
Abstract
The study of salt tolerance mechanisms in halophyte plants can provide valuable information for crop breeding and plant engineering programs. The aim of the present study was to investigate whole transcriptome analysis of Aeluropus littoralis in response to salinity stress (200 and 400 mM NaCl) by de novo RNA-sequencing. To assemble the transcriptome, Trinity v2.4.0 and Bridger tools, were comparatively used with two k-mer sizes (25 and 32 bp). The de novo assembled transcriptome by Bridger (k-mer 32) was chosen as final assembly for subsequent analysis. In general, 103290 transcripts were obtained. The differential expression analysis (log2FC > 1 and FDR < 0.01) showed that 1861 transcripts expressed differentially, including169 up and 316 down-regulated transcripts in 200 mM NaCl treatment and 1035 up and 430 down-regulated transcripts in 400 mM NaCl treatment compared to control. In addition, 89 transcripts were common in both treatments. The most important over-represented terms in the GO analysis of differentially expressed genes (FDR < 0.05) were chitin response, response to abscisic acid, and regulation of jasmonic acid mediated signaling pathway under 400 mM NaCl treatment and cell cycle, cell division, and mitotic cell cycle process under 200 mM treatment. In addition, the phosphatidylcholine biosynthetic process term was common in both salt treatments. Interestingly, under 400 mM salt treatment, the PRC1 complex that contributes to chromatin remodeling was also enriched along with vacuole as a general salinity stress responsive cell component. Among enriched pathways, the MAPK signaling pathway (ko04016) and phytohormone signal transduction (ko04075) were significantly enriched in 400 mM NaCl treatment, whereas DNA replication (ko03032) was the only pathway that significantly enriched in 200 mM NaCl treatment. Finally, our findings indicate the salt-concentration depended responses of A. littoralis, which well-known salinity stress-related pathways are induced in 400 mM NaCl, while less considered pathways, e.g. cell cycle and DNA replication, are highlighted under 200 mM NaCl treatment.
Similar content being viewed by others

Introduction
Soil salinity is an important challenge for food security and sustainable agriculture in the world1. Salt stress negatively influences the growth, development, and yield of agricultural products2,3,4. Higher plants adapt to different environmental stresses by complex molecular, cellular, and physiological mechanisms. The transcriptional alteration of stress-related genes is one of the key approaches at molecular level to cope with adverse conditions5,6. Plants employ complicated strategies such as ion homeostasis, osmotic regulation, antioxidant systems, and signaling pathways of phytohormones and MAPK to tolerate salinity stress6. Although our knowledge on this area is increasing continuously, but the molecular basis of salt resistance is not still well understood. Various omics technologies such as transcriptomics, proteomics, genomics and metabolomics are used for investigation of plant molecular responses7,8. Amongst these techniques, transcriptomics provides comprehensive insights into salt tolerance mechanisms in glyo- and halophyte plants. RNA sequencing (RNA-seq) is an accurate and high-throughput technique, which is widely used in transcriptome analysis studies9,10. Based on the availability of reference genome, de novo and genome-guided RNA-seq are used to evaluate the transcriptome. De novo RNA-seq is an innovative technique typically used for non-model plants that their genome is not available for transcriptome reconstruction10.
Halophyte plants are adapted for living in extreme environments and constitute about 1% of the Earth’s flora3. These plants are ideal candidates for studying and exploring the complex physiological and molecular mechanisms of salt resistance11. Aeluropus littoralis, a halophyte plant, is native to Iran and belongs to Poaceae family (grasses). This plant has C4 photosynthetic system and tolerates salinity up to 600 mM12. The small genome size, 342 Mb, and high salt tolerance ability of A. littoralis, make it a suitable candidate for salinity studies in monocotyledons. The transcriptome analysis of A. littoralis in different salt stress conditions can extend the molecular knowledge and identifying undercover mechanisms of salt resistance in halophytes. These findings can be subsequently used for improving economically important crop plants in genetic engineering and plant breeding programs. This study is the first report of high-throughput analysis (de novo RNA-Seq) of A. littoralis transcriptome that provides new insights at molecular responses of halophytes to salinity.
Results
Biochemical assessments
After applying salt stress, the accumulation of salt crystals on the leaf surfaces of A. littoralis was apparently observed in both treatments (Fig. 1a). The variations in protein content and enzyme activities of salt treated samples in comparison with control plants clearly confirmed the incidence of salt stress. The results showed that protein content in both 200 and 400 mM NaCl treatments was significantly increased compared to control as well as compared to each other (Fig. 1b). Moreover, the SOD enzyme activity was considerably increased compared to control, however, no significant difference was observed between two salt treatments (Fig. 1c). In contrast, CAT enzyme activity was significantly decreased in both salt treatments compared to control and each other (Fig. 1d).

The effect of salinity on A. littoralis after 72 h of NaCl treatment (0, 200 and 400 mM) under hydroponic condition. (a) salt crystals on the leaf surfaces of A. littoralis, (b) leaf protein content of treated plants, (c) SOD enzyme activity and, (d) CAT enzyme activity of plant leaf samples. Data are presented as the mean of three biological replicates with error bars indicating SEM (n = 3). Different letters indicate significant differences at P ≤ 0.05.
Na+ and K+ concentrations
The Na+ concentration in A. littoralis leaves considerably increased in 200 and 400 mM NaCl treatments compared to the control (P < 0.05) after 72 h. However, the K+ content of leaf tissues was remained almost the same and no significant effect of NaCl concentration was observed. NaCl treatment showed a drastic effect on the K+/Na+ ratio in the salt treated plants and the ratio was significantly lower than control plants, though the difference between 200 and 400 mM salt treated plants was not considerable (Fig. 2).

The Na+ and K+ contents and the K+/Na+ ratio in leaf samples of A. littoralis treated with different NaCl concentrations (0, 200 and 400 mM) after 72 h. Data are presented as the mean of three biological replicates with error bars indicating SEM (n = 3). Different letters indicate significant differences at P ≤ 0.05.
RNA sequencing andde novotranscriptome assembly
For RNA sequencing, nine cDNA libraries from three treatments (0, 200 and 400 mM NaCl) were generated, and a total of 364937505 clean reads were assembled using Trinity and Bridger tools with 25 and 32 k-mer sizes. The detailed information of the read numbers in different samples is provided in Supplementary File 2.
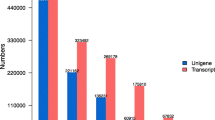
The results of assembly methods comparison showed that the 32 k-mer size is better in both assemblers. Summary of assembly statistics and BUSCO results are shown in Table 1 and Supplementary File 1 (Fig. S1), respectively. Assembly quality metrics revealed that Bridger (k-mer 32) transcriptome is the most appropriate, with the largest N50 value (2076). In addition, assembly completeness evaluation by BUSCO showed better results for Bridger (k-mer 32) transcriptome and 88.1% of 1440 conserved plant genes were present in the assembled contigs. Whereas, Trinity (k-mer 32) assembled transcriptome includes 85.2% of the conserved plant-specific orthologous genes. Therefore, the de novo transcriptome generated by Bridger (k-mer 32) was selected for subsequent analysis. This transcriptome includes 103290 transcripts (isoforms) and 83344 genes with a GC content of 49.72%. Length distribution of assembled contigs by Bridger (k-mer 32) is shown in Supplementary File1 (Fig. S2). By considering transcript expression, 17487 transcripts represented 90% of the total expression data (Ex90) and had an N50 of 2254 bp (Ex90N50).
Functional annotation of the A. littoralis transcriptome
Selected assembled transcripts were annotated by different tools (BLAST, HMMER, TransDecoder, RNAMMER v1.2, TMHMM v2.0, SignalP v4.1, and KASS) and public databases (UniProtKB, Rfam, miRbase, Pfam, Rnammer, tmHMM, signalp, GO and KEGG). Candidate coding regions in A. littoralis transcriptome (103290 transcripts) were identified by TransDecoder and 142168 ORFs and 62586 probable coding sequences were predicted. Sequence homology search results against the UniprotKB/SwissProt database by BLASTP (E-value > 1e−5, for predicted protein sequences) and BLASTX (E-value > 1e−5, for assembled transcripts) were 40809 (39.5%), and 42710 (41.34%) annotated contigs, respectively. The BLASTX against the uniprot_trEMBL_plants database revealed that A. littoralis transcripts have highest similarity with Poales and Poaceae family (92%). The similarity species distribution of Poaceae family was as Arundo donax (15%), Setaria viridis (12%), Panicum miliaceum (9%), Zea mays (7%), Sorghum bicolor (7%), Setaria italica (6%) and other Poaceae (14%) (Supplementary File1, Fig. S3).
A total of 155341 probable domains in A. littoralis transcriptome were identified by searching against Pfam database and, these domains were classified into 9775 unique domain groups. The four most abundant domain groups were protein kinase, protein tyrosine kinase, phosphotransferase enzyme family, and PPR repeat. The homology search against the miRBase database resulted in the annotation of 105 transcripts as potential miRNAs. In general, 53262 contigs (51.5% of all transcripts) annotated within at least one functional database. The Venn diagram of overlapping annotated transcripts in A. littoralis transcriptome against different databases is shown in Supplementary File 1 (Fig. S4). Detailed information on the annotations is represented in Supplementary File 3.
Functional annotation was followed by GO analysis and 41764 transcripts were categorized into 76 GO terms. The number of transcripts in three main categories of biological process (BP), molecular function (MF) and cellular component (CC) was 34385 (33.2%), 35949 (34.8%) and 36074 (34.9%), respectively. The most dominant GO term in the biological process category was “zinc ion binding,” with 12517 transcripts, followed by “sequence-specific DNA binding” (7015 transcripts) and “endonuclease activity” (4127 transcripts). In the molecular function category, “RNA modification” (13625 transcripts), “defense response” (8808 transcripts) and, “cell division” (3955 transcripts) were the most prominent. In the cellular component category, “mitochondria” (22119 transcripts), “plant-type cell wall” (5881 transcripts) and “integral component of membrane” (4596 transcripts) were the most abundant terms (Supplementary File 1 & Supplementary Files 4–6). KEGG enrichment analysis was performed to identify active metabolic processes in A. littoralis transcriptome. In conclusion, 17832 transcripts were assigned to 390 KEGG pathways and 42630 transcripts were assigned to at least one KEGG pathway or GO term (Supplementary File 3).
PCA and differential expression analysis
Results of PCA analysis revealed the distinct differences in transcript expression patterns among the treatments. As can be seen in Fig. 3, the first two principal components contain 71.18% of the information and the treatments are grouped in separate clusters that highlights particular response of A. littoralis to different salt concentrations. The first principal component contains 52.35% of the variance and the second principal component contains 18.83% of the variance.

Principal component analysis (PCA) on the read counts of A. littoralis in response to different NaCl concentrations (0, 200 and 400 mM).
For finding the differentially expressed transcripts, two pair-wise comparisons, 200 mM vs. control and 400 mM vs. control, were carried out (log2FC > 1 and FDR < 0.01). A total of 485 differentially expressed transcripts were identified in the 200 mM vs. control comparison, including 169 up and 316 down-regulated transcripts. Moreover, 1465 differentially expressed transcripts were found in the pair-wise comparisons between 400 mM and control, including 1035 up-regulated and 430 down-regulated transcripts. In up-regulated transcripts (1204 transcripts), 89 transcripts were common in both salt treatments, 944 and 80 transcripts were specifically up-regulated in 400 and 200 mM salt treatments, respectively. On the other hand, in 746 down-regulated transcripts, 49 transcripts were common in both salt treatments, 381 and 265 transcripts were specifically down-regulated in 400 and 200 mM NaCl treatments, respectively. Interestingly, two transcripts with different expression patterns (up-regulation in 400 and down-regulation in 200 mM) were identified in both treatments. The detailed information of the differentially expressed transcripts in each of pair-wise comparisons is provided in Supplementary File 7.
By homology search of the differentially expressed transcripts against the PlantTFDB database, a total of 67 transcripts were annotated and categorized into 20 families. ERF and WRKY were the most abundant TF families with 24 and 9 transcripts, respectively. In 400 mM NaCl treatment, 48 TFs were up and 17 TFs were down-regulated, while in 200 mM salt treatment, 2 TFs were up and 4 TFs were down-regulated (Supplementary File 8).
Functional enrichment of differentially expressed genes
To investigate the function of differentially expressed genes (DEGs), GO enrichment analysis was performed using goseq package. Among the 1035 up-regulated DEGs in 400 mM NaCl treatment, 514 genes were significantly assigned to 120 GO terms (FDR < 0.05). Amongst 68 GO terms in the biological process (BP), “chitin response” (child GO terms; response to abscisic acid, regulation of jasmonic acid mediated signaling pathway, stress-activated protein kinase signaling cascade, response to lipid, ethylene-activated signaling pathway and else), “polysaccharide catabolism” (child GO terms; phosphatidylcholine biosynthetic process, oxidation-reduction process, polysaccharide catabolic process, else), and “response to stimulates” were the most important terms. Furthermore, 50 GO terms were involved in molecular function (MF), in which “hydrolyzing O-glycosyl compounds” and “amylase activity” were the dominant categories. The “PRC1 cell complex” and “vacuole” terms were also enriched in cellular component (CC). Moreover, 145, 149, and 72 out of 430 down-regulated DEGs in 400 mM NaCl treatment were significantly assigned to 12 BP, 27 MF and 6 CC, respectively (FDR < 0.05).
Up-regulated DEGs in 200 mM NaCl treatment (169 DEGs) were grouped in 7 BP (including “cell cycle”, “cell division” and “mitotic cell cycle process”), 27 MF and 6 CC terms. Also, 2 MF and 2 CC terms in 200 mM NaCl treatment were enriched by 316 down-regulated DEGs, while no significant BP terms were found in this treatment (Supplementary File 9). The top five over-represented GO terms in BP, MF and CC categories (FDR < 0.05) are reported in Table 2.
KEGG enrichment
Generally, DEGs were assigned to five categories in KEGG database, pathways related to metabolism, genetic information processing, environmental information processing, cellular process, and organismal systems. Results of GOseq analysis revealed that in 200 mM NaCl treatment just one pathway, DNA replication proteins (ko03032), was significantly enriched in up-regulated genes, and no pathways were enriched in down-regulated genes. While, in 400 mM NaCl treatment, six and three pathways were significantly enriched with up and down-regulated genes, respectively (Table 3). Two pathways of plant MAPK signaling pathway (ko04016), and plant hormone signal transduction (ko04075) were significantly enriched in both up and down-regulated genes in 400 mM NaCl treatment.
Phytohormone signal transduction
This pathway was significantly enriched in both up and down-regulated genes in 400 mM NaCl treatment. Moreover, our results showed that the signaling pathway of most plant hormones including auxin, gibberellin, abscisic acid, ethylene, jasmonic acid, brassinosteroid, and salicylic acid was affected by the treatment of 400 mM NaCl treatment. In this regard, the PYL gene was down-regulated and PP2C, ABF and, SNRK2 genes up-regulated in the ABA signaling pathway (Fig. 4). In addition, in the signaling pathway of auxin hormone, all three classes of primary auxin-response genes (IAA, GH3 and SAUR) were strongly affected by 400 mM NaCl. Hence, GH3 and SAUR genes were over-expressed and IAA gene was down-regulated. Also, the down-regulation of IAA and PYL genes were observed in 200 mM NaCl treatment (Fig. 4). For additional details see Supplementary File 10.

The effect of 200 and 400 mM NaCl treatment on phytohormone signal transduction pathway (Ko04075). Auxin-responsive protein IAA (AUX/IAA), auxin responsive GH3 gene family (GH3), SAUR family protein (SAUR), abscisic acid receptor PYR/PYL family (PYR-PYL), protein phosphatase 2 C (PP2C), serine/threonine-protein kinase SRK2 (SNRK2), ABA-responsive element binding factor (ABF), EIN3-binding F-box protein (EBF1_2), xyloglucan: xyloglucosyl transferase TCH4 (TCH4), jasmonate ZIM domain-containing protein (JAZ), transcription factor TGA (TGA) and pathogenesis-related protein 1 (PR-1). Plant Hormone Signal Transduction Pathway adapted from KEGG77.
MAPK signaling pathway
Another important pathway of message transduction, which significantly enriched in 400 mM treatments with DEGs (up and down-regulated) was MAPK signaling pathway. Whereas the MAPK signaling pathway was not significantly enriched in 200 mM treatment, but two genes (ACS6 and PYL) were significantly down-regulated in the pathway (Fig. 5). The ACS6 showed the highest down-regulation (238.85 fold, log2FC = −7.9) in both 200 and 400 mM NaCl treatments. On the other hand, the PP2C (protein phosphatase 2C) showed the highest up-regulation (32 fold, log2FC = 5) in the 400 mM NaCl treatment (Supplementary File 10).

The effect of 200 and 400 mM NaCl on plant MAPK signaling pathway (Ko04016). WRKY transcription factor 33 (WRKY33), 1-aminocyclopropane-1-carboxylate synthase 1/2/6 (ACS6), abscisic acid receptor PYR/PYL family (PYR-PYL), protein phosphatase 2 C (PP2C), EIN3-binding F-box protein (EBF1_2), basic endochitinase B (ChiB) and calmodulin (CaM4). MAPK Signaling Pathway-Plant adapted from KEGG77.
DNA replication
In the 200 mM NaCl treatment, only DNA replication-associated proteins with up-regulated genes were significantly enriched. However, no pathway was found for the down-regulated genes. Subunit I DNA polymerase epsilon, factor MCM3 of helicase, the large subunit of DNA primase, and replication factor A1 of RPA were significantly up-regulated. Subunit I DNA polymerase epsilon was the only component that was identified in both salt treatments (Supplementary File 10). In the 400 mM NaCl treatment, no transcripts related to DNA replication pathway were significantly enriched.
Validation of DEGs by qRT-PCR
To validate RNA-seq results, four differentially expressed transcripts in the RNA-seq experiment were randomly selected for qRT-PCR. The results showed that the DEGs selected for validation exhibited similar up or down-regulation patterns as those shown from RNA-seq data analysis (Fig. 6), although the observed relative expression differences between RNA-seq and qRT-PCR is owing to intrinsic features of these methods.

qRT-PCR validation of RNA-seq data. Four differentially expressed genes in the RNA-seq experiment were analyzed by qRT-PCR. Data are shown as the fold change at 200 and 400 mM NaCl relative to the control (No NaCl). The expression data were normalized to gapdh as a reference gene. Relative expression was calculated based on the 2−ΔΔCt method and represent the mean expression value ± SEM (n = 3) relative to the control. (*) represents a statistically significant difference (P ≤ 0.05) when compared to the control.
Discussion
Salt stress affects various biochemical, physiological and molecular processes in plants. The halophytes, such as A. littoralis, have a variety of well-adapted mechanisms to encounter with the saline conditions and mitigate the adverse effects of salinity on the plant growth and development.
It is thought that Na+ and K+ homeostasis is crucial for salt-tolerance in plants13. In our study the Na+ content of A. littoralis leaves at 200 and 400 mM NaCl increased in comparison with the control plants. However, the Na+ concentration difference in 200 and 400 mM salt treated plants was not significant. This could be the result of excretion of elevated levels of Na+ from the salt glands on the plant leaf surface. Similarly, accumulation of Na+ in the shoots and roots of A. littoralis14,15 and A. lagopoides16 in different salt stress treatments has been reported. It has shown that halophyte grasses like Aeluropus spp. may employ more efficient mechanisms for K+ homeostasis and Na+ exclusion than ion sequestration mechanisms generally used by dicot halophytes17,18.
Study of the salt tolerance mechanisms in halophyte plants provides valuable information for designing crop breeding programs. Besides, de novo transcriptome assembly method offers a notable opportunity to reconstruct reference transcriptome in non-model organisms without reference genome sequence as well as allows the discovery of novel genes. In the present study, the transcriptional response of A. littoralis, a halophyte plant (Poaceae), was evaluated under two salt concentrations in 72 h after stress using de novo RNA-seq analysis. To reconstruct the reference transcriptome, two assemblers including Trinity and Bridger, which are the most reliable and widely used assembly tools, were chosen19,20,21. The Trinity package and Bridger have already been used to assemble the transcriptome of several plant species21,22,23. The key idea of Bridger’s algorithm is to create a bridge between the reference genome-based assembly tools (e.g. Cufflinks) and those which assemble without the reference genome (e.g. Trinity)24. The results of this study showed that the contigs obtained from Bridger with a length of 32 k-mer provides better data for performing downstream analyses. Although Trinity had more assembled bases than Bridger, length-related statistics indicated the higher quality of Bridger in both k-mers. Furthermore, results of BUSCO analysis showed the superiority of Bridger to Trinity, which is in agreement with previous studies (Table 1 and Supplementary File 1 (Fig. S1))25,26. Therefore, the de novo assembled transcriptome by Bridger (32 k-mer) was used for further analysis. The GC content of the reference transcriptome was 49.72%, which is in the range of previous studies in Festuca arundinacea (49.49%) and Arundo donax (49%) transcriptomes (both belongs to Poaceae family)27,28.
To provide a biological interpretation of assembly results, the differential expression analysis and functional annotation of 103290 transcripts were done. Results of GO enrichment of DEGs revealed that the phosphatidylcholine biosynthetic process was the only common term between two salt treatments in BP category. Accordingly, most of the well-known terms related to stress such as oxidation-reduction process, ROS metabolic process, response to abscisic acid and regulation of jasmonic acid mediated signaling pathway were induced under 400 mM NaCl treatment. In contrast, cellular-related biological processes like cell cycle and cell division were induced under 200 mM NaCl (Supplementary File 9). These results indicated that A. littoralis responses to 200 and 400 mM NaCl in the completely different manners. Hence, it can be stated that 200 mM NaCl is not detected as stress condition, but 400 mM NaCl induces the most stress-related terms in the A. littoralis. Our results support previous findings that 200 mM NaCl enhances the biomass of A. littoralis in liquid MS culture medium29 and also increases the root length in comparison with control plants30.
Phosphatidylcholine is a dominant phospholipid in the cell membrane and forms 40 to 60% of membrane lipids31. It seems that in higher plants this compound, due to its role in maintaining the cell membrane integrity, is a major contributor to the adaptation response to environmental stresses32. In the present study, two transcripts encoding phosphoethanolamine N-methyl transferase (NMT, EC:2.1.1.103) and choline-phosphate cytidylyl transferase (PCYT1, EC:2.7.7.15) were up-regulated under both salt treatments in the phosphatidylcholine biosynthetic process. Up-regulation of these genes under salt stress was also reported in the comprehensive transcriptome analysis of Sporobolus virginicus halophyte plant33. Mou et al. (2002) reported that inactivation of NMT gene in Arabidopsis caused a high sensitivity to salt stress, therefore they suggested that this enzyme plays an important role in the salt stress tolerance34.
Interestingly, our results indicated that chromatin modification plays an important role in the response to salinity stress in A. littoralis. In this regard, the PRC1 complex which plays a major role in chromatin remodeling was enriched in up-regulated genes in 400 mM NaCl treatment (Supplementary File 9). In Drosophila melanogaster, as a model organism in chromatin studies, the PRC1 complex mediates the ubiquitination of the lysine residue in histone H2A and is involved in maintaining long-term transcriptional suppression and plays role in the epigenetic alteration of chromatin remodeling35. It has long been thought that PRC1 complex is absent in plants, but this opinion was changed by the discovery of the ubiquitinated residue of H2A histone in A. thaliana. In plants, PRC1 complex has five subunits and is related to gene deactivation36. Furthermore, other evidences such as the presence of nucleosome/chromatin assembly factor group D13 (HMGB13) and structural maintenance of chromosomes 4 (SMC4) genes in top 20 over-expressed genes in both salt treatments (Supplementary File 7) directed us to conclude about the importance of chromatin modification in response to salinity stress. HMGBs involves in DNA-related cellular processes such as duplication, transcription, and chromatin assembly37. The Damage-associated molecular pattern-like activities of HMGB3 was reported in Arabidopsis36. Besides, expression changes of some members of the HMG family have been reported in response to abiotic and biotic stresses in Arabidopsis38,39. The SMC4 is one of the catalytic members of the chromatin condensing complex which participates in the chromatin condensation during mitosis and chromosome segregation in meiosis38. It has been reported that SMC4 has an important role in the formation of Arabidopsis epigenome and collaborates with DNA methylation to assemble suppressor chromatin complexes40. Different expression pattern of ALP1 (protein-antagonist of like heterochromatin protein 1), another gene that involves in chromatin remodeling, was observed in 200 mM (log2FC = −8.72) and 400 mM (log2FC = 3.6) NaCl treatments (Supplementary File 7). The role of ALP1 in opposing PcG protein-mediated chromatin silencing in Arabidopsis has been reported41. Altogether, these results indicated that the mentioned pathways and genes may have led to chromatin modification in response to salinity stress in A. littoralis.
Plants as immobile organisms are constantly subjected to the environmental stimuli, so they need an efficient and effective signaling system to response to these stimuli. Based on the KEGG enrichment results, the plant MAPK signaling and phytohormone signaling pathways were enriched with both up and down-regulated transcripts under 400 mM NaCl treatment. Although, these pathways were not enriched under 200 mM NaCl treatment, but some transcripts including IAA, PYL, and ACS6 were showed notable differential expression compared to control (Fig. 4 and 5).
MAPK cascades are known as a key signaling pathway involved in response to various stresses42,43. Although in our results, kinase proteins of this pathway were not detected, but some of downstream genes such as WRKY33 showed differential expression after 72 h salt stress. Expression of WRKY33 transcription factor was up-regulated in 400 mM NaCl treatment, and no change was observed in 200 mM treatment. Salt stress sensitivity has been increased in wrky33 null and wrky25wrky33 double mutants of Arabidopsis. However, overexpression of PcWRKY33 (Polygonum cuspidatum) has decreased salt stress tolerance in Arabidopsis42.
Phytohormones play an important role in regulation of plant response to different stresses. In our study, signaling pathways of the abscisic acid (ABA), auxin, ethylene, Brassinosteroids, jasmonic acid, and salicylic acid were affected by 400 mM NaCl treatment.
Stimulation of plant hormone signaling pathways is shown in response to salinity stress44. ABA is known as stress hormone and plays a critical role in response to various stresses45,46. All key genes of ABA signaling pathway, including PYL and PP2C, were induced under 400 mM salt treatment, but only PYL was affected by 200 mM NaCl treatment. PYL was down-regulated in both salt treatments, while PP2C was up-regulated in 400 mM NaCl treatment (Fig. 4 & Supplementary File 10). Our results showed that up-regulation of both PYL and PP2C genes is not crucially necessary for steadiness of ABA signaling pathway, that is in consistent with previous findings46.
Auxin signaling genes classified into three main classes including auxin response genes, auxin response factors, and primary auxin-response genes (including IAA, GH3 and SAUR). Our results showed that all three primary auxin-response genes were strongly affected by 400 mM NaCl (Fig. 4 & Supplementary File 10). In several studies up-regulation of various members of the GH3 family in sorghum, chickpea, medicago and rice under salt and drought stress is reported47,48,49. For instance, it has shown that the majority of OsGH3 genes, namely OsGH3-2 and OsGH3-8 were up-regulated in rice under salt stress49.
The DNA replication pathway was the only enriched pathway in 200 mM treatment, and all related genes (including PRI2, POLA, MCM3, and RAF1) were up-regulated. To the best of our knowledge, the reports on the effect of salt treatments on the genes involved in DNA replication pathway are limited. It has been shown that a component of the MCM2–7 complex may function as a DNA helicase which is necessary for replication initiation and elongation per cell cycle in eukaryotic cells50. Transcriptome analysis of seagrass Zostera marina L. by RNA-seq showed that DNA replication pathway was significantly enriched by up-regulated genes such as the MCM6 gene. The enhanced salinity stress tolerance in tobacco plants due to overexpression of MCM6 has been reported. In addition, it is shown that promoter region of this gene contains responsive elements to salinity and cold stresses51.
Among salinity stress-responsive genes, TFs play a key role in regulating the molecular responses at the transcriptional level50. In this study, 67 differentially expressed TFs were found within 20 different families, in which the ERF, WRKY, C2H2, NAC, bZIP, MYB-related and MYB were the largest families (Supplementary File 8). Previous studies in plants have been reported the response of same TF families to the salt stress52,53. The comparison between the two salt treatments showed that the number of families and their members in the 400 mM treatment was remarkably higher than 200 mM treatment. Most of the differentially expressed TFs in both treatments were related to ERF family. In this regard, 16 and 7 members of this family were up and down-regulated in the 400 mM NaCl treatment, respectively. In 200 mM NaCl treatment, all four down-regulated TFs were belongs to ERF family (Supplementary File 8). It is well known that the activation of a large number of stress-responsive genes is mediated through specific TFs, which play important roles in the regulation of secondary metabolites biosynthesis in plants. The role of ERF family in initiation of appropriate response to salt stress has been confirmed in previous studies54,55,56. Significant induction of ERF76 gene has been reported in the leaves, roots, and stems of Populus simonii × Populus nigra under salt stress. Overexpression of the ERF76 gene increased the tolerance of transgenic poplar to salt stress55, indicating that ERF family could be considered as an important family of TFs in response to stress. Additionally, members of GRF family (lesser-known in salt stress), was identified in 200 mM NaCl treatment (Supplementary File 8). In contrast to the ERF, GRF is a small family of plant-specific TFs that involves in growth, development, and reproduction and differentiation processes57. Recently a few studies reported the role of this family in coordination plant growth in abiotic and biotic stress conditions57,58,59. For example, seven genes of GRFs family were differentially expressed in the tomato leaves at different time points under NaCl stress57.
It should be noted that a little proportion of up and down-regulated transcripts in 400 mM and up-regulated transcripts in 200 mM salt treatment were categorized as unknown function (NA) transcripts. On the other hand, more than 50% of down-regulated transcripts in 200 mM NaCl treatment with high expression value categorized as NA (Supplementary File 7).
Conclusion
In this study the A. littoralis responses to salinity stress at transcriptome level were investigated by de novo RNA-seq analysis. Our findings highlighted the well-known salt tolerance mechanisms such as MAPK and phytohormones signaling, synthesis of osmolytes, and ionic homeostasis under 400 mM NaCl treatment. In contrast, the processes related to growth such as cell division and DNA replication were enriched in GO and KEGG analysis by up-regulated transcripts under 200 mM NaCl treatment. Several pieces of evidence supported the key role of chromatin remodeling in response to salinity stress in A. littoralis. Finally, our findings suggest that A. littoralis responses to salt stress are greatly salt concentration dependent. It seems likely that 200 mM NaCl is not serves as stress condition and actually enhances the plant growth, but 400 mM NaCl induces plant tolerance mechanisms. This study has gone some way towards increasing our understanding of salt tolerance molecular mechanisms in halophyte plants. Future studies will focus on lesser-known processes and pathways and their relation with salinity tolerance in A. littoralis.
Materials and Methods
Plant material
The A. littoralis seedlings were grown under hydroponic culture in Hoagland solution60 (pH = 5.8) and greenhouse condition (photoperiod 16 h light and 8 h darkness, 27 °C and 65% relative humidity). To minimize the possible genetic variation of plant material, a single plant was propagated by successive cutting. After obtaining adequate plant materials, rooted cuttings were grown in the pots. Aeration was carried out using air pumps, and Hoagland solution was replaced with fresh solution every two weeks. The one-month seedlings were subjected to different concentrations of NaCl (200 and 400 mM). After 72 h of salt treatment, leaf, stem, and root samples were harvested from the control (0 mM NaCl) and salt-treated plants and were stored in −80 °C.
Preparation of protein extract
For preparing the plant protein extract, 200 mg of fresh leaf samples were homogenized with 1800 μl of 50 mM potassium phosphate buffer (pH 7.0). The homogenates were centrifuged at 20000×g for 20 min at 4 °C. The supernatant was dispensed in aliquots and stored at −80 °C for subsequent protein content determination and enzyme activity assays.
Protein content measurement
Protein content of leaf samples (three replicates) was determined according to the method of Bradford61. Briefly, 200 μl of protein extract, 1800 μl of distilled water and 500 μl of Bradford solution were mixed and the absorbance was recorded at 595 nm. Protein concentration is calculated from standard curve of Bovine Serum Albumin (BSA).
SOD enzyme activity assay
The activity of SOD enzyme was measured by the evaluation of its inhibitory effect on nitrobluetetrazolium (NBT)62. The 2 ml assay reaction mixture contained phosphate buffer (50 mM), methionine (0.013 mM), EDTA (0.1 μM) and riboflavin (2 μM). Immediately after adding riboflavin, reaction mixture was poured into the test tube containing 50 μl of protein extract and exposed to the light for 15 min. The absorbance of samples was measured at 560 nm. The activity of SOD enzyme was calculated based on the enzyme unit per g fresh weight (U gFW−1).
Catalase enzyme activity assay
CAT enzyme activity was determined by measuring the decrease in absorbance at 240 nm per unit time due to decomposition of H2O2. The reaction mixture consisted of 1000 μl of 100 mM phosphate buffer (pH = 7) and 750 µl of 15 mM H2O2. The reaction was started by adding 75 μl of the enzyme extract. The enzyme activity was expressed as enzyme unit per g fresh weight (U gFW−1)63.
Na+ and K+ determination
The plant leaves were thoroughly rinsed with deionized water and then dried at 80 °C for 72 h. 500 mg of dried leaf samples were kept in a furnace at 550 °C for 6 h and the resulting ash was extracted with 5 ml 2 N HCl for 30 min. The solutions were diluted with deionized water and after filtration, the amount of Na+ and K+ was determined with a PFP7 Flame Photometer, GENWAY, UK.
Statistical Analysis
Effect of different salinity treatments on protein content, the SOD, and catalase enzymes activity were analyzed using the GLM procedure of SAS statistical software version 9.1 according to the following statistical model:
Yij = μ + Ti + eij
Which, Yij is the E-value of the studied trait, μ is mean of studied trait, Ti is the effect of treatments and eij are random errors. The mean of the treatments was compared using Duncan’s multiple range test.
RNA extraction and Illumina sequencing
Total RNA was isolated using RNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s protocol and treated with DNase I (Fermentas). RNA quantity and quality were evaluated using spectrophotometry, Agilent 2100 Bioanalyzer and 1% agarose gel. RNA samples with a ratio of 260/280 nm and 28 S/18 S greater than 1.8 and RNA integrity number (RIN) > 8 were selected for subsequent processes. Subsequently, nine cDNA libraries were constructed and each cDNA library was sequenced using the Illumina HiSeq 2500 platform to generate 150 bp paired-end reads.
Quality control and de novo assembly
The quality of the raw reads was evaluated using the FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)64 and the low-quality bases (<20) and adapter contamination were removed using Trimmomatic v0.3865. The trimmed data were again evaluated by FastQC to confirm the quality of reads.
To obtain the most appropriate de novo transcriptome assembled results and increase the confidence of data quality, two different transcriptome assembly approaches including Trinity v2.4.022,66 and Bridger24 were compared. Each assembler was run with two k-mer sizes (25 and 32 base pair). While Trinity employs the De Bruijn method to perform assembly25,26, Bridger applies a rigorous mathematical model to construct splicing graphs27. Following, the four contig datasets, Trinity (25-mer), Trinity (32-mer), Bridger (25-mer) and Bridger (32-mer) reassembled by Cap3 tool67 to perform a meta-assembly, reducing redundancy and generating more complete consensus sequences (cutoff 95%). Finally, the results were evaluated by different quality metrics including N50 length of the transcriptome assemblies, the total number of bps in the assembly and BUSCO v2.0.1 analysis using OrthoDB v9.1 ‘embryophyta’ database as a reference to assess the assembly and annotation completeness68.
Functional annotation
The best-assembled transcriptome was annotated using several approaches. First, TransDecoder tool69 was used to find the probable coding regions of transcripts and the open reading frames (ORFs) with a minimum length of 100 amino acids. Then, functional annotation of the transcripts was performed using BLASTX against UniProtKB/SwissProt databases and uniprot_trEMBL_plants database (E-value <1e−5). Moreover, a homology search based on the BLASTP was performed using the predicted proteins as query against UniProtKB/SwissProt databases (E-value <1e−5). To recognize homologous non-coding RNAs as well as miRNAs, transcript sequences were aligned against the miRbase and Rfam databases using BLASTN tool (E-value <1e−5). To determine the protein domains, all transcripts with predicted ORFs were used for searching conserved functional domains against Pfam database by HMMER tool (version 3.1b2) at an E-value <1e−5. Signal peptide and transmembrane domain prediction were performed by signalP (version 4.1) and TMHMM server (version 2.0) tools, respectively. The assignment of Gene Ontology (GO) terms to transcripts were performed based on UniProtKB/SwissProt database to assign unigenes to functional categories. Moreover, online KEGG Automatic Annotation Server (KAAS; http://www.genome.jp/kegg/kaas/) was used to assign pathway information to the transcripts based on 38 plant species and single-directional best hit (SBH) method.
Differential expression analysis
RSEM tool (http://deweylab.biostat.wisc.edu/RSEM)70 was applied to estimate the expression level of transcripts. Initially, each of the cDNA libraries was separately aligned to the reference transcriptome using the Bowtie 2.071. Then, the expression level of each transcript was normalized based on the library size (sequencing depth) and transcripts length and reported in fragments per kb per million (FPKM). To summarize the results and provide statistical tests for pairwise comparisons, the differential expression analysis was performed using edgeR package72 (FDR = 0.01 and log2FC = 1). Principal component analysis (PCA) was performed to assess the correlation between replicates and treatments. As well, to identify differentially expressed transcription factors (TFs), the homology search was performed against the plant TFs database (PlantTFDB-all_TF_pep) using plantTFDBv4.0 prediction server (http://planttfdb_v4.cbi.pku.edu.cn/prediction.php).
Gene ontology and KEGG enrichment
To investigate the functional enrichment of the differentially expressed transcripts and visualize the results, the GOseq73 and GOplot74 packages were used, respectively. The pathways annotation of up and down-regulated transcripts was performed by KAAS (http://www.genome.jp/kegg/kaas/)75. The significantly over-represented pathways were screened using the GOseq package (FDR < 0.05).
RNA-seq results validation by qRT-PCR
Four differentially expressed transcripts in RNA-seq experiment were randomly selected for RNA-seq validation by qRT-PCR. The Primer3 software was used to design the specific primer pairs (Supplementary File 11), and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a reference gene. qRT-PCR was done using Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific). The 15 μl reaction mixture contained 1.0 μl of diluted cDNA sample, 0.5 μl of each forward and reverse primers (10 pM), 10 μl real-time SYBR Green master mix and 3 μl distilled water. The cycling conditions were as initial denaturation at 95 °C for15 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s. Each sample was quantified in three biological replicates. The relative gene expression was quantified by Livack and Schmittgen (2001) method76.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Koushafar, M., Khoshgoftarmanesh, A. H., Moezzi, A. A. & Mobli, M. Effect of dynamic unequal distribution of salts in the root environment onperformance and crop per drop (CPD) of hydroponic-grown tomato. Sci Hortic. 131, 1–5 (2011).
Zhu, J. K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant. Biol. 53, 247–273 (2002).
Flowers, T. J. & Colmer, T. D. Salinity tolerance in halophytes. New Phytol. 179, 945–963 (2008).
Wang, J. et al. Transcriptomic profiling of the salt-stress response in the halophyte Halogeton glomeratus. BMC Genomics. 16, 169 (2015).
Bartels, D. & Sunkar, R. Drought and salt tolerance in plants. Crit. Rev. Plant. Sci. 24, 23–58 (2005).
Hanin, M., Ebel, C., Ngom, M., Laplaze, L. & Masmoudi, K. New insights on plant salt tolerance mechanisms and their potential use for breeding. Front plant sci. 7, 1787 (2016).
Yamaguchi-Shinozaki, K. & Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant. Biol. 57, 781–803 (2006).
Pennisi, E. Genomics. ENCODE project writes eulogy for junk DNA. Science 337, 1161 (2012).
Flintoft, L. Transcriptomics: Digging deep with RNA-Seq. Nat Rev Genet. 9(8), 568–568 (2008).
Wang, Z., Gerstein, M. & Snyder, M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10, 57–63 (2009).
Bose, J., Rodrigo-Moreno, A. & Shabala, S. ROS homeostasis in halophytes in the context of salinity stress tolerance. J. Exp. Bot. 65, 1241–1257 (2014).
Gulzar, S. & Khan, E. Seed germination of a halophytic grass Aeluropus logopoides. Ann. Botany. 87(3), 319–324 (2001).
Rouached, H., Secco, D. & Arpat, B. A. Regulation of ion homeostasis in plants: current approaches and future challenges. Plant Signal. Behav. 5(5), 501–502 (2010).
Moshaei, M. R., Nematzadeh, G. A., Askari, H., Nejad, A. S. M. & Pakdin, A. Quantitative gene expression analysis of some sodium ion transporters under salinity stress in Aeluropus littoralis. Saudi. J. Biol. Sci. 21(5), 394–399 (2014).
Fakhrfeshani, M., Shahriari-Ahmadi, F., Niazi, A., Moshtaghi, N. & Zare-Mehrjerdi, M. The effect of salinity stress on Na+, K+ concentration, Na+/K+ ratio, electrolyte leakage and HKT expression profile in roots of Aeluropus littoralis. J. Plant Mol. Breed. 3(2), 1–10 (2015).
Sanadhya, P., Agarwal, P. & Agarwal, P. K. Ion homeostasis in a salt-secreting halophytic grass. AoB Plants 7, 1–15 (2015).
Rozema, J. & Schat, H. Salt tolerance of halophytes, research questions reviewed in the perspective of saline agriculture. Environ. Exp. Bot. 92, 83–95 (2013).
Munns, R. & Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 59, 651–681 (2008).
Rana, S. B., Zadlock, F. J. IV, Zhang, Z., Murphy, W. R. & Bentivegna, C. S. Comparison of de novo transcriptome assemblers and k-mer strategies using the killifish, Fundulus heteroclitus. PLoS One. 11(4), e0153104 (2016).
Honaas, L. A. et al. Selecting superior de novo transcriptome assemblies: lessons learned by leveraging the best plant genome. PLoS One. 11(1), e0146062 (2016).
Wang, S. & Gribskov, M. Comprehensive evaluation of de novo transcriptome assembly programs and their effects on differential gene expression analysis. Bioinformatics. 33(3), 327–333 (2017).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech. 29(7), 644 (2011).
Bankar, K. G., Todur, V. N., Shukla, R. N. & Vasudevan, M. Ameliorated de novo transcriptome assembly using Illumina paired end sequence data with Trinity Assembler. Genom. Data. 5, 352–359 (2015).
Chang, Z. et al. Bridger: a new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol. 16(1), 30 (2015).
Ragionieri, L. et al. Identification of mature peptides from pban and Capa genes of the moths Heliothis peltigera and Spodoptera littoralis. Peptides 94, 1–9 (2017).
Zhang, G. H. et al. De novo sequencing and transcriptome analysis of Pinellia ternata identify the candidate genes involved in the biosynthesis of benzoic acid and ephedrine. Front Plant Sci. 7, 1209 (2016).
Amombo, E. et al. Comprehensive Transcriptome Profiling and Identification of Potential Genes Responsible for Salt Tolerance in Tall Fescue Leaves under Salinity Stress. Genes. 9(10), 466 (2018).
Evangelistella, C. et al. De novo assembly, functional annotation, and analysis of the giant reed (Arundo donax L.) leaf transcriptome provide tools for the development of a biofuel feedstock. Biotechnol Biofuels. 10(1), 138 (2017).
Fatemi, F., Hashemi-petroudi, S. H., Nematzadeh, G., Askari, H. & Abdollahi, M. R. Exploiting Differential Gene Expression to Discover Ionic and Osmotic-Associated Transcripts in the Halophyte Grass Aeluropus littoralis. Biol. Proced. Online 21(1), 14 (2019).
Zouari, N. et al. Identification and sequencing of ESTs from the halophyte grass Aeluropus littoralis. Gene 404(1-2), 61–69 (2007).
Bolognese, C. P. & McGraw, P. The isolation and characterization in yeast of a gene for arabidopsis s-adenosylmethionine: Phospho-ethanolaminen-methyltransferase. Plant Physiol. 124(4), 1800–1813 (2000).
17. Tasseva, G., Richard, L., & Zachowski, A. Regulation of phosphatidylcholine biosynthesis under salt stress involves choline kinases in Arabidopsis thaliana. FEBS letters, 566, 115-120 (2004).
Yamamoto, N. et al. Comprehensive analysis of transcriptome response to salinity stress in the halophytic turf grass Sporobolus virginicus. Front. Plant Sci. 6, 241 (2015).
Mou, Z. et al. Silencing of phosphoethanolamine N-methyltransferase results in temperature-sensitive male sterility and salt hypersensitivity in Arabidopsis. Plant Cell. 14(9), 2031–204 (2002).
Shao, Z. et al. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 98(1), 37–46 (1999).
Berke, L. & Snel, B. The plant Polycomb repressive complex 1 (PRC1) existed in the ancestor of seed plants and has a complex duplication history. BMC Evol. Biol. 15(1), 44 (2015).
Reeves, R. Nuclear functions of the HMG proteins. Biochim Biophys Acta Gene Regul Mech. 1799, 3–14 (2010).
Kwak, K. J., Kim, J. Y., Kim, Y. O. & Kang, H. Characterization of transgenic Arabidopsis plants overexpressing high mobility group B proteins under high salinity, drought or cold stress. Plant Cell Physiol. 48(2), 221–231 (2007).
Lildballe, D. L. et al. The expression level of the chromatin-associated HMGB1 protein influences growth, stress tolerance, and transcriptome in Arabidopsis. J Mol Biol. 384(1), 9–21 (2008).
Wang, J. et al. Mutation of Arabidopsis SMC4 identifies condensin as a corepressor of pericentromeric transposons and conditionally expressed genes. Gene Dev 31(15), 1601–1614 (2017).
Liang, S. C. et al. Kicking against the PRCs–a domesticated transposase antagonises silencing mediated by Polycomb group proteins and is an accessory component of Polycomb repressive complex 2. PLoS Genet. 11(12), e1005660 (2015).
Jiang, Y. & Deyholos, M. K. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 69(1-2), 91–105 (2009).
Bao, W., Wang, X., Chen, M., Chai, T. & Wang, H. A WRKY transcription factor, PcWRKY33, from Polygonum cuspidatum reduces salt tolerance in transgenic Arabidopsis thaliana. Plant Cell Rep. 37, 1033–1048 (2018).
Fahad, S. et al. Phytohormones and plant responses to salinity stress: a review. Plant Growth Regul 75(2), 391–404 (2015).
Zhang, F. et al. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 6, 20582 (2016).
Park, S. Y. et al. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 324, 1068–1071 (2009).
Wang, S. et al. Auxin-related genefamilies in abiotic stress response in Sorghum bicolor. Funct. Integr. Genom. 10, 533–546 (2010).
Singh, V. K., Jain, M. & Garg, R. Genome-wide analysis and expression profiling suggest diverse roles of GH3genes during development and abiotic stress responses in legumes. Front. Plant. Sci. 5, 789 (2015).
Kong, W. et al. Evolutionary Analysis of GH3 Genes in Six Oryza Species/Subspecies and Their Expression under Salinity Stress in Oryza sativa ssp. japonica. Plants. 8(2), 30 (2019).
Stevens, R. et al. Two E2F sites in the Arabidopsis MCM3 promoter have different roles in cell cycle activation and meristematic expression. J. Biol. Chem. 277, 32978–32984 (2002).
Dang, H. Q., Tran, N. Q., Tuteja, R. & Tuteja, N. Promoter of a salinity and cold stress-induced MCM6 DNA helicase from pea. Plant Signal Behav. 6(7), 1006–1008 (2011).
Golldack, D., Lüking, I. & Yang, O. Plant tolerance to drought and salinity: stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep. 30(8), 1383–1391 (2011).
Tanaka, H., Suzuki, R., Okabe, N., Suzuki, T. & Kodama, Y. Salinity stress-responsive transcription factors in the liverwort Marchantia polymorpha. Plant Biotech. 35(3), 281–284 (2018).
Nakano, T., Suzuki, K., Fujimura, T. & Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant physiol. 140(2), 411–432 (2006).
Licausi, F., Ohme‐Takagi, M. & Perata, P. APETALA 2/Ethylene Responsive Factor (AP 2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol. 199(3), 639–649 (2013).
Thirugnanasambantham, K. et al. Role of ethylene response transcription factor (ERF) and its regulation in response to stress encountered by plants. Plant Mol. Biol. Report. 33(3), 347–357 (2015).
Kim, J. S. et al. Arabidopsis GROWTH-REGULATING FACTOR7 Functions as a Transcriptional Repressor of Abscisic Acid–and Osmotic Stress–Responsive Genes, Including DREB2A. The Plant Cell. 24(8), 3393–3405 (2012).
Hewezi, T., Maier, T. R., Nettleton, D. & Baum, T. J. The Arabidopsis microRNA396-GRF1/GRF3 regulatory module acts as a developmental regulator in the reprogramming of root cells during cyst nematode infection. Plant physiol. 159(1), 321–335 (2012).
Khatun, K. et al. Molecular characterization and expression profiling of tomato GRF transcription factor family genes in response to abiotic stresses and phytohormones. Int. J. Mol. Sci. 18(5), 1056 (2017).
Hoagland, D. R. & Arnon, D. I. M. Antioxidant responses of two barley varieties to saline stress. California Agricul. Exp. Station Circ. 347, 1–32 (1995).
Bradford, M. M. A rapid and sensitive method for quantitation of microgram of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254 (1976).
Giannopolitis, C. N. & Ries, S. K. Superoxide dismutases: I. Occurrence in higher plants. Plant Physiol. 59(2), 309–314 (1977).
Aebi, H. Catalase in vitro. Methods Enzymol. 105, 121–126 (1984).
Andrews, S. A quality control tool for high throughput sequence data. GPL v3 (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15), 2114–2120 (2014).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat protoc. 8(8), 1494 (2013).
Huang, X. & Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 9(9), 868–877 (1999).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31(19), 3210–321 (2015).
Haas, B., & Papanicolaou, A. TransDecoder (find coding regions within transcripts) (2016).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 12(1), 323 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat methods. 9(4), 357 (2012).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26(1), 139–140 (2010).
Young, M. D., Wakefield, M. J., Smyth, G. K., & Oshlack, A. goseq: Gene Ontology testing for RNA-seq datasets. Gene 1–21 (2010).
Walter, W., Sánchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31(17), 2912–2914 (2015).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 25(4), 402–408 (2001).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Acknowledgements
The authors would like to acknowledge the financial and infrastructure support of Genetics and Agricultural Biotechnology Institute of Tabarestan (GABIT), Sari Agricultural Sciences and Natural Resources University. Besides, this study has been partially funded by the grant No: 950514 of the Biotechnology Development Council of the Islamic Republic of Iran.
Author information
Authors and Affiliations
Contributions
Elham Younesi-Melerdi, Ghorban-Ali Nematzadeh and Ali Pakdin-Parizi conceived and designed the research. Ghorban-Ali Nematzadeh provided funding acquisition. Elham Younesi-Melerdi and Ali Pakdin-Parizi prepared the materials and performed the experiments. Elham Younesi-Melerdi, Seyed Abolfazl Motahari, Mohammad Reza Bakhtiarizadeh and Ali Pakdin-Parizi participated in data analyses. Elham Younesi-Melerdi, Ali Pakdin-Parizi and Mohammad Reza Bakhtiarizadeh wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Younesi-Melerdi, E., Nematzadeh, GA., Pakdin-Parizi, A. et al. De novo RNA sequencing analysis of Aeluropus littoralis halophyte plant under salinity stress. Sci Rep 10, 9148 (2020). https://doi.org/10.1038/s41598-020-65947-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65947-5
- Springer Nature Limited
This article is cited by
-
Transcriptome Sequencing of Rectretohalophyte Aeluropus lagopoides Revealed Molecular Insight of Salt Stress Adaptation
Journal of Plant Growth Regulation (2024)
-
Transcriptome and de novo analysis of Rosa xanthina f. spontanea in response to cold stress
BMC Plant Biology (2021)