
Abstract
Clostridioides difficile is the leading cause of nosocomial infections and a worldwide urgent public health threat. Without doubt, there is an urgent need for new effective anticlostridial agents due to the increasing incidence and severity of C. difficile infection (CDI). The aim of the present study is to investigate the in vivo efficacy of auranofin (rheumatoid arthritis FDA-approved drug) in a CDI mouse model and establish an adequate dosage for treatment. The effects of increased C. difficile inoculum, and pre-exposure to simulated gastric intestinal fluid (SGF) and simulated intestinal fluid (SIF), on the antibacterial activity of auranofin were investigated. Auranofin’s in vitro antibacterial activity was stable in the presence of high bacterial inoculum size compared to vancomycin and fidaxomicin. Moreover, it maintained its anti-C. difficile activity after being exposed to SGF and SIF. Upon testing in a CDI mouse model, auranofin at low clinically achievable doses (0.125 mg/kg and 0.25 mg/kg) significantly protected mice against CDI with 100% and 80% survival, respectively. Most importantly, auranofin (0.125 mg/kg and 0.25 mg/kg) significantly prevented CDI recurrence when compared with vancomycin. Collectively, these results indicate that auranofin could potentially provide an effective, safe and quick supplement to the current approaches for treating CDI.
Similar content being viewed by others

Introduction
Clostridioides difficile is the worldwide leading cause of nosocomial infections and antibiotic-associated diarrhea1. A recent report released by the Centers for Disease Control and Prevention (CDC) stated that about 223,900 patients were hospitalized with C. difficile infections (CDI) in the United States in 2017, which was associated with around 12,800 mortality cases and in excess of $1 billion healthcare cost2.
CDI symptoms range from mild to severe watery diarrhea to more severe life threatening complications such as pseudomembranous colitis, toxic megacolon, colon perforation, sepsis, systemic inflammatory response syndrome and shock3. Disease manifestations are attributed to the toxin-mediated damage elicited by the two major toxins TcdA and TcdB. These toxins catalyze inactivation of host GTPases (Rac, Rho and CDC42) and perturbation of actin cytoskeleton, ultimately causing intense inflammation, loss of tight junctions of the intestinal mucosal layer, enormous fluid secretion, cell rounding and finally necrosis and apoptosis of the colonic mucosal cells4,5. The incidence and severity of CDI has increased dramatically due to the overuse of antibiotics and the emergence of hypervirulent epidemic strains such as, but not limited to, pulsed-field gel type North American pulsotype 1 (NAP1) or PCR ribotype 027, which were responsible for several outbreaks globally6,7. Moreover, the clinical management of CDI is hindered by the ability of C. difficile to produce spores which are highly resistant to environmental conditions, antibiotics and disinfection processes. Spores can persist on unsuitable environments for long periods and spread in the environment8. Once ingested by susceptible hosts, these spores germinate, in response to bile acids in the gut, into vegetative cells that colonize in the intestine, produce toxins and establish infection9. Consequently, C. difficile spores serve as the major cause CDI dissemination and recurrence.
Even though the overuse of antibiotics is responsible for CDI, the management of CDI requires antibiotic administration. Currently, only two drugs are approved for treatment of both non-severe and severe CDI; vancomycin and fidaxomicin. While metronidazole is not FDA-approved for treatment of CDI, it was previously recommended as a first-line therapeutic option for CDI in adults. It use is now restricted to non-severe CDI cases when patients are unable to obtain or be treated with vancomycin or fidaxomicin10. Vancomycin or metronidazole treatments are limited by the high treatment failure (22% with metronidazole, and 14% with vancomycin), and the high recurrence rate (25–30%)7,11. Furthermore, fidaxomicin has lower recurrence rate due to its less disturbance effect on gut microbiota; yet, its high cost restricts its use12,13,14. Further compounding the CDI problem is the emerging resistance or reduced susceptibility to these antibiotics13,15. Thus, the critical and the unmet need for developing new anti-CDI therapeutics cannot be overemphasized.
Auranofin is an FDA-approved anti- rheumatoid arthritis drug, with a well-studied safety profile for human use16,17. Recently, auranofin has gained interest in repurposing for treatment of bacterial and parasitic infections18,19,20,21,22,23. Furthermore, it is undergoing Phase II clinical trials for the treatment of amoebic dysentery, giardiasis (NCT02736968) and tuberculosis (NCT02968927). Auranofin possesses strong antibacterial and antifungal activities17,22,23,24,25. We previously demonstrated that auranofin has a potent anticlostridial activity with strong inhibition of both toxins and spores production in vitro24. We hypothesized that auranofin’s potent antibacterial and antivirulence activity against C. difficile would be beneficial in treating mice infected with C. difficile in an in vivo CDI mouse model. The main objective of the present study was to investigate the in vivo efficacy of auranofin treatment in a CDI mouse model and to study the ability of auranofin to prevent CDI recurrence. In addition, this study established the doses needed to achieve 100% protection in CDI mouse model and prevent recurrence. The impact of increasing C. difficile inoculum, and its pre-exposure to simulated gastric fluid and simulated intestinal fluid, on the antibacterial activity of auranofin were also investigated.
Results and Discussion
The effect of C. difficile inoculum size on the antibacterial activity of auranofin
C. difficile is known to colonize the intestinal tract in large populations. Additionally, a higher inoculum (~106 CFU/mL) is often used to infect animals in in vivo CDI models. It was reported that the bacterial burden recovered from cecal and fecal contents of infected mice had averages of ~106 to 107 CFU/g26,27,28. The dependence of the antibacterial activity of anticlostridial drugs on the inoculum effect is an important consideration, especially for a weakly absorbed drug like auranofin (85% of the administered dose is not absorbed and recovered in feces)29. After being administered orally, auranofin will be localized in the gut, and target the colonizing C. difficile populations. However, the standard antibacterial susceptibility assays typically evaluate test agents at a lower inoculum size (~105 CFU/mL). Thus, we evaluated the impact of the high C. difficile inoculum (HI, ~5 × 107 CFU/mL), compared with the standard inoculum (SI, 5 × 105 CFU/mL), on the antibacterial activity of auranofin. Upon testing against SI, auranofin exhibited a potent in vitro activity against the C. difficile strains tested with MIC values ranging from 0.25–1 µg/mL (Table 1), in agreement with a previous study24. Furthermore, auranofin’s antibacterial activity was identical to or one-fold higher, as the inoculum size increased from 105 CFU/mL to 107 CFU/mL (Table 1), suggesting that its activity was not impacted by increasing the inoculum size. Its MIC90 was not affected by the increase in the inoculum size. Fidaxomicin MICs, in agreement with a previous study30 were not affected by increasing the C. difficile inoculum size (MICs of HI were equal to or one-fold higher than SI MICs except the MICs against C. difficile NR-49278 that increased by three-fold). Additionally, its MIC90 with the HI was the same as that of the SI. Conversely, vancomycin’s activity was negatively impacted by the increased inoculum size (MIC increased three-fold against C. difficile NR-49278, C. difficile NR-49281, and C. difficile NR-49284), in accordance with a previous report30. Additionally, its MIC90 with the HI was one-fold higher than that of the SI.
The effect of simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) on the antibacterial activity of auranofin
It is important to analyze the stability of drugs, especially those intended for oral administration, in harsh conditions of the gastrointestinal tract (GIT). The stability of a drug in gastric and intestinal fluids provides evidence whether it is prone to degradation process by the effect of GIT fluids prior to absorption31. Drugs stability in presence of the GIT fluids can be investigated by incubating the drug in simulated gastric fluid (for 1-2 hours) and simulated intestinal fluids (for 3-4 hours) to mimic the in vivo drug exposure to these fluids31,32. To investigate the effect of SGF and SIF on the antibacterial activity of auranofin against C. difficile, auranofin, and vancomycin and fidaxomicin (control antibiotic) were incubated with SGF and SIF for 2, 4 and 24 hours and their MICs against 2 clinical C. difficile strains were determined. As depicted in Table 2A, after incubation with SGF, the MICs of auranofin did not increase against C. difficile ATCC BAA 1870, even after 24 hours exposure, and increased by one-fold only against C. difficile ATCC 43255 after 24 hours exposure. This result suggests that auranofin was stable after exposure to the gastric pH and was not affected by the enzymes of gastric fluids. Similarly, vancomycin and fidaxomicin MICs (after exposure to SGF) were similar to or one-fold higher than their corresponding MICs in absence of SGF. Furthermore, auranofin MICs, after incubation with SIF up to 24 hours, were equal to or one-fold higher than its MIC without incubation with SIF (Table 2B), suggesting that auranofin was not affected by exposure to the intestinal fluids. The antibacterial activity of vancomycin and fidaxomicin also, were not affected by incubation with SIF (MICs are equal to or one-fold higher than their corresponding MICs without exposure to SIF) (Table 2B). This result came in coincidence with a previous report30.
In vivo efficacy of auranofin in a CDI mouse model
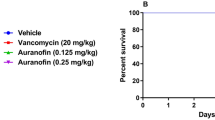
The potent antibacterial and antivirulent activities of auranofin against C. difficile24 in addition to its stability in SGF and SIF prompted us to investigate its efficacy in a CDI mouse model and its potential to protect mice from CDI recurrence. In our study, CDI was established first before treatment. Three groups of mice were treated with (0.125 mg/kg, 0.25 mg/kg and 0.5 mg/kg) of auranofin. Two additional groups were used, positive control (vancomycin) and negative control (vehicle) groups. Mice were treated with the corresponding drugs for 5 days and monitored for disease symptoms. As shown in Fig. 1, vancomycin (10 mg/kg) protected 100% of mice up to 5 days, as previously reported27,33. In addition, auranofin, at low clinically achievable concentration (0.125 mg/kg), was able to protect 100% of the mice against C. difficile during the 5-days treatment period. Interestingly, the higher doses (0.25 mg/kg and 0.5 mg/kg) protected only 80% and 40% of mice, respectively. There was no significant difference in survival between vancomycin-treated, and auranofin (0.25 mg/kg)- treated groups. This effect (lower dose more effective) could be due to the potent anticommensal activity of auranofin against the human gut intestinal microbiota34, which could increase by increasing the administered dose leading to establishment of C. difficile colonization in the intestine and higher mortality. This activity was also, reported for niclosamide against C. difficile where the lower dose (2 mg/kg) protected mice from CDI more effectively than higher doses (10 mg/kg and 50 mg/kg)35. The effectiveness of lower dose of auranofin could also be explained by their potent anti-inflammatory activity. It was reported that the anti-inflammatory drug, indomethacin, increased the severity of C. difficile infection in mice36. Additionally, in previous study investigating the efficacy of auranofin against vancomycin-resistant enterococci peritonitis, lower doses of auranofin provided the best protection (100%)37. Moreover, Fig. 2 depicts the mean relative daily weight for all mice groups. The control group (vehicle-treated) showed weight loss starting day 2 after infection and their weight continued to decrease till day 4. Conversely, vancomycin-treated mice did not show weight loss till day 5. Similarly, auranofin-treated mice maintained a stable body weight with a minor weight reduction till day 5 (Fig. 2).

Auranofin protects mice against CDI. Mice were treated with auranofin (0.125 mg/kg, 0.25 mg/kg, and 0.5 mg/kg), vancomycin (10 mg/kg), or the vehicle for 5 days after infection with C. difficile spores. Kaplan–Meier survival curves were analyzed using a log-rank (Mantel–Cox) test. Asterisks (*) denote statistical significant difference between mice treated with either auranofin, or vancomycin in comparison with the vehicle-treated mice.

Average relative weight of all surviving mice. Infected mice were treated with auranofin (0.125 mg/kg, 0.25 mg/kg, and 0.5 mg/kg), vancomycin (10 mg/kg), or the vehicle for 5 days and weighed daily till the end of the experiment. The data are presented as percent relative weight (mean ± standard deviation) for each group.
Symptomatic recurrence of CDI occurs in approximately 20% of patients and is challenging to treat38,39,40,41,42. In addition to subsequent prolongation of C. difficile shedding and transmission, 1 out of every 5 patients experienced C. difficile recurrence episode died within 30 days of diagnosis43. Then, we sought to investigate this promising activity of auranofin in preventing C. difficile recurrence. Mice were infected and treated for 5 days and mice were monitored for survival and possible C. difficile recurrence until the 20th day. Vancomycin-treated mice, in accordance with a previous study33, were susceptible to C. difficile recurrence where 60% of mice died after stopping vancomycin treatment. In contrast, auranofin (0.125 mg/kg and 0.25 mg/kg), significantly protected mice from CDI recurrence with 100% and 80% survival, respectively after 20 days (Fig. 3). Additionally, the relative body weight results (Fig. 4) showed that vehicle-treated group started to lose weight on day 2 and the weight loss continued till day 4. Afterwards, the surviving mice showed clinical recovery and started to gain weight till they returned to the normal weight. Vancomycin-treated mice, in coincidence with a previous report33, maintained their weight till the start of recurrence after the treatment discontinuation. By day 9, mice started to lose weight which continued to decrease until day 12. Thereafter, the average weight of surviving mice (40%) started to increase till they reached the normal weight. In contrast, auranofin-treated (0.125 mg/kg and 0.25 mg/kg) groups maintained a stable body weight along the duration of the experiment.

Efficacy of auranofin against CDI recurrence. Mice were treated with auranofin (0.125 mg/kg, and 0.25 mg/kg), vancomycin (10 mg/kg), or the vehicle for 5 days after infection with C. difficile spores and the treatments were stopped afterwards. Mice were monitored for survival. Kaplan–Meier survival curves were analyzed using a log-rank (Mantel–Cox) test. Asterisks (*) denote statistical significant difference between mice treated with either auranofin, or vancomycin in comparison with vehicle-treated mice.

Average relative weight of all surviving mice in C. difficile recurrence experiment. Infected mice were treated with auranofin (0.125 mg/kg and 0.25 mg/kg), vancomycin (10 mg/kg), or the vehicle for 5 days and treatments were stopped thereafter. Mice were weighed daily till the end of the experiment. The data are presented as percent relative weight (mean ± standard deviation) for each group.
A point worth noting, auranofin doses used in this study are achievable clinically. The recommended long term dosing regimen of auranofin in adult patients is 6-9 mg daily, and 0.1–0.25 mg/kg/day for children, in a single dose or divided doses44,45. Consequently, the most effective dose in this study, (0.125 mg/kg), is within range of doses administered clinically to humans. In addition, the therapeutic benefits and toxicity profile of auranofin have been monitored in clinical trials in more than 5,000 rheumatoid arthritis patients taking the drug and some of whom were monitored for more than 7 years. Auranofin did not show any evidence of cumulative toxicity and it was approved by the FDA for long-term treatment of rheumatoid arthritis in 198546. Furthermore, auranofin is approved for long-term treatment of rheumatoid arthritis, a much greater course than would be expected for anticlostridial therapeutics.
In conclusion, this study investigated the effectiveness of auranofin, at clinically achievable doses, as a CDI therapeutic. Auranofin’s in vitro antibacterial activity was stable in the presence of high bacterial inoculum size compared to vancomycin and fidaxomicin. Moreover, it maintained its anti-C. difficile activity after being exposed to SGF and SIF. Interestingly, it significantly protected mice against CDI at low doses (0.125 mg/kg and 0.25 mg/kg). Most importantly, auranofin (0.125 mg/kg and 0.25 mg/kg) significantly prevented CDI recurrence. These results indicate that auranofin warrants further investigation as a new CDI treatment option.
Materials and Methods
Bacterial strains, media and reagents
All experiments were performed following the relevant guidelines and regulations of the Purdue University Institutional Biosafety Committee. C. difficile strains (Table 3) were obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources) (Manassas, VA, USA), and the American Type Culture Collection (ATCC) (Manassas, VA, USA). Brain heart infusion broth was purchased from Becton, Dickinson and Company (Cockeysville, MD, USA). Hemin and vitamin K were obtained from Sigma-Aldrich (Saint Louis, MO, USA). Yeast extract, sucrose and L-cysteine were purchased from Fisher Scientific (Fail Lawn, NJ, USA). Phosphate buffered saline (PBS) (Corning, Manassas, VA, USA), pepsin from porcine gastric mucosa, pancreatin from porcine pancreas, hydrochloric acid (HCl), sodium chloride (NaCl), sodium hydroxide (NaOH), bovine serum albumin (Sigma-Aldrich, Saint Louis, MO, USA), monobasic potassium phosphate (KH2PO4) (Macron chemicals, Center Valley, PA, USA), vancomycin hydrochloride, gentamicin sulfate, kanamycin monosulfate, taurocholic acid (Chem-Impex, Wood Dale, IL, USA), metronidazole (Alfa Aesar, Ward Hill, MA, USA), and colistin sulfate, fidaxomicin (Cayman Chemical, Ann Arbor, MI, USA) were purchased commercially.
Evaluation of the effect of C. difficile inoculum size on the antibacterial activity of auranofin
The broth microdilution assay was used to determine the impact of C. difficile inoculum size on the minimum inhibitory concentrations (MICs) of auranofin and control antibiotics, as described previously24,47,48. Briefly, standard inoculum (SI: ~5 × 105 CFU/mL) and high inoculum (HI: ~5 × 107 CFU/mL) of each C. difficile strain were prepared in brain heart infusion supplemented broth (BHIS) and tested against auranofin and control antibiotics. Plates were then, incubated anaerobically at 37 °C for 48 hours. MICs reported are the lowest drug concentration that completely suppressed the growth of bacteria, as observed visually.
Activity of auranofin after exposure to simulated gastric fluid (SGF) and simulated intestinal fluid (SIF)
Simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) were prepared as described earlier32,49. Briefly, SGF (pH = 1.2) was prepared by dissolving NaCl (2 g) and pepsin (3.2 g) in 7 mL of concentrated HCl and deionized water was subsequently added to make up a final volume of 1 L. Then, the pH was adjusted to 1.2. To prepare SIF (pH = 6.8), 6.8 g of KH2PO4 was dissolved in 250 mL of water, and 77 mL of 0.2 N NaOH and 500 mL of deionized water were added. Afterwards, 10 g of pancreatin was added, and the pH of the resulting solution was adjusted to 6.8.
The broth microdilution assay24,47,48 was used to determine the MICs of auranofin and control antibiotics in presence of SGF and SIF. Briefly, auranofin and control drugs were incubated with each of SGF and SIF for 2, 4 and 24 hours. After the corresponding times, broth microdilution assay was performed to determine the MICs of the tested drugs.
Preparation of C. difficile spores for mice infection
C. difficile spores were prepared as described earlier50. Briefly, C. difficile ATCC 43255 was inoculated onto BHIS agar and incubated anaerobically for 5 days. Spores were collected anaerobically using PBS containing 10% bovine serum albumin, heated at 70 °C for 20 minutes to get rid of vegetative cells and counted by dilution and plating onto BHIS supplemented with 0.1% taurocholic acid. Spores were then, stored at 4°C overnight before infecting mice.
In vivo efficacy of auranofin in a CDI mouse model
CDI mouse model
The study was reviewed, approved and performed following the guidelines of the Purdue University Animal Care and Use Committee (PACUC) and according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Mice were housed in individually ventilated autoclaved cages and received sterile food and water ad libitum throughout the duration of the experiment. CDI mouse model was performed as described previously33 with modifications. Since disruption of microbiota depends on mice drinking naturally, we performed three modifications: (1) increasing the concentrations of antibiotics to ensure microbiota disruption, (2) adding 7.5% sucrose to the drinking water containing antibiotics to overcome the very bitter taste of the antibiotics in drinking water, as mice are expected to decrease their rate of water consumption due to its bitter taste, and (3) extending the duration of administering antibiotic cocktail in drinking water to 5 days to ensure microbiota disruption. Eight-week-old female pathogen-free C57BL/6 mice (Jackson, ME, USA) were pre-treated with an antibiotic cocktail in sterile drinking water to disrupt the mice normal intestinal microflora, reducing the colonization resistance and facilitating infection with the toxigenic strain of C. difficile. The cocktail contained kanamycin (1.2 mg/mL), gentamicin (0.105 mg/mL), colistin (2550 U/mL), metronidazole (0.645 mg/mL), vancomycin (0.135 mg/mL) and sucrose (75 mg/mL) for 5 days. Afterwards, mice were switched to regular autoclaved water for 2 days and they received a single dose of clindamycin (10 mg/kg) intraperitoneally 1 day prior to C. difficile challenge.
For infection, mice were restrained and infected intragastrically with 1.3 ×106 spores of C. difficile ATCC 43255 via oral gavage using a ball tipped metal feeder. Number of spores used were re-counted after infection to confirm the infected dose.
In vivo efficacy of different doses of auranofin in a CDI mouse model
Following infection, mice were randomly allocated into groups (n = 5) for treatment. Two hours post-infection, three groups were treated orally with auranofin (0.125 mg/kg, 0.25 mg/kg and 0.5 mg/kg), one group was treated with vancomycin (10 mg/kg) via oral gavage, and one group was treated orally with the vehicle (10% DMSO in PBS). Treatments were continued once daily for five days and mice were checked (6 times daily) for disease signs (including weight loss, behavioral changes, hunched posture, decreased activity, wet tail and diarrhea).
In vivo efficacy of auranofin in C. difficile recurrence
In order to investigate the activity of auranofin in preventing C. difficile recurrence, mice were infected, as described above and two groups were treated orally with auranofin (0.125 mg/kg and 0.25 mg/kg), one group was treated with vancomycin (10 mg/kg) via oral gavage, and one group was treated orally with the vehicle (10% DMSO in PBS) for 5 days. Treatments were stopped after 5 days and mice were monitored (6 times daily) for disease signs and recurrence of infection till the 20th day. Then, mice were humanely euthanized at 21st day post-infection using CO2 asphyxiation.
Statistical analyses
The survival data were analyzed by Log-rank (Mantel-Cox) test utilizing GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA, USA).
Ethical approval
All animal housing and experiments were reviewed, approved and performed under the guidelines of the Purdue University Animal Care and Use Committee and carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Data availability
Data presented in this study are available from the corresponding author upon a proper request.
References
Zhang, S. et al. Cost of hospital management of Clostridium difficile infection in United States-a meta-analysis and modelling study. BMC Infect Dis 16, 447, https://doi.org/10.1186/s12879-016-1786-6 (2016).
Centers for Disease Control and Prevention (CDC). Antibiotic / Antimicrobial Resistance (AR/AMR), Biggest Threats and Data, https://www.cdc.gov/drugresistance/biggest-threats.html (2019).
Kachrimanidou, M. & Malisiovas, N. Clostridium difficile Infection: A Comprehensive Review. Crit Rev Microbiol 37, 178–187, https://doi.org/10.3109/1040841X.2011.556598 (2011).
Davies, A. H., Roberts, A. K., Shone, C. C. & Acharya, K. R. Super toxins from a super bug: structure and function of Clostridium difficile toxins. Biochem J 436, 517–526, https://doi.org/10.1042/BJ20110106 (2011).
Chumbler, N. M. et al. Clostridium difficile Toxin B Causes Epithelial Cell Necrosis through an Autoprocessing-Independent Mechanism. Plos Pathog 8, https://doi.org/10.1371/journal.ppat.1003072 (2012).
He, M. et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 45, 109–113, https://doi.org/10.1038/ng.2478 (2013).
Smits, W. K., Lyras, D., Lacy, D. B., Wilcox, M. H. & Kuijper, E. J. Clostridium difficile infection. Nat Rev Dis Primers 2, 16020 (2016).
Awad, M. M., Johanesen, P. A., Carter, G. P., Rose, E. & Lyras, D. Clostridium difficile virulence factors: Insights into an anaerobic spore-forming pathogen. Gut Microb 5, 579–593, https://doi.org/10.4161/19490976.2014.969632 (2014).
Viswanathan, V. K., Mallozzi, M. J. & Vedantam, G. Clostridium difficile infection: An overview of the disease and its pathogenesis, epidemiology and interventions. Gut Microb 1, 234–242, https://doi.org/10.4161/gmic.1.4.12706 (2010).
McDonald, L. C. et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis 66, E1–E48, https://doi.org/10.1093/cid/cix1085 (2018).
Vardakas, K. Z. et al. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents 40, 1–8, https://doi.org/10.1016/j.ijantimicag.2012.01.004 (2012).
Orenstein, R. Fidaxomicin failures in recurrent Clostridium difficile infection: a problem of timing. Clin Infect Dis 55, 613–614, https://doi.org/10.1093/cid/cis495 (2012).
Cornely, O. A., Miller, M. A., Louie, T. J., Crook, D. W. & Gorbach, S. L. Treatment of first recurrence of Clostridium difficile infection: fidaxomicin versus vancomycin. Clin Infect Dis 55(Suppl 2), S154–161, https://doi.org/10.1093/cid/cis462 (2012).
Zhanel, G. G., Walkty, A. J. & Karlowsky, J. A. Fidaxomicin: A novel agent for the treatment of Clostridium difficile infection. Can J Infect Dis Med 26, 305–312, https://doi.org/10.1155/2015/934594 (2015).
Baines, S. D. & Wilcox, M. H. Antimicrobial Resistance and Reduced Susceptibility in Clostridium difficile: Potential Consequences for Induction, Treatment, and Recurrence of C. difficile Infection. Antibiotics 4, 267–298, https://doi.org/10.3390/antibiotics4030267 (2015).
Roder, C. & Thomson, M. J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R&D 15, 13–20, https://doi.org/10.1007/s40268-015-0083-y (2015).
AbdelKhalek, A., Abutaleb, N. S., Elmagarmid, K. A. & Seleem, M. N. Repurposing auranofin as an intestinal decolonizing agent for vancomycin-resistant enterococci. Sci Rep-Uk 8, https://doi.org/10.1038/S41598-018-26674-0 (2018).
Capparelli, E. V., Bricker-Ford, R., Rogers, M. J., McKerrow, J. H. & Reed, S. L. Phase I Clinical Trial Results of Auranofin, a Novel Antiparasitic Agent. Antimicrob Agents Chemother 61, https://doi.org/10.1128/AAC.01947-16 (2017).
da Silva, M. T. et al. In vivo and in vitro auranofin activity against Trypanosoma cruzi: Possible new uses for an old drug. Exp Parasitol 166, 189–193, https://doi.org/10.1016/j.exppara.2015.05.012 (2016).
Harbut, M. B. et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc Natl Acad Sci U S A 112, 4453–4458, https://doi.org/10.1073/pnas.1504022112 (2015).
Tejman-Yarden, N. et al. A reprofiled drug, auranofin, is effective against metronidazole-resistant Giardia lamblia. Antimicrob Agents Chemother 57, 2029–2035, https://doi.org/10.1128/AAC.01675-12 (2013).
Thangamani, S. et al. Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci Rep 6, 22571, https://doi.org/10.1038/srep22571 (2016).
Thangamani, S., Mohammad, H., Abushahba, M. F., Sobreira, T. J. & Seleem, M. N. Repurposing auranofin for the treatment of cutaneous staphylococcal infections. Int J Antimicrob Agents 47, 195–201 (2016).
AbdelKhalek, A., Abutaleb, N. S., Mohammad, H. & Seleem, M. N. Antibacterial and antivirulence activities of auranofin against Clostridium difficile. Int J Antimicrob Agents 53, 54–62, https://doi.org/10.1016/j.ijantimicag.2018.09.018 (2019).
Thangamani, S. et al. Repurposing approach identifies auranofin with broad spectrum antifungal activity that targets Mia40-Erv1 pathway. Front Cell Infect Microbiol 7, 4 (2017).
Fletcher, J. R., Erwin, S., Lanzas, C. & Theriot, C. M. Shifts in the Gut Metabolome and Clostridium difficile Transcriptome throughout Colonization and Infection in a Mouse Model. Msphere 3, https://doi.org/10.1128/mSphere.00089-18 (2018).
Hutton, M. L. et al. Repurposing auranofin as a Clostridioides difficile therapeutic. J Antimicrob Chemother (2019).
Battaglioli, E. J. et al. Clostridioides difficile uses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Sci Transl Med 10, https://doi.org/10.1126/scitranslmed.aam7019 (2018).
Gottlieb, N. L. Pharmacology of Auranofin - Overview and Update. Scand J Rheumatol, 19-28 (1986).
Babakhani, F., Seddon, J., Robert, N., Shue, Y. K. & Sears, P. Effects of Inoculum, pH, and Cations on the In Vitro Activity of Fidaxomicin (OPT-80, PAR-101) against Clostridium difficile. Antimicrob AgentsChemother 54, 2674–2676, https://doi.org/10.1128/Aac.01842-09 (2010).
Asafu-Adjaye, E. B. et al. Validation and application of a stability-indicating HPLC method for the in vitro determination of gastric and intestinal stability of venlafaxine. J Pharmaceut Biomed 43, 1854–1859, https://doi.org/10.1016/j.jpba.2006.12.035 (2007).
Yellepeddi, V. K. et al. Biopharmaceutical Characterization and Oral Efficacy of a New Rapid Acting Antidepressant Ro 25-6981. J Pharm Sci-Us 107, 2472–2478, https://doi.org/10.1016/j.xphs.2018.05.005 (2018).
Chen, X. H. et al. A Mouse Model of Clostridium difficile-Associated Disease. Gastroenterol 135, 1984–1992, https://doi.org/10.1053/j.gastro.2008.09.002 (2008).
Maier, L. et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–+, https://doi.org/10.1038/nature25979 (2018).
Tam, J. et al. Host-targeted niclosamide inhibits C. difficile virulence and prevents disease in mice without disrupting the gut microbiota. Nat Commun 9, https://doi.org/10.1038/S41467-018-07705-W (2018).
Munoz-Miralles, J. et al. Indomethacin increases severity of Clostridium difficile infection in mouse model. Future Microbiol 13, 1271–1281, https://doi.org/10.2217/fmb-2017-0311 (2018).
Abutaleb, N. S. & Seleem, M. N. Antivirulence activity of auranofin against vancomycin-resistant enterococci: in vitro and in vivo studies. Int J Antimicrob Agents, https://doi.org/10.1016/j.ijantimicag.2019.10.009 (2019).
Eyre, D. W. et al. Predictors of first recurrence of Clostridium difficile infection: implications for initial management. Clin Infect Dis 55(Suppl 2), S77–87, https://doi.org/10.1093/cid/cis356 (2012).
Cornely, O. A., Miller, M. A., Louie, T. J., Crook, D. W. & Gorbach, S. L. Treatment of first recurrence of Clostridium difficile infection: fidaxomicin versus vancomycin. Clin Infect Dis 55(Suppl 2), S154–161, https://doi.org/10.1093/cid/cis462 (2012).
Petrella, L. A. et al. Decreased cure and increased recurrence rates for Clostridium difficile infection caused by the epidemic C. difficile BI strain. Clin Infect Dis 55, 351–357, https://doi.org/10.1093/cid/cis430 (2012).
Jung, K. S. et al. Risk Factors for Treatment Failure and Recurrence after Metronidazole Treatment for Clostridium difficile-associated Diarrhea. Gut and liver 4, 332–337, https://doi.org/10.5009/gnl.2010.4.3.332 (2010).
Kelsen, J. R. et al. Recurrence rate of Clostridium difficile infection in hospitalized pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis 17, 50–55, https://doi.org/10.1002/ibd.21421 (2011).
Centers for Disease Control and Prevention (CDC). Antibiotic Resistance Threats in the United States, 2013. Centers for Disease Control and Prevention. p. 1-114., (2013).
Furst, D. E. Mechanism of action, pharmacology, clinical efficacy and side effects of auranofin. An orally administered organic gold compound for the treatment of rheumatoid arthritis. Pharmacotherapy 3, 284–298 (1983).
Fantini, F. et al. Changes of Immunological Parameters during Auranofin Treatment in Children Affected with Juvenile Chronic Arthritis. Int J Clin Pharm Res 6, 61–67 (1986).
Blodgett, R. C. & Pietrusko, R. G. Long-Term Efficacy and Safety of Auranofin - a Review of Clinical-Experience. Scand J Rheumatol, 67-78 (1986).
Mody, D., Athamneh, A. I. M. & Seleem, M. N. Curcumin: A natural derivative with antibacterial activity against Clostridium difficile. J Glob Antimicrob Resist, https://doi.org/10.1016/j.jgar.2019.10.005 (2019).
Shao, X. et al. Chemical Space Exploration Around Thieno[3,2-d]pyrimidin-4(3H)-one Scaffold led to a Novel Class of Highly Active Clostridium difficile Inhibitors. J Med Chem, https://doi.org/10.1021/acs.jmedchem.9b01198 (2019).
Pan, X. M., Li, J., Gan, R. & Hu, X. N. Preparation and in vitro evaluation of enteric-coated tablets of rosiglitazone sodium. Saudi Pharm J 23, 581–586, https://doi.org/10.1016/j.jsps.2015.02.018 (2015).
Edwards, A. N. & McBride, S. M. Isolating and Purifying Clostridium difficile Spores. Methods Mol Biol 1476, 117–128, https://doi.org/10.1007/978-1-4939-6361-4_9 (2016).
Acknowledgements
The authors would like to thank Haroon Mohammad and Marwa Alhashimi for their kind help in this work. Research reported in this publication was supported by the NIAID/NIH (Grant number R01AI130186).
Author information
Authors and Affiliations
Contributions
N.S.A. designed and performed the in vitro experiments, and in vivo mice infection experiments, collected the data, managed the statistics, prepared the figures and wrote the manuscript. M.N.S. provided reagents and items for the study, supervised the conduct of the study and data collection, assisted with results interpretation and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abutaleb, N.S., Seleem, M.N. Auranofin, at clinically achievable dose, protects mice and prevents recurrence from Clostridioides difficile infection. Sci Rep 10, 7701 (2020). https://doi.org/10.1038/s41598-020-64882-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64882-9
- Springer Nature Limited
This article is cited by
-
Promiscuous, persistent and problematic: insights into current enterococcal genomics to guide therapeutic strategy
BMC Microbiology (2024)
-
Inhibition of selenoprotein synthesis is not the mechanism by which auranofin inhibits growth of Clostridioides difficile
Scientific Reports (2023)
-
Investigating auranofin for the treatment of infected diabetic pressure ulcers in mice and dermal toxicity in pigs
Scientific Reports (2021)
-
In vivo efficacy of auranofin in a hamster model of Clostridioides difficile infection
Scientific Reports (2021)