by randomly sampling one read per position of each of the samples with ANGSD. The tree was built using the program RapidNJ36. FigTree v.1.4.4 (Rambaut, 2012; https://tree.bio.ed.ac.uk/software/figtree/) was used to visualize the tree. To compare the genetic diversity of our dataset with other published honeybee datasets, we built two haplotype networks using mitochondrial DNA sequences. In addition to the modern and historical samples, we used other Apis mellifera mitochondrial sequences downloaded from the NCBI website (Table 1). All mitochondrial sequences were aligned using MAFFT37. Cytochrome b (CytB) and the sequence spanning from the beginning of the Cytochrome Oxidase I to the end of Cytochrome Oxidase II (COI-COII) were extracted separately from the alignment, according to the NCBI sequence KM458618.138. Haplotype networks were reconstructed using TempNet39. The procedure was repeated using the non-admixed individuals from subsets as defined in Supplementary Table S1.
Genetic variability and neutrality tests
Based on NGSadmix results for K = 5 and geographical location, we grouped individuals according to the most likely lineage they belonged to: lineage A in South Africa; lineage Y in Kingdom of Saudi Arabia and Yemen ; lineage C in Malta , Italy, Croatia, Slovenia, Austria (AT1971), Switzerland, Bulgaria, Germany, and Denmark; lineage M in Austria, The Netherlands, Sweden, England, France, Luxembourg, Portugal, Spain, Scotland, and Poland; and lineage O in Jordan and Lebanon (see Supplementary Table S1, and Fig. 1 for details). As sampling was done ad-hoc and lineages only ascertained a posteriori, our study does not include modern individuals from lineage O, and ancient ones from lineages A and Y. However, lineages C and M are represented with both historical and modern individuals (Supplementary Table S1). We divide each group further, including four unmixed or less mixed individuals, based on geographic location (Supplementary Table S1).
We estimated the population scaled mutation rate θ and the neutrality test statistic Tajima's D according to the method described in Korneliussen et al.40. ANGSD was also used to estimate Watterson and Pairwise θ. We used F-statistics to investigate the genetic distance between the populations observed in NGSadmix. Reynold weighted FST41 was calculated using ANGSD, for each of the subgroups defined above. We looked for outlier levels of FST to identify loci that have probably undergone geographically restricted positive selection42. We performed 45 pairwise comparisons, focused on modern versus historical samples in lineages C and M: (i) Historic vs Modern C, (ii) Historic vs Modern M North, and (iii) Historic vs Modern M South.
Results
In this study, we sequenced 46 historical honeybees from 17 different European countries. Depth of coverage in the targeted regions was between 0 × and 59 ×, with an average of 25.75 × (1.43 × genome-wide [0.0 × to 5.02 ×]). Depth of coverage within the targeted regions was on average 20 times higher than in other sites in the genome, ranging from a threefold to a 66-fold increase (Table 2). Five historical honeybees with depth of coverage below 0.5 × in target regions were excluded from further analysis. Depth of coverage was 7 × in the Apis cerana specimen used as outgroup, while in the rest of the modern specimens, depth of coverage ranged from 12 to 27 × (Table 2).
Quality control
Sequences from modern specimens showed relatively low error rates between 0.01% and 0.16%. Historical samples presented higher error rates (between 0.1 and 0.7%), which correspond mainly to post-mortem deamination C->T and G->A (Supplementary Figure S1). These affected mostly the beginning and end of reads, which is to be expected in museum preserved samples. After masking transitions with mapDamage, error rates were halved.
Phylogeny
Phylogenetic relationships between all the individuals is represented in Fig. 2A, in parallel with the admixture plot for K = 8 (see next section). This value of K was chosen as it mirrors the clades inferred from the phylogenetic tree. There are two main branches: the top one includes the two African lineages: A and Y. The next lineage to split is lineage O, followed by C and M. Finally, lineage C further subdivides in two groups.

Phylogenetic tree and admixture plot of honeybees (A) Neighbor-Joining tree of historic and contemporary (denoted by an asterisk) European honeybees. Apis cerana is used as an outgroup. Colour of labels correspond to the five evolutionary lineages: pink—lineage A, green—lineage Y, violet—lineage O, orange—lineage C, and gray—lineage M (B) Admixture plot for K = 8. Colour of labels as in (A). Coloured bars represent proportion of membership to each K group. When possible, colours match colours in (A), such as in lineages A, Y, and O.
The haplotype networks obtained from cytochrome b and COI-COII region can be seen in Fig. 3. Both networks show some degree of clustering according to lineages, with individuals belonging to lineages C and M sharing some haplotypes.

Haplotype network based on (a) cytochrome b (CYTB) and (b) cytochrome c oxidase 1 and 2 (COI-COII). Size of circles is proportional to number of individuals with that haplotype; black circles represent haplotypes not present in our sampling. Coloured areas represent the evolutionary lineages where enclosed haplotypes have been found.
Admixture was widespread
Admixture proportions of both modern and historical samples are represented in Fig. 2B (admixture proportions for other values of K are shown in Supplementary Figure S2). Admixture was high for many historical European samples. Individuals from Scandinavia, and some from the Netherlands, England, and France have high levels of admixture. The level of admixture observed for modern samples was low as previously reported in Harpur et al.28, but this does not mean admixture does not occur, as individuals were chosen for that study because they were not admixed.
Nucleotide diversity was higher in historic populations
Supplementary Figure S3 shows the distribution of Watterson theta across the different chromosomes, and a plot with the values of Tajima's D can be seen in Supplementary Figure S4. Nucleotide diversity is highest in historical O lineage individuals and lowest in the Y lineage samples. Individuals belonging to lineages A and M seem to have similar levels of nucleotide diversity. On the other hand, lineage C has lower levels of diversity but not as low as lineage Y. Boxplots with theta Watterson (Fig. 4A) and Tajima’s D (Fig. 4B) were made using the sites from the haploid sampling and with the group division explained above. The highest level of diversity can be seen in the historical samples from the O lineage. The modern A group has an intermediate level of diversity, whereas the groups formed by historic C and M are close to this A group. Groups formed by modern C and M have low diversity levels. The modern Y group has a slightly higher diversity than the latter.

Boxplot of (A) Watterson’s estimator and (B) Tajima’s D values per chromosome for several groups of European honeybees. Colour indicates historic (red) or contemporary (blue) populations. Alphanumeric codes represent groups of 4 (3 in lineage O) least admixed individuals from a combination geographical area, lineage and time, as described in supplementary Table S1.
Fixation indices follow lineage distributions
Pairwise FST values are shown in Table 3. The highest values are in the pairwise comparisons of lineage Y with the other lineages. Moderately high are the FST values for the pairwise comparisons with the modern C group. The distance between the C groups and the M groups is also high despite geographic proximity. Lineage A is more like the historical populations of the C and M groups and to the O group, according to FST values. Modern samples are more distant to lineage A. The historical C samples are also close to the O group, while the historical M samples from the North are close to all other M samples modern and historical. Furthermore, historical M samples from the North of Europe are most like the modern M samples from Poland, which makes sense geographically. And, similarly, historical M individuals from the South of Europe are like modern M samples from Spain. On the other hand, modern M specimens from Spain and Poland are also close to each other, according to FST values.
Genes under selection
We also looked at selection over time. Histograms of number of gene regions per FST value are shown in Supplementary Figure S5. The top 5% genes with higher FST values for three pairwise comparisons (historical vs modern C, historical vs modern M east, and historical vs modern M west) were selected (Supplementary Table S3). Two genes are common in the three comparisons: MRJP7, major royal jelly protein, and an unnamed gene that synthesizes a membrane protein (Table 4).
Discussion
Historic data supports five genomic lineages in honeybees
The topology of the Apis mellifera phylogenetic tree which includes both modern and historical samples corroborates previous phylogenetic inferences and its division in five lineages28,43,44. In this analysis, we did not compare our data with the sequences from Wallberg et al.43, as they used SOLiD sequencing chemistry, and combining data from two different sequencing technologies might have caused biases in our estimates. Nevertheless, Cridland et al.44 successfully generated a phylogenetic tree which included representatives of A, C, M, O, and Y lineages based on whole genomes that, globally, is congruent with the phylogeny generated in this study based on targeted regions (Fig. 2A). In both phylogenomic reconstructions, lineage Y seems highly divergent, originating from lineage A shortly after the separation of A/Y from C/M/O. Wallberg et al.43 found evidence of admixture in Jordanian O samples, originating from A populations. However, when including Y-lineage samples, Cridland et al. found that this admixture was from lineage Y rather than A44. Our specimens from Jordan and Lebanon do not show any sign of admixture when K = 5 or more and are basal in the Neighbor-Joining tree in regard to the European lineages. This would imply that admixture from lineage A is more recent than 1976, when our Jordanian samples were collected, or reflect different sampling localities. Harpur et al.28 did not find any significant admixture between lineages in their data; however, this was to be expected giving their sampling strategy to avoid admixed individuals.
Decrease in genetic diversity in modern honeybees compared to historic populations
Despite the limited sample size, we think that our estimates of genetic diversity are reliable, as both simulation and empirical studies indicate that a large sample size is not required when analysing a large number of SNP markers45,46. Our results suggest that genetic diversity has decreased in European honeybees over the last century. This is supported by the lower nucleotide diversity found in modern C-M samples (π ≅ 0.001), compared to historical ones (π ≅ 0.003). Genetic diversity influences a wide range of phenotypes in honeybee colonies, from expression of antimicrobial compounds, resistance to pathogens, thermoregulation, foraging behaviour and colony defence8, all essential to colony survival, and response to environmental stress, with lower genetic diversity reducing the variation of these phenotypes as well. Tasks within a colony, such as defence and hygienic behaviour, are performed by a small subset of workers descendent from only some patrilineal lines8. Differences in propensity for certain tasks are believed to be influenced by genetics. For example, hygienic and non-hygienic colonies have a difference in gene expression in Cytochrome P450 gene and a limited number of other genes47. This means that when genetic diversity is decreased the number of workers in a colony performing some tasks may decrease or less specialized workers will perform such tasks, decreasing the efficiency of the colony48. This may originate from high selection pressure selecting for traits based only on queen performance but ignoring the genetic contribution of drones or failing to maintain sufficient levels of genetic diversity within a colony.
We hypothesize that management practices that increase relatedness between colonies, as well as a reduction of number and density of colonies due to, for example, a decrease of suitable habitat availability, are the main factors contributing to the observed decrease of genetic diversity. At the population level, genetic diversity can be affected by selective sweeps, background selection, temporal fluctuations in the direction of selection on segregating alleles49, and the level of genetic recombination48.
In addition, decreasing density or fewer colonies, demographic expansions, as well as habitat fragmentation would also lower genetic diversity. Colony density in wild populations in Europe is much lower than in African savannahs, despite harsher environmental conditions. This has been associated with more intensive beekeeping in Europe50. It has also been found that abiotic factors, such as temperature and land use, are associated with both density of colonies and genetic diversity51.
Domestication and professional breeding aim at selecting individuals with specific traits, consciously or unconsciously narrowing genetic variation. Artificial selection on managed hives, however, would only have an indirect effect on wild colonies when drones from managed hives breed with wild queens, or new queens from a managed hive establish a new colony in the wild. This is very frequent, as beekeepers do not track all bees in their colonies. In any case, in many European countries, there are much less wild colonies than managed ones due to the lack of suitable nesting places, so gene flow between them has probably masked any genetic difference between them.
Our results seem to contradict those of a study from 201252 where Harpur and colleagues find the within-colony diversity to be higher in managed colonies than in wild ones, while our results show global patterns of decline within lineages. It should be emphasized that the two are studying different levels of genetic diversity, theirs within-colony diversity in admixed populations, and ours on a meta-population level looking only at non-admixed individuals. It is not surprising that Harpur et al. found higher diversity within managed colonies, as beekeepers will bring in stock from other parts of the world, what they called the progenitor populations, and these will admix with the local populations despite beekeeper’s intentions, creating higher diversity descendants as de la Rua highlights in a reply to that work53. Our results indicate that despite this within colony gain, global patterns of diversity decreased within a short time span (from 1960–1984 to 2013 in lineage C and 1917–1973 to 2013 in lineage M).
Signs of positive selection within the MRJP gene family
We detected signs of selection in MRJP7 and in another gene of unknown function. In a recent genomic analysis, Harpur et al. also detected positive selection in MRJP 7 and MRJP 428, another gene of the same family, in current honeybees. Major Royal Jelly Protein is a family of nine genes and one pseudogene located in tandem on a 60 kb cluster located on chromosome 1154. This family of genes encodes secretory proteins that are the major protein content of Royal Jelly, a nutrient-rich substance produced by nurse bees used to feed the larvae, which is only found in some genus of Hymenoptera55. MRJP seems to have evolved recently deriving from the Yellow family of genes and it seems to have diversified independently in each species where it has been found. In honeybees, MRJP is mostly expressed in workers (particularly nurses) but also other castes56,57, and besides being involved in the production of Royal Jelly, it has also been associated with brain function58, caste determination and many aspects of eusociality54. These functions seem to derive from Royal Jelly’s function in establishing division of labour in the colonies through determining the development of larvae into queens and worker. However, their biochemical function is not determined at the moment. MRJP 4 is down regulated in honeybee heads after infection54. Given that Royal Jelly Proteins affect many aspects of behaviour, nutrition and development, and that this pattern of selection is found not only when analysing modern bees alone28, but also when comparing historical bees to modern bees, we speculate that domestication can be responsible for the selection signal. While MRJP7 is but one gene within the MRJP/Yellow family, we speculate that selection on this gene, due to its association with nutrition and development, could be caused by selective pressure from beekeeping practices such as the desire for higher honey production or more fertile queens.
The observed decrease in genetic diversity could potentially have an impact on the ability of colonies to react and survive to current and upcoming threats, such as pathogens, pesticides, and climate change. The distribution of evolutionary lineages and admixture proportions in honeybees is not fully understood yet. Several geographical regions are under-sampled, such as most of Africa and areas of the Middle East. The fragmented nature of the sampling carried out in most honeybee genetic studies, has made the lineage nomenclature inconsistent, making comparisons among studies difficult, unreliable or impossible. Mapping with better precision the distribution of each lineage and areas of current and past admixture would help us to better understand the population dynamics of honeybees. Natural history collections with proper annotations of sampling locality and date prove once again to be an essential resource to study temporal trends and provide a glimpse of evolutionary processes occurring in historical times.
Data availability
The BAM files of the sequence data mapped to the Apis mellifera genome v4.5 have been deposited in the Short Read Archive under BioProject PRJNA505606. Custom scripts used to analyse the data can be accessed in https://github.com/LuciaRT/code-bees.
References
Potts, S. G. et al. Declines of managed honey bees and beekeepers in Europe. J. Apic. Res. 49, 15–22 (2010).
Kohl, P. L. & Rutschmann, B. The neglected bee trees: European beech forests as a home for feral honey bee colonies. PeerJ 6, e4602 (2018).
Kritsky, G. Beekeeping from antiquity through the middle ages. Annu. Rev. Entomol. 62, 249–264 (2017).
Bloch, G. et al. Industrial apiculture in the Jordan valley during Biblical times with Anatolian honeybees. Proc. Natl. Acad. Sci. USA 107, 11240–11244 (2010).
Crane, E. Honeybees. In Evolution of domesticated animals (ed. Mason, I.L.) 403–415 (Longman Group, London, 1984).
Phillips, E. F. Beekeeping (Applewood Books, Carlisle, 1918).
Meixner, M. D. et al. Conserving diversity and vitality for honey bee breeding. J. Apic. Res. 49, 85–92 (2010).
Oxley, P. R. & Oldroyd, B. P. The genetic architecture of honeybee beeding. In Advances in Insect Physiology Vol. 39 (ed. Simpson, S. J.) (Elsevier, Amsterdam, 2010).
Oldroyd, B. P. Domestication of honey bees was associated with expansion of genetic diversity. Mol. Ecol. 21, 4409–4411 (2012).
De la Rúa, P., et al. Apis mellifera. In IUCN 2014. IUCN Red List of Threatened Species. Version 2018–1 e.T42463639A42463665 (2014).
Ruttner, F. Biogeography and Taxonomy of Honeybees. Springer, Berlin. https://doi.org/10.1007/978-3-642-72649-1 (1988).
Engel, M. M. S. The taxonomy of recent and fossil honey bees (Hymenoptera: Apidae; Apis). J. Hymenopt. Res. 8, 165–196 (1999).
Ruttner, F. & Louveaux, J. Biometrical-statistical analysis of the geographic variability of Apis mellifera L.. Apidologie 9, 363–381 (1978).
Franck, P. et al. Genetic diversity of the honeybee in Africa: Microsatellite and mitochondrial data. Heredity (Edinb). 86, 420–430 (2001).
Whitfield, C. W., Cziko, A.-M. & Robinson, G. E. Gene expression profiles in the brain predict behavior in individual honey bees. Science 302, 296–299 (2003).
Whitfield, C. W. et al. Genomic dissection of behavioral maturation in the honey bee. In PNAS (2006).
Weinstock, G. M. et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443, 931–949 (2006).
Evans, J. D. Beepath: An ordered quantitative-PCR array for exploring honey bee immunity and disease. J. Invertebr. Pathol. 93, 135–139 (2006).
Robertson, H. M. & Wanner, K. W. The chemoreceptor superfamily in the honey bee, Apis mellifera: Expansion of the odorant, but not gustatory, receptor family. Genome Res. 16, 1395–1403 (2006).
Gilbert, M. T. P., Moore, W., Melchior, L. & Worobey, M. DNA extraction from dry museum beetles without conferring external morphological damage. PLoS One 2, e272 ST-DNA Extraction from Dry Museum Beetles (2007).
Campos, P. F. & Gilbert, T. M. P. DNA Extraction from Keratin and Chitin 43–49 (Humana Press, Totowa, 2012). https://doi.org/10.1007/978-1-61779-516-9_6.
Heintzman, P. D., Elias, S. A., Moore, K., Paszkiewicz, K. & Barnes, I. Characterizing DNA preservation in degraded specimens of Amara alpina (Carabidae: Coleoptera). Mol. Ecol. Resour. 14, 606–615 (2014).
Campos, P. F. et al. Ancient DNA sequences point to a large loss of mitochondrial genetic diversity in the saiga antelope (Saiga tatarica) since the Pleistocene. Mol. Ecol. 19, 4863–4875 (2010).
Themudo, G. E., Rufino, C. & Campos, P. F. Complete mitochondrial DNA sequence of the endangered giant sable antelope (Hippotragus niger variani): Insights into conservation and taxonomy. Mol. Phylogenet. Evol. 83, 242–249 (2014).
Cruz-Dávalos, D. I. et al. Experimental conditions improving in-solution target enrichment for ancient DNA. Mol. Ecol. Resour. 17, 508–522 (2017).
Elsik, C. G. et al. Finding the missing honey bee genes: Lessons learned from a genome upgrade. BMC Genomics 15, 86 (2014).
Evans, J. D. et al. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol. Biol. 15, 645–656 (2006).
Harpur, B. A. et al. Population genomics of the honey bee reveals strong signatures of positive selection on worker traits. Proc. Natl. Acad. Sci. https://doi.org/10.1073/pnas.1315506111 (2014).
Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 5, 337 (2012).
Schubert, M. et al. Improving ancient DNA read mapping against modern reference genomes. BMC Genomics 13, 178 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014).
Jónsson, H., Ginolhac, A. & Schubert, M. mapDamage2. 0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Skotte, L., Korneliussen, T. S. & Albrechtsen, A. Estimating individual admixture proportions from next generation sequencing data. Genetics 195, 693–702 (2013).
Saitou, N. & Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987).
Simonsen, M., Mailund, T. & Pedersen, C. N. S. Rapid Neighbour-Joining. In Algorithms in Bioinformatics 113–122 (Springer, Berlin, 2008). https://doi.org/10.1007/978-3-540-87361-7_10.
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Hu, P. et al. Complete mitochondrial genome of the Algerian honeybee, Apis mellifera intermissa (Hymenoptera: Apidae). Mitochondrial DNA A DNA Mapp Seq Anal 27, 1791–1792 (2016).
Prost, S. & Anderson, C. N. K. TempNet: A method to display statistical parsimony networks for heterochronous DNA sequence data. Methods Ecol. Evol. 2, 663–667 (2011).
Korneliussen, T. S., Moltke, I., Albrechtsen, A. & Nielsen, R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics 14, 289 (2013).
Reynolds, J., Weir, B. S. & Cockerham, C. C. Estimation of the coancestry coefficient: Basis for a short-term genetic distance. Genetics 105, 767–779 (1983).
Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 39, 197–218 (2005).
Wallberg, A. et al. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat. Genet. 46, 1081–1088 (2014).
Cridland, J. M., Tsutsui, N. D. & Ramírez, S. R. The complex demographic history and evolutionary origin of the western honey bee, Apis mellifera. Genome Biol. Evol. 9, 457–472 (2017).
Nazareno, A. G., Bemmels, J. B., Dick, C. W. & Lohmann, L. G. Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 17, 1136–1147 (2017).
Willing, E.-M., Dreyer, C. & van Oosterhout, C. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 7, e42649 (2012).
Boutin, S., Alburaki, M., Mercier, P. L., Giovenazzo, P. & Derome, N. Differential gene expression between hygienic and non-hygienic honeybee (Apis mellifera L.) hives. BMC Genomics 16, 500 (2015).
Oldroyd, B. P. & Fewell, J. H. Genetic diversity promotes homeostasis in insect colonies. Trends Ecol. Evol. 22, 408–413 (2007).
Charlesworth, B. Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet. Res. 68, 131–149 (1996).
Moritz, R. F. A., Kraus, F. B., Kryger, P. & Crewe, R. M. The size of wild honeybee populations (Apis mellifera) and its implications for the conservation of honeybees. J. Insect Conserv. 11, 391–397 (2007).
Jaffe, R. et al. Estimating the density of honeybee colonies across their natural range to fill the gap in pollinator decline censuses. Conserv. Biol. 24, 583–593 (2010).
Harpur, B. A., Minaei, S., Kent, C. F. & Zayed, A. Management increases genetic diversity of honey bees via admixture. Mol. Ecol. 21, 4414–4421 (2012).
De la Rúa, P. et al. Conserving genetic diversity in the honeybee: Comments on Harpur et al. (2012). Mol. Ecol. 22, 3208–3210 (2013).
Drapeau, M. D., Albert, S., Kucharski, R., Prusko, C. & Maleszka, R. Evolution of the Yellow/Major Royal Jelly Protein family and the emergence of social behavior in honey bees. Genome Res. 16, 1385–1394 (2006).
Buttstedt, A., Moritz, R. F. A. & Erler, S. Origin and function of the major royal jelly proteins of the honeybee (Apis mellifera) as members of the yellow gene family. Biol. Rev. 89, 255–269 (2014).
Dobritzsch, D., Aumer, D., Fuszard, M., Erler, S. & Buttstedt, A. The rise and fall of major royal jelly proteins during a honeybee (Apis mellifera) workers’ life. Ecol. Evol. 9, 8771–8782 (2019).
Buttstedt, A., Moritz, R. F. A. & Erler, S. More than royal food—major royal jelly protein genes in sexuals and workers of the honeybee Apis mellifera. Front. Zool. 10, 72 (2013).
Kucharski, R., Maleszka, R., Hayward, D. C. & Ball, E. E. A royal jelly protein is expressed in a subset of Kenyon cells in the mushroom bodies of the honey bee brain. Naturwissenschaften 85, 343–346 (1998).
Gibson, J. D. & Hunt, G. J. The complete mitochondrial genome of the invasive Africanized Honey Bee, Apis mellifera scutellata (Insecta: Hymenoptera: Apidae). Mitochondrial DNA 27, 561–562. https://doi.org/10.3109/19401736.2014.905858 (2016).
Crozier, R. H. & Crozier, Y. C. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics 133, 97–117 (1993).
Haddad, N. J. Mitochondrial genome of the Levant Region honeybee, Apis mellifera syriaca (Hymenoptera: Apidae). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 27, 4067–4068. https://doi.org/10.3109/19401736.2014.1003846 (2016).
Fuller, Z. L. et al. Genome-wide analysis of signatures of selection in populations of African honey bees (Apis mellifera) using new web-based tools. BMC Genomics. https://doi.org/10.1186/s12864-015-1712-0 (2015).
Acknowledgements
We are grateful for the collaboration of museum curators and collection managers that provided access and permission to sample their honeybee collections, namely Alexandra Marçal and Luís Filipe Lopes from the Museu de História Natural de Lisboa, Cristina Rufino from Museu da Ciência da Universidade de Coimbra, Frederique Bakker from Naturalis Biodiversity Center, Lars Vilhelmsen from Natural History Museum of Denmark, University of Copenhagen. Without their time and knowledge, this study would not have been possible. GET also wishes to thank Tom Gilbert from the EvoGenomics research group, Natural History Museum, University of Copenhagen for his generosity during this project. This project was funded by FEDER through Programa Operacional Competitividade e Internacionalização—COMPETE (FCOMP-01-0124-FEDER-028307), and by FCT—Fundação para a Ciência e a Tecnologia (PTDC/BIA-BIC/4177/2012). RRF thanks the Danish National Research Foundation for its support of the Center for Macroecology, Evolution, and Climate (grant DNRF96) and the Villum Fonden for the Young Investigator Grant VKR023446.
Author information
Authors and Affiliations
Contributions
G.E.T., P.F.C., A.B.J. designed research. G.E.T., A.R.I., P.F.C. performed research. G.E.T, L.R.T, R.R.F. analysed data. G.E.T., A.R.I., L.R.T., R.R.F., P.F.C. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Espregueira Themudo, G., Rey-Iglesia, A., Robles Tascón, L. et al. Declining genetic diversity of European honeybees along the twentieth century. Sci Rep 10, 10520 (2020). https://doi.org/10.1038/s41598-020-67370-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67370-2
- Springer Nature Limited
This article is cited by
-
SIMplyBee: an R package to simulate honeybee populations and breeding programs
Genetics Selection Evolution (2023)
-
Landfill fire impact on bee health: beneficial effect of dietary supplementation with medicinal plants and probiotics in reducing oxidative stress and metal accumulation
Environmental Science and Pollution Research (2023)
-
Genetic Characterizations of the Iranian Honey Bee (Apis mellifera meda Skorikov 1929) Populations Using the Microsatellite DNA Markers
Biochemical Genetics (2023)
-
Honey bee (Apis mellifera ligustica) acetylcholinesterase enzyme activity and aversive conditioning following aluminum trichloride exposure
BMC Zoology (2022)
-
A genotyping by sequencing approach can disclose Apis mellifera population genomic information contained in honey environmental DNA
Scientific Reports (2022)