
Abstract
Symptoms of Staphylococcus lugdunensis infection are often similar to those of Staphylococcus aureus infection, including skin and soft-tissue lesions, bacteremia and infective endocarditis. Despite the severity of these infections, S. lugdunensis is regarded as a less important pathogen than drug-resistant S. aureus. To investigate its ability to cause infectious diseases, a methicillin-resistant S. lugdunensis (MRSL) strain JICS135 was isolated from a patient with bacteremia and subjected to whole genome sequencing. Similar to most strains of methicillin-resistant S. aureus (MRSA), this MRSL strain possessed the staphylococcal cassette chromosome mec (SCCmec) located close to the origin of replication. However, the SCCmec in this MRSL strain, with three ccr complexes, was structurally unique and currently untypable. Moreover, the SCCmec of this MRSL strain was found to carry two genes encoding microbial surface components recognizing adhesive matrix molecules (MSCRAMM)-like proteins accompanied by glycosyl transferases, one of which may have been derived from S. aureus and the other from S. epidermidis, indicating that this MRSL evolved to carry virulence factors from other staphylococci. The emergence of this strain, the first MRSL strain whose genome has been sequenced completely, may be of public concern.
Similar content being viewed by others

Introduction
In 1988, two new coagulase-negative species, Staphylococcus lugdunensis and S. schleiferi, were isolated from human clinical specimens1. One of these human pathogens, S. lugdunensis, initially isolated from an axillary lymph node sample1, has become a coagulase-negative Staphylococcus species (C-NS) of significant interest2. Similar to S. aureus, S. lugdunensis is a skin-commensal species and a pathogen responsible for hospital- and community-acquired infections2. S. lugdunensis causes skin and soft tissue infections, bloodstream infections, and infective endocarditis3.
Methicillin-resistant S. lugdunensis (MRSL) was first isolated in 2003 from premature neonate in Singapore with a catheter-associated bloodstream infection4. Since then, MRSL has been isolated from patients in several countries, including Ethiopia5, Hong Kong6, Italy7, Singapore8, Taiwan9,10,11,12 and the USA13,14. A prospective study in Singapore8 showed that five (4.7%) of 106 clinical isolates of S. lugdunensis collected from 2004 to 2006 were resistant to methicillin and harbored mecA. The mecA gene encodes an alternative penicillin-binding protein 2 (PBP 2a) which has lowered affinity to β-lactam reagents, preventing bacterial growth retardation by the drugs. Dissemination of MRSL was detected in nephrology centers in Hong Kong; of 252 patients, 21 (8.3%) were MRSL carriers6. Subsequently, MRSL was isolated from three (42.8%) of seven patients with S. lugdunensis bacteremia in Japan15 and from seven (46.6%) of 15 patients with S. lugdunensis bacteremia in Iraq16. The increased recognition of MRSL among S. lugdunensis isolates suggests an emerging public health problem. A molecular epidemiological study demonstrated that MRSL isolates containing staphylococcal cassette chromosome mec (SCCmec) V structure while harboring an additional ccrAB2 locus were emerging in central Taiwan10. To date, however, the complete genome sequence of a MRSL strain has not been determined. The present study reports the comparative whole genome analysis of a clinical MRSL isolate from Japan that caused a bloodstream infection. The structure of its SCCmec was determined and its characteristics analyzed.
Methods
Statement on ethics control and appropriateness of the experiments
All of the methods and the experimental protocols employed in this study were performed in accordance with relevant guidelines and regulations, and were approved by the Juntendo University School of Medicine Research Ethics Committee (permission #2019041) and the Saiseikai Yokohamashi Tobu Hospital Ethics Committee (permission #2018065). Informed consent was obtained from all participants. Prior to the start of this study, all researchers who performed these experiments had completed an ethics training course provided by the Association for the Promotion of Research Integrity, Tokyo, Japan.
Bacterial isolates and patient characteristics
JICS135 was isolated from one of two sets of blood culture taken from an inpatient in 2014. The inpatient was a 77 year-old man with chronic kidney disease who required a long-term internal catheter. Blood cultures were processed using the BacTAlert system (bioMe´rieux, Basingstoke, UK) at Saiseikai Yokohamashi Tobu Hospital in Japan. Identification and minimal inhibitory concentrations (MICs) of antibiotics were determined by DxM 1096 MicroScan WalkAway (Beckman Coulter, U.S.) based on Clinical and Laboratory Standards Institute (CLSI) guidelines (M100S, 26th edition).
DNA manipulation and species identification
Strain JICS135 was grown on sheep blood agar (Kyokuto Pharmaceutical Industrial Co., Ltd., Japan), subjected to Microflex Biotyper matrix-assisted laser desorption ionization/time of flight mass spectrometry (MALDI-TOF MS)17 and identified by comparison with a database complete as of March 2018 (Bruker, Billerica, MA, USA). The complete genome determination performed in this study, followed by comparisons of its 16S ribosomal RNA gene sequence with identical sequences in the database and average nucleotide identity (ANI) analysis18,19 employing ANI calculator20 in EZbiocloud homepage (https://www.ezbiocloud.net/tools/ani) confirmed that JICS135 was S. lugdunensis.
Genome sequencing, annotation and comparisons with other S. lugdunensis strains
The genomic DNA of JICS135 was subjected to whole-genome sequencing using PacBio RS II (Pacific Biosciences, Menlo Park, CA). A total of 1163.8 Mbp (433x coverage) sequencing reads were assembled with HGAP 2.021, followed by circularization with Minimus 222. The RAST automated annotation servers23 were used for primary coding sequence (CDS) extraction and initial functional assignment. The CDS annotations were confirmed by one-to-one visual comparisons on InSilico Molecular Cloning (IMC) software (In Silico Biology, Inc., Kanagawa, Japan), which assists in evaluating the prevalence of the annotated sequence by comparison of each CDS with its homologues registered in databases. IMC software as also used for circular genome display and comparative analyses of the JICS135 genome with the genomes of the S. lugdunenis strains HKU09-01 and N920143 (Figs. 1–3). The sequence and annotation have been deposited in the databases with accession number AP021848.

Functional genomic organization of the chromosome of Staphylococcus lugdunensis strain JICS135. The first and second outermost circles show open reading frames on the plus and minus strands, respectively. Colors are explained in a table to the right of the figure. The third circle shows G + C contents, with purple indicating higher than average, and the fourth circle shows GC-skew (light green means higher than average). Positions of SCCmecMRSL-JICS135 (Fig. 5) and νSa4MRSL-JICS135 and νSl1 (Fig. 6) are also indicated. A gray arc with arrowheads outside represents a region with low homology to S. aureus genomes as shown in Fig. 2.

Comparison of chromosomal GC-skew of Staphylococcus lugdunensis strain JICS135 to other staphylococci. Arrows indicate positions where the GC-skew trend change near presumable replication termination sites. Changes in GC-skew values of S. aureus genomes are mostly symmetric across the vertical axis on the genome map, whereas those of coagulase-negative staphylococci including S. lugdunensis JICS135 are not.

Homologous regions of JICS135 to other staphylococcal chromosomes by dot-plots. When compared to S. aureus genomes, large-scale rearrangement of chromosome is seen in coagulase-negative staphylococci including S. lugdunensis JICS135.
Other computer-based genome analyses
Multilocus sequence typing (MLST) was determined by depositing the complete genome sequence of JICS135 in the Center for Genomic Epidemiology (CGE) website24. S. aureus virulence genes were identified using VirulenceFinder 2.025 of the CGE server with thresholds of 90% nucleotide sequence identity and 60% minimum length. Drug resistant genes were initially identified by ResFinder 3.226,27 of the CGE server, followed by one-to-one visual inspection of annotated genes. Phylogenetic relationship among sequenced S. lugdunensis strains was analyzed by CSI Phylogeny 1.428 of the CGE server, that is based on single nucleotide polymorphism (SNP) among genomes, allowing to draw a maximum-likelihood phylogenetic tree29. With a NEWICK-format file from result of analysis by the CSI Phylogeny, a tree was re-drawn as a radial layout with centered root by FigTree ver. 1.4.4 software (http://tree.bio.ed.ac.uk/, personally distributed by Professor Andrew Rambaut at Institute of Evolutionary Biology, University of Edinburgh). The IMC software described above was employed for GC-contents, GC-skew analyses and dot plots to identify homologous regions in two genomes.
Results
After complete genome determination of strain JICS135, the sequence was subjected to average nucleotide identity (ANI) analysis with whole genome sequences of S. lugdunensis strains HKU09-0130 and N92014331. The Ortho ANIu values20 to the genomes were 99.30% and 99.41%, respectively. These values are sufficient to conclude that the strain JICS135 is S. lugdunensis.
Figure 1 shows the overall features of the entire genome of MRSL strain JICS135. The chromosome of this strain contained 2,687,768 base pairs, encoding 2,498 proteins, six sets of ribosomal RNA genes and 61 transfer RNA genes. The GC contents (33.72%) did not differ markedly from those of other genomes of well-known human-pathogenic staphylococci, including S. lugdunensis reference genome strain HKU09-0130 (33.87%), S. saprophyticus type strain ATCC 1530532 (33.24%), S. haemolyticus type strain ATCC29970 (32.93%) [accession #CP035291], S. epidermidis type strain ATCC 14990 (32.25%) [accession #CP035288], and S. aureus type strain DSM 2023133 (32.86%). Plasmids were not detected in JICS135, indicating that all drug-resistance genes are on its chromosome. Its MLST was found to be ST3.
Interestingly, direction of gene transcription of the JICS135 genome was not symmetric with respect to the vertical axis of the genome map, with the change in direction observed around 7 o’clock, a finding supported by the GC-skew values (Fig. 1). This finding suggests that the replication termination site of JICS135 is located around the 7 o’clock position on the genome map. Genomes of S. lugdunensis HKU09-01, S. saprophyticus ATCC 15305, S. haemolyticus ATCC 29970 and S. epidermidis ATCC 14990 showed similar biased GC-skew, whereas S. aureus strains DSM 20231 (without SCCmec) and N315 (with SCCmec)34 were symmetric across the vertical axis (Fig. 2). The genomes of coagulase-negative staphylococci including S. lugdunensis JICS135 showed large-scale chromosomal rearrangements when compared with the genomes of S. aureus strains, and a non-homologous region of JICS135 to genomes of other staphylococcal species was found across the replication origin (ori) (Fig. 3). Approximate position of the non-homologous region is indicated in Fig. 1 as an arc of outer circle with arrows.
JICS135 contained three drug-resistance genes, the β-lactam-resistant genes mecA and blaZ, and an aminoglycoside-resistant gene aac(6′)-aph(2″). The nucleotide sequence of mecA in JICS135 was 99.90% identical to the mecA sequence of S. aureus strain N315. The minimum inhibitory concentrations (MIC) of various antibiotics to JICS135 are shown in Table 1. JICS135 was resistant to oxacillin and methicillin, suggesting its resistance to all β-lactams tested. JICS135 was susceptible to antibacterial agents showing activity against MRSA, such as linezolid (LZD), vancomycin (VCM), daptomycin (DAP), and arbekacin (ABK), as well as to levofloxacin (LVFX), but showed intermediate resistance to gentamicin. These results support the finding, that the JICS135 genome encoded only β-lactam- and aminoglycoside-resistance genes.
Figure 4 shows the chromosomal regions of JICS135 similar to those of two other S. lugdunensis strains, HKU09-0130 and N92014331. The gapped regions appearing in each strain are candidates of specific insertions occurring in its genome. JICS135 contained a large insertion, which was absent from strains HKU09-01 and N920143. This insertion was located close to the origin of replication of the JICS135 genome. This insertion included complexes of genes encoding the methicillin-resistant determinant mec and the DNA recombinase ccr, clearly indicating that this domain is the Staphylococcal Cassette Chromosome mec (SCCmec), which confers β-lactam resistance onto staphylococcal species35. Figure 5 illustrates the structure of the SCCmec of strain JICS135, which has been designated SCCmecMRSL-JICS135. This region had direct repeats at both ends, located exactly at the boundaries of the inserted region of the JICS135 genome (Fig. 4). The distance between the repeats at the ends was 92,958 bps. Other repeats were observed in the middle parts of the SCCmecMRSL-JICS135 and ccr complexes, suggesting that SCCmecMRSL-JICS135 was formed by multiple insertions of SCCs of different origins. Due to its complicated structure, we were unable to type SCCmecMRSL-JICS135 using established procedures36. SCCmecMRSL-JICS135 contained a mecA gene, which was not flanked by the sensor gene mecR or the repressor gene mecI, but no other determinants of drug resistance. Rather, SCCmecMRSL-JICS135 contained two genes encoding large proteins similar to staphylococcal microbial surface components recognizing adhesive matrix molecules (MSCRAMMs)37,38,39. One of these, designated lwrC1 (gene 1 of S. lugdunensis cell wall-anchored with specific repeats in cassette chromosome), encoded a protein containing repeats of the sequence STSDSESHSDSESDSDSE, whereas the other, designated lwrC2, encoded a protein containing repeats of the sequence SDADSD (where S, T, D, E, A and H represent serine, threonine, glutamine, glutamic acid, alanine and histidine, respectively). The C-termini of both of these products possessed LPXTG cell wall sorting signals40,41, suggesting that they attach to molecules composed of cell surfaces of infected human tissues. Interestingly, the lwrC1 and lwrC2 genes were accompanied by transglycosylases and related genes. The lwrC1 gene was flanked by homologues of gtfA and gtfB, which are required for the glycosylation of a gspB gene product and enhances attachment of Streptococcus gordonii to platelets42. Similar glycosylation of a sraP gene product of S. aureus enhances its attachment to host tissues43. The lwrC2 gene is also located close to transglycosylation-related genes, suggesting that the lwrC2 products may interact with tissues.

Genome rearrangement map of Staphylococcus lugdunensis strain JICS135 compared with S. lugdunensis strains HKU09-01 and N920143. Regions of >90% nucleotide identity are shown with red lines, illuminating gaps representing regions specific to each strain. Major insertions of JICS135 are indicated in the figure.

Structure of SCCmecMRSL-JICS135 compared with closely-related structures. Arrows indicates orfs and their directions. Three sets of ccr genes, responsible for the integration and excision of SCC, were identified. The mecA gene in SCCmecMRSL-JICS135 was the only drug resistant determinant. Two novel genes, lwrC1 and lwrC2, characteristic of MSCRAMMs, were accompanied by glycosylation-related genes. The sequences of the direct repeats (DR) were: DR1, 5′-GAAGGGTATCATAAATAA-3′; DR2, 5′-GAAGCGTATCATAAATAA-3′; DR3, 5′-GAAGCATATCATAAATGA-3′; DR4, 5′-GAAGCATATCATAAGTGA-3′; and DR5, 5′-GAAGCGTATCATAAGTGA-3′. Closely-related structures found by BLAST analyses were aligned in parallel with colors representing homology: a, 0.0 ≤ e-value <1.0E-100, overlap ≥ 90.0%, identity ≥ 20.0%; b: 1.0E-100 ≤ e-value <1.0E-50, overlap ≥ 40.0%, identity ≥ 20.0%; c: 1.0E-100 ≤ e-value <1.0E-10, overlap ≥ 30.0%, identity ≥ 20.0%; d: 1.0E-10 ≤ e-value <1.0E-2, overlap ≥ 20.0%, identity ≥ 20.0%.
MRSL strains isolated in Hong Kong and Taiwan were found to have SCCmecs with structures similar to SCCmecMRSL-JICS13544,45. Draft genome information has indicated that SCCmec6756 and SCCmec558044 each possesses three ccr complexes, two MSCRAMM-like genes and glycosyltransferases, although there were significant differences in regions between orfX and the mec complex. SCCmec6756 and SCCmec5580 have been designated SCCmec types V.4.1.3 and V.4.2.2, respectively. SCCmecMRSL-JICS135 was less similar to SCCmecs of S. lugdunensis strains CMUH-22 and CMUH-2545 than to SCCmec6756 and SCCmec5580, but was highly homologous to a region between the hsdR gene encoding a restriction endonuclease of a restriction-modification system and the downstream end of SCCmecMRSL-JICS135. Because the sequences of SCCmec6756 and SCCmec5580 have not been available in databases, a comparison of structure of the SCCmec of strain CMUH-25 with that of SCCmecMRSL-JICS135 is shown in Fig. 5. Other analyses have shown that the region including lwrC1 was most similar to part of the arginine catabolic mobile element (ACME) region of S. epidermidis strain I14OR146, whereas the lwrC2 region was most similar to the SCCmecs of S. lugdunensis, followed by part of the SCCmec of S. aureus strain COL47. Indeed, the product of the lwrC1 gene was most similar to the SdrH protein of S. epidermidis whereas the product of the lwrC2 gene was highly similar to a hypothetical protein of S. aureus. These findings suggest that SCCmecMRSL-JICS135 consists of multiple domains originating from other staphylococcal species.
In addition to SCCmecMRSL-JICS135, JICS135 had at least four other inserted regions relative to strains HKU09-01 and N920143, containing blaZRI, νSa4MRSL-JICS135, aac(6′)-aph(2″) and νSl1 (Fig. 4). The blaZRI gene confers resistance to β-lactams, and the insertion contained a transposase similar to Tn554, indicating that the drug-resistance gene was inserted together with the transposon, as the blaZRI-Tn554 combination is widely seen in staphylococci. The aminoglycoside-resistance gene aac(6′)-aph(2″) was accompanied by IS256 (Fig. 4), which is also widely seen in staphylococci. JICS135 also contained two genomic islands, νSa4MRSL-JICS135 and νSl1 (Figs. 4 and 6), similar to the S. aureus genomic island νSa448, which often carries the tst gene encoding the protein toxic shock syndrome toxin 1 (TSST-1). In comparison with νSa4 of S. aureus strain N315, which contains the genes sel, sec. 3 and tst, encoding the superantigens TSST-1 and staphylococcal enterotoxins L and C3, respectively34, νSa4MRSL-JICS135 contained a gene encoding a ferrichrome-binding protein, which is involved in iron acquisition, and νSl1 contained cadmium resistance genes (Fig. 6). The sequence of an integrase for νSa4MRSL-JICS135 was 96% identical to that of νSa4 of S. aureus strain N315, with sequences for the direct repeats at both ends included in those of N315 (Fig. 6), strongly suggesting that νSa4MRSL-JICS135 and S. aureus νSa4 shared a common origin. In contrast to νSa4MRSL-JICS135, the integrase for νSl1 had only 29% sequence identity to that of νSa4 of S. aureus strain N315. We failed to identify direct repeats at both ends. Although νSl1 and νSa4 of S. aureus strain N315 had several genes in common, their lineages may differ. Because a database search showed that S. lugdunensis strains Klug93G-449, FDAARGOS141, FDAARGOS377, and FDAARGOS381, with accession numbers CP014022, CP023539 and CP023970, respectively, had elements 99.9% identical to that of JICS135, we designated this element as νSl1 (i.e. the first ν element identified in S. lugdunensis). S. lugdunensis strains HKU09-01 and N920143 did not possess integrases identical to those for νSa4MRSL-JICS135 and νSl1.

Structure of νSa4MRSL-JICS135 and νSa1 in comparison with νSa4 of S. aureus strain N315. The sequence of direct repeats for S. aureus νSa4 (DRνSa4) was 5′-GTTTTACATCATTCCCGGCAT-3′, whereas that for S. lugdunensis DRνSa4MRSL-JICS135 was 5′-TTTTACATCATACCTGGCAT-3′. The parallelogram with colors representing homology values are the same as those in Fig. 4.
A comparative analysis also revealed unique insertions in strains HKU09-01 and N920143. The former possessed a restriction-modification system (hsdSMR) and a prophage-like insertion with integrase and phage component genes, whereas the latter possessed an insertion of prophage ϕSL1 (Fig. 4). The JICS135 genome did not have hsdSMR at the site corresponding to that of HKU09-01; however, this gene cluster was found in SCCmecMRSL-JICS135 (Fig. 5).
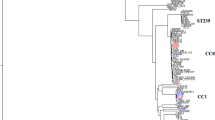
In order to see relationship among sequenced S. lugdunensis strains, a whole genome-wide phylogenetic analysis based on single nucleotide polymorphism (SNP) was performed (Fig. 7). JICS135 was relatively close to strain Klug93G-4 isolated in Hong Kong. The two Asian isolates JICS135 and Klug92G-4 (red) were also phylogenetically close to the north American ones (green), whereas the European isolates (blue) seemed to form a few clades that had distance from the one with JICS135 and some north American strains. On the other hand, S. lugdunensis whole genome sequence reference strain HKU09-01 isolated in Hong Kong belonged to one of the European clades. In addition to JICS135, only UCIM6116 had mecA and ccrC genes, suggesting that the strain has SCCmec among the strains shown in Fig. 7. However, the sequence of UCIM6116 lacked ccrAB, ccrA2B2, lwrC1 and lwrC2, indicating that the strain does not have similar element to SCCmecMRSL-JICS135.

Whole genome-wide phylogenetic relationship among S. lugdunensis strains. Maximum-likelihood tree based on SNPs extracted from the genomes by mapping them to reference sequence of S. lugdunensis strain HKU09-0130 is presented. The branch length indicates proportions of SNPs to the total 23,055 ones called among the 33 genomes (refer to the scale bar). S. lugdunensis strains that whole genome sequences are available in databases, or those of strains HKU09-01, M23590 (BioSample #SAMN00139437), N92014331, VCU139 (BioSample #SAMN02436593), VCU148 (BioSample #SAMN00116837), VCU150 (BioSample #SAMN00116873), ASC-027-V-Sch2 (BioSample #SAMN02463890), UCIM6116 (BioSample #SAMN02381740), MJR7738 (BioSample #SAMN03948580), FDAARGOS_14157, FDAARGOS_14357, FDAARGOS_22257, FDAARGOS_37757, FDAARGOS_38157, VISLISI_2158, VISLISI_2258, VISLISI_2558, VISLISI_2758, VISLISI_3358, VISLISI_3758, C_3358, Klug93G-449, NCTC7990 (accession #LS483312), NCTC12217 (BioSample #SAMN06177169), E7 (BioSample #SAMN10735326), SL13 (accession #CP041722.1), SL29 (accession #CP041723.1), SL55 (accession #CP041724.1), SL117 (accession #CP041725.1), SL118 (accession #CP041726.1), SL122 (accession #CP041727.1), APC3758 (accession #CP038807.1) and JICS135 (this study) were subjected to CSI Phylogeny 1.428 with parameters of default values (minimum depth at SNP positions: 10, relative depth at SNP positions: 10, minimum distance between SNPs: 10, minimum SNP quality: 30, minimum read mapping quality: 25, minimum Z-score: 1.96 with ignoring heterozygous SNPs). Strains isolated in Asia, Europe and north America are indicated in red, blue and green, respectively. Strains without information about locations of isolation are indicated in gray.
Discussion
The complete sequencing of the genome of methicillin-resistant S. lugdunensis strain JICS135 showed that the size of its genome was approximately the same as other coagulase-negative staphylococci, and smaller than that of S. aureus. Due mainly to the insertion of SCCmec, the genome of JICS135 was larger than that of the genomes of methicillin-susceptible S. lugdunensis strains described to date. Unlike S. aureus genomes, the transcriptional direction of genes was not symmetric across the vertical axis of the genomic map of JICS135, suggesting that the replication termination site is not located opposite the site of the origin of replication on the circular chromosome, but at about the 7 o’clock position on the genome map, a finding supported by GC-skew results. This asymmetry was initially thought to be due to the insertion of SCCmecMRSL-JICS135 slightly downstream of the origin of replication. Similar findings, however, were observed in other S. lugdunensis genomes without SCCmecs, suggesting that insertion of SCCmecMRSL-JICS135 was not responsible for this asymmetry. S. haemolyticus strain JCSC1435 also shows asymmetry in GC-skew50, with similar phenomena observed in other coagulase-negative staphylococci (Fig. 2). Dot plots have shown that coagulase-negative staphylococci had large-scale genome inversions when compared with S. aureus genomes. It was also notable that regions around the origins of replication of S. aureus genomes and JICS135 were not homologous, with these regions extending 100 kbp upstream and 500 kbp downstream of the origin of the JICS135 genome, as shown in Fig. 1. Because the non-homologous region was shifted to the right side of the genome map, that side could include sequences that lead to the GC-skew bias. We also found that frequently-transcribed genes that can affect replication speed, such as those encoding ribosomal RNAs, ribosomal proteins and tRNAs, located on the right side of the genome map were concentrated in JICS135. In contrast, those located on the left side were scattered, delaying replication of the left side relative to the right side and leading to the replication termination site being located at about the 7 o’clock position.
ST3 was found to be the most frequently isolated (20.7%) S. lugdunensis MLST in France, Belgium and Slovenia, but MRSL was not found51. In contrast, Taiwan, ST6 (19.0%)52 and ST38 (77.8%)53 were the most common MRSLs in Taiwan. Our finding, that JICS135 belonged to ST3, indicates that the JICS135 lineage is distinct from the strains isolated in other countries.
In contrast to other S. lugdunensis genomes, the JICS135 genome apparently contained no prophages, as no insertions 40–50 kbp in length with phage integrase accompanied by major phage component genes were detected in JICS135. However, JICS135 had several insertions of mobile genetic elements, the most striking being as large as 93 kbp of the SCCmec element, SCCmecMRSL-JICS135, close to the origin of replication. The insertion of SCCmecMRSL-JICS135 occurred in orfX gene, encoding 23S rRNA (pseudouridine [1915]-N[3])-methyltransferase RlmH54, as seen in other staphylococcal species. SCCmecMRSL-JICS135 also contained a mec complex and ccr recombinase genes. Despite their common features, the structure of SCCmecMRSL-JICS135 differs significantly from those found in other Staphylococci. SCCmecMRSL-JICS135 was as large as 93 kbp in size and contained multiple sets of ccr recombinases and other unique features. We found that SCCmecMRSL-JICS135 contained the lwrC1 and lwrC2 genes, encoding large cell-wall anchored proteins with unique repeats, a feature observed in MSCRAMMs, surface adhesive proteins typically found in S. aureus, suggesting that the products of the lwrC1 and lwrC2 genes likely play important roles in attachment to host cells and tissues37. This hypothesis is consistent with the finding that JICS135 was isolated from a patient with bloodsteam infection. Moreover, the lwrC1 and lwrC2 genes were accompanied by transglycosylases and related genes. In other genera, these glycosylated proteins are secreted by specific mechanisms involving SecA2 and SecY2 that are distinct from general secretory mechanisms. These secreted proteins subsequently attach covalently to the bacterial cell walls, increasing their affinity to platelets42. Because strain JICS135 does not contain genes encoding SecA2 or SecY2, the mechanism by which the lwrC1 and lwrC2 gene products translocate through the cytoplasmic membrane is unclear. Although the targets of these gene products have not been determined, SCCmecMRSL-JICS135 containing these MSCRAMM genes may increase the virulence of S. lugdunensis strains. Investigations to identify the molecules targeted by the lwrC1 and lwrC2 gene products are ongoing.
Analysis also revealed that the lwrC1 locus has the highest homology to S. epidermidis, whereas the lwrC2 locus is most similar to S. aureus, suggesting that SCCmecMRSL-JICS135 is a hybrid of staphylococcal strains resulting from multiple gene crossovers. Because both S. epidermidis and S. aureus are included in normal human flora, MSSL strains can acquire both higher virulence and drug resistance by the incorporation of elements such as SCCmecMRSL-JICS135. In addition to the lwrC1 and lwrC2 genes, other MSCRAMM genes have been detected in S. lugdunensis31. The combination of these MSCRAMMs and SCCmec can enhance the virulence of MRSL strains. Further analyses will be required to prove whether SCCmecMRSL-JICS135 confers higher affinity to fibronectin to JICS135 and the enhanced affinity leads to increase of virulence of the strain.
Three genes responsible for antibiotic resistance were identified in JICS135. The mecA gene in SCCmecMRSL-JICS135 was not accompanied by mecR and mecI, whereas the β-lactamase blaZ gene was accompanied by the sensor gene blaR and the repressor gene blaI. These findings suggested that blaZ gene expression correlates with the concentration of β-lactam reagents, and that mecA gene expression is under the control of β-lactams via blaR55. The MICs for β-lactams indicate that of JICS135 is resistant to these antibiotics, which may be a consequence of the combined effects of mecA and blaZ. The aac(6′)-aph(2″) gene, which is responsible for aminoglycoside resistance, was also functional, because the MIC for gentamycin indicates intermediate resistance of JICS135. Similar to many MRSA strains, the aac(6′)-aph(2″) gene is inserted into the JICS135 genome along with the transposon Tn554, indicating inter-species horizontal transfer among staphylococci, probably under selective pressure of aminoglycoside reagents.
No other known S. aureus virulence factors other than these MSCRAMM proteins were detected in S. lugdunensis JICS135 genome. Genetic methods are required to identify S. lugdunensis genes associated with virulence. For example, virulence can be evaluated in a library subjected to transposon-insertion mutagenesis using a model organism56. This approach may provide more information needed to understand the pathogenicity of S. lugdunensis.
Because fewer people have been infected by S. lugdunensis than by S. aureus and other major coagulase-negative staphylococci, little is known about the molecular epidemiology of S. lugdunensis infection. This drug-resistant S. lugdunensis strain containing a complex SCCmecMRSL-JICS135 structure may become more widespread, suggesting the need for continuous surveys of S. lugdunensis isolates, including their drug resistance properties and their association with patient symptoms. Our analysis using whole genome sequences of S. lugdunensis in Fig. 7 showed that phylogenetically close strains to JICS135 are present. In addition to JICS135, however, only strain UCIM6116 (the sequencing has not been completed) seemed to have SCCmec which structure was not likely to be similar to SCCmecMRSL-JICS135. Further analyses will elucidate relationship among carriage of SCCmecs, their structures and sequence types of other parts of chromosomes, and the information would provide us evolutional pathways of MRSL strains.
This comparative analysis of S. lugdunensis genomes, including JICS135, revealed variations in their mobile genetic elements, which are responsible for the drug resistance and virulence of these strains. These findings suggested that novel types of S. lugdunensis strains that differ in drug resistance and virulence emerge as causes of hospital- and community-acquired infections.
References
Freney, J. et al. Staphylococcus lugdunensis sp. nov. and Staphylococcus schleiferi sp. nov., Two Species from Human Clinical Specimens. International Journal of Systematic and Evolutionary Microbiology 38, 168–172, https://doi.org/10.1099/00207713-38-2-168 (1988).
Frank, K. L., Del Pozo, J. L. & Patel, R. From clinical microbiology to infection pathogenesis: how daring to be different works for Staphylococcus lugdunensis. Clin Microbiol Rev 21, 111–133, https://doi.org/10.1128/CMR.00036-07 (2008).
Zinkernagel, A. S. et al. Significance of Staphylococcus lugdunensis bacteremia: report of 28 cases and review of the literature. Infection 36, 314–321, https://doi.org/10.1007/s15010-008-7287-9 (2008).
Tee, W. S. N., Soh, S. Y., Lin, R. & Loo, L. H. Staphylococcus lugdunensis Carrying the mecA Gene Causes Catheter-Associated Bloodstream Infection in Premature Neonate. Journal of Clinical Microbiology 41, 519–520, https://doi.org/10.1128/jcm.41.1.519-520.2003 (2003).
Tibebu, M. Severe hospital acquired pneumonia and septicemia due TO methicillin resistant Staphylococcus lugdunensis in a newborn in Northwestern Ethiopia. Ethiopian medical journal 52, 99–101 (2014).
Ho, P. L. et al. Carriage niches and molecular epidemiology of Staphylococcus lugdunensis and methicillin-resistant S. lugdunensis among patients undergoing long-term renal replacement therapy. Diagn Microbiol Infect Dis 81, 141–144, https://doi.org/10.1016/j.diagmicrobio.2014.10.004 (2015).
Spanu, T. et al. Ventriculitis due to Staphylococcus lugdunensis: two case reports. J Med Case Rep 2, 267, https://doi.org/10.1186/1752-1947-2-267 (2008).
Tan, T. Y., Ng, S. Y. & He, J. Microbiological characteristics, presumptive identification, and antibiotic susceptibilities of Staphylococcus lugdunensis. J Clin Microbiol 46, 2393–2395, https://doi.org/10.1128/JCM.00740-08 (2008).
Wu, A. B. et al. Clinical and microbiological characteristics of community-acquired Staphylococcus lugdunensis infections in Southern Taiwan. J Clin Microbiol 49, 3015–3018, https://doi.org/10.1128/JCM.01138-11 (2011).
Tseng, S. P. et al. Genotypes and phenotypes of Staphylococcus lugdunensis isolates recovered from bacteremia. J Microbiol Immunol Infect 48, 397–405, https://doi.org/10.1016/j.jmii.2013.11.006 (2015).
Lin, J. F. et al. Clinical experience and microbiologic characteristics of invasive Staphylococcus lugdunensis infection in a tertiary center in northern Taiwan. J Microbiol Immunol Infect 48, 406–412, https://doi.org/10.1016/j.jmii.2013.12.010 (2015).
Yen, T. Y. et al. Emergence of oxacillin-resistant Staphylococcus lugdunensis carrying staphylococcal cassette chromosome mec type V in central Taiwan. J Microbiol Immunol Infect 49, 885–891, https://doi.org/10.1016/j.jmii.2014.11.018 (2016).
Bhumbra, S., Mahboubi, M. & Blackwood, R. A. Staphylococcus lugdunensis: Novel Organism causing Cochlear Implant Infection. Infect Dis Rep 6, 5406, https://doi.org/10.4081/idr.2014.5406 (2014).
Mehmood, M. & Khasawneh, F. A. Staphylococcus lugdunensis gluteal abscess in a patient with end stage renal disease on hemodialysis. Clin Pract 5, 706, https://doi.org/10.4081/cp.2015.706 (2015).
Ainoda, Y. et al. Multicenter Study of the Clinical Presentation of Staphylococcus lugdunensis Bacteremia in Japan. Jpn J Infect Dis 70, 405–407, https://doi.org/10.7883/yoken.JJID.2016.130 (2017).
Al-Charrakh, A. H. & Obayes, M. H. First record of isolation and characterization of methicillin resistant Staphylococcus lugdunensis from clinical samples in Iraq. Biomed Res Int 2014, 736259, https://doi.org/10.1155/2014/736259 (2014).
Szabados, F., Woloszyn, J., Richter, C., Kaase, M. & Gatermann, S. Identification of molecularly defined Staphylococcus aureus strains using matrix-assisted laser desorption/ionization time of flight mass spectrometry and the Biotyper 2.0 database. J Med Microbiol 59, 787–790, https://doi.org/10.1099/jmm.0.016733-0 (2010).
Goris, J. et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57, 81–91, https://doi.org/10.1099/ijs.0.64483-0 (2007).
Richter, M. & Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106, 19126–19131, https://doi.org/10.1073/pnas.0906412106 (2009).
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S. & Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286, https://doi.org/10.1007/s10482-017-0844-4 (2017).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563, https://doi.org/10.1038/nmeth.2474, https://www.nature.com/articles/nmeth.2474#supplementary-information (2013).
Sommer, D. D., Delcher, A. L., Salzberg, S. L. & Pop, M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8, 64, https://doi.org/10.1186/1471-2105-8-64 (2007).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75, https://doi.org/10.1186/1471-2164-9-75 (2008).
Larsen, M. V. et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50, 1355–1361, https://doi.org/10.1128/JCM.06094-11 (2012).
Joensen, K. G. et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol 52, 1501–1510, https://doi.org/10.1128/JCM.03617-13 (2014).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Clausen, P., Aarestrup, F. M. & Lund, O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19, 307, https://doi.org/10.1186/s12859-018-2336-6 (2018).
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M. & Lund, O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. Plos One 9, e104984, https://doi.org/10.1371/journal.pone.0104984 (2014).
Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17, 368–376, https://doi.org/10.1007/bf01734359 (1981).
Tse, H. et al. Complete genome sequence of Staphylococcus lugdunensis strain HKU09-01. J Bacteriol 192, 1471–1472, https://doi.org/10.1128/JB.01627-09 (2010).
Heilbronner, S. et al. Genome sequence of Staphylococcus lugdunensis N920143 allows identification of putative colonization and virulence factors. FEMS Microbiol Lett 322, 60–67, https://doi.org/10.1111/j.1574-6968.2011.02339.x (2011).
Kuroda, M. et al. Whole genome sequence of Staphylococcus saprophyticus reveals the pathogenesis of uncomplicated urinary tract infection. Proc Natl Acad Sci U S A 102, 13272–13277, https://doi.org/10.1073/pnas.0502950102 (2005).
Shiroma, A. et al. First Complete Genome Sequences of Staphylococcus aureus subsp. aureus Rosenbach 1884 (DSM 20231T), Determined by PacBio Single-Molecule Real-Time Technology. Genome Announc 3, https://doi.org/10.1128/genomeA.00800-15 (2015).
Kuroda, M. et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet (London, England) 357, 1225–1240, https://doi.org/10.1016/s0140-6736(00)04403-2 (2001).
Katayama, Y., Ito, T. & Hiramatsu, K. A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 44, 1549–1555, https://doi.org/10.1128/aac.44.6.1549-1555.2000 (2000).
Kondo, Y. et al. Combination of multiplex PCRs for staphylococcal cassette chromosome mec type assignment: rapid identification system for mec, ccr, and major differences in junkyard regions. Antimicrob Agents Chemother 51, 264–274, https://doi.org/10.1128/AAC.00165-06 (2007).
Patti, J. M., Allen, B. L., McGavin, M. J. & Hook, M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol 48, 585–617, https://doi.org/10.1146/annurev.mi.48.100194.003101 (1994).
Deivanayagam, C. C. et al. A novel variant of the immunoglobulin fold in surface adhesins of Staphylococcus aureus: crystal structure of the fibrinogen-binding MSCRAMM, clumping factor A. EMBO J 21, 6660–6672 (2002).
Foster, T. J., Geoghegan, J. A., Ganesh, V. K. & Hook, M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol 12, 49–62, https://doi.org/10.1038/nrmicro3161 (2014).
Schneewind, O., Model, P. & Fischetti, V. A. Sorting of protein A to the staphylococcal cell wall. Cell 70, 267–281 (1992).
Schneewind, O., Mihaylova-Petkov, D. & Model, P. Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J 12, 4803–4811 (1993).
Bensing, B. A. & Sullam, P. M. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol 44, 1081–1094 (2002).
Siboo, I. R., Chaffin, D. O., Rubens, C. E. & Sullam, P. M. Characterization of the accessory Sec system of Staphylococcus aureus. J Bacteriol 190, 6188–6196, https://doi.org/10.1128/JB.00300-08 (2008).
Liu, M. C.-J. et al. Structures of SCC mec elements in methicillin-resistant Staphylococcus lugdunensis are closely related to those harboured by community-associated methicillin-resistant Staphylococcus aureus. Journal of Medical Microbiology 68, 1367–1372, https://doi.org/10.1099/jmm.0.001013 (2019).
Chang, S. C. et al. Characterization of two novel variants of staphylococcal cassette chromosome mec elements in oxacillin-resistant Staphylococcus lugdunensis. J Antimicrob Chemother 72, 3258–3262, https://doi.org/10.1093/jac/dkx291 (2017).
O’Connor, A. M., McManus, B. A. & Coleman, D. C. First description of novel arginine catabolic mobile elements (ACMEs) types IV and V harboring a kdp operon in Staphylococcus epidermidis characterized by whole genome sequencing. Infect Genet Evol 61, 60–66, https://doi.org/10.1016/j.meegid.2018.03.012 (2018).
Gill, S. R. et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol 187, 2426–2438, https://doi.org/10.1128/JB.187.7.2426-2438.2005 (2005).
Baba, T. et al. Genome and virulence determinants of high virulence community-acquired MRSA. The Lancet 359, 1819–1827, https://doi.org/10.1016/s0140-6736(02)08713-5 (2002).
Ho, P. L. et al. Emergence of ileS2-Carrying, Multidrug-Resistant Plasmids in Staphylococcus lugdunensis. Antimicrob Agents Chemother 60, 6411–6414, https://doi.org/10.1128/AAC.00948-16 (2016).
Takeuchi, F. et al. Whole-genome sequencing of Staphylococcus haemolyticus uncovers the extreme plasticity of its genome and the evolution of human-colonizing staphylococcal species. J Bacteriol 187, 7292–7308, https://doi.org/10.1128/JB.187.21.7292-7308.2005 (2005).
Chassain, B. et al. Multilocus sequence typing analysis of Staphylococcus lugdunensis implies a clonal population structure. J Clin Microbiol 50, 3003–3009, https://doi.org/10.1128/JCM.00988-12 (2012).
Cheng, C. W. et al. Persistence of a major endemic clone of oxacillin-resistant Staphylococcus lugdunensis sequence type 6 at a tertiary medical centre in northern Taiwan. Int J Infect Dis 36, 72–77, https://doi.org/10.1016/j.ijid.2015.05.022 (2015).
Yeh, C. F. et al. Clinical Features, Outcomes, and Molecular Characteristics of Community- and Health Care-Associated Staphylococcus lugdunensis Infections. J Clin Microbiol 54, 2051–2057, https://doi.org/10.1128/JCM.00847-16 (2016).
Boundy, S. et al. Characterization of the Staphylococcus aureus rRNA methyltransferase encoded by orfX, the gene containing the staphylococcal chromosome Cassette mec (SCCmec) insertion site. J Biol Chem 288, 132–140, https://doi.org/10.1074/jbc.M112.385138 (2013).
Liu, P., Xue, H., Wu, Z., Ma, J. & Zhao, X. Effect of bla regulators on the susceptible phenotype and phenotypic conversion for oxacillin-susceptible mecA-positive staphylococcal isolates. J Antimicrob Chemother 71, 2105–2112, https://doi.org/10.1093/jac/dkw123 (2016).
Bae, T. et al. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci U S A 101, 12312–12317, https://doi.org/10.1073/pnas.0404728101 (2004).
Sichtig, H. et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nat Commun 10, 3313, https://doi.org/10.1038/s41467-019-11306-6 (2019).
Argemi, X. et al. Whole-Genome Sequencing of Seven Strains of Staphylococcus lugdunensis Allows Identification of Mobile Genetic Elements. Genome Biol Evol 9, https://doi.org/10.1093/gbe/evx077 (2017).
Acknowledgements
This work was mainly supported by Grant-in-aid for special research in subsidies for ordinary expenses of private schools from the Promotion and Mutual Aid Corporation for Private Schools of Japan. The work was also supported by grants from the Research Program on Emerging and Re-emerging Infectious Diseases from the Japan Agency for Medical Research and Development (Grant number 19fk0108061h0302). We greatly appreciate support by Drs. Yuh Morimoto in Center of Excellence for Infection Control Science, Graduate School of Medicine, Juntendo University, Tokyo, Japan and Takashi Sasaki in Sapporo Medical University for bacterial strain preparations and critical suggestion on computer-based genome analyses.
Author information
Authors and Affiliations
Contributions
R. Shibuya, Y. Uehara and T. Baba made the conception and design of the study. R. Shibuya and T. Baba prepared the article and figures. K. Teruya, K. Satou and T. Hirano performed analyses and interpretation of data. T. Kirikae and K. Hiramatsu contributed to critical writing and reviewing of this manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shibuya, R., Uehara, Y., Baba, T. et al. Complete genome sequence of a methicillin-resistant Staphylococcus lugdunensis strain and characteristics of its staphylococcal cassette chromosome mec. Sci Rep 10, 8682 (2020). https://doi.org/10.1038/s41598-020-65632-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65632-7
- Springer Nature Limited