
Abstract
Hepatocellular carcinoma (HCC) is closely associated with liver fibrosis. Hepatic stellate cells (HSC) and cancer-associated myofibroblasts are key players in liver fibrogenesis and hepatocarcinogenesis. Overexpression of fibroblast growth factor (FGF) receptors contributes to HCC development and progression. This study aimed to elucidate the role of FGFs in the HSC-HCC crosstalk. Analysis of the expression of the fifteen paracrine FGF-members revealed that FGF9 was only expressed by HSC but not by HCC cells. Also in human HCC tissues, HSC/stromal myofibroblasts were identified as cellular source of FGF9. High expression levels of FGF9 significantly correlated with poor patient survival. Stimulation with recombinant FGF9 induced ERK- and JNK-activation combined with significantly enhanced proliferation, clonogenicity, and migration of HCC cells. Moreover, FGF9 significantly reduced the sensitivity of HCC cells against sorafenib. Protumorigenic effects of FGF9 on HCC cells were almost completely abrogated by the FGFR1/2/3 inhibitor BGJ398, while the selective FGFR4 inhibitor BLU9931 had no significant effect. In conclusion, these data indicate that stroma-derived FGF9 promotes tumorigenicity and sorafenib resistance of HCC cells and FGF9 overexpression correlates with poor prognosis in HCC patients. Herewith, FGF9 appears as potential prognostic marker and novel therapeutic target in HCC.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is one of the most aggressive malignancies and one of the most common causes of cancer death worldwide1. In most cases, HCC is diagnosed in already advanced stages, and currently, there are very limited options for HCC treatment. The multi-tyrosin kinase inhibitor sorafenib is the first-line treatment for advanced-stage HCC patients, however, the benefits are at best modest and transient1,2. Therefore, better understanding of the drivers of HCC development and progression and new therapeutic targets are highly needed.
HCC is strongly associated with liver fibrosis and cirrhosis, suggesting that the environment in which HCC arises may influence cancerogenesis3. The activation of hepatic stellate cells (HSC) into extracellular matrix (ECM)-producing myofibroblasts is the key event of hepatic fibrosis4. Several studies using genetic cell fate mapping have provided strong evidence that HSC are also the major precursors of myofibroblasts in the HCC microenvironment3, and that HSC/stromal myofibroblasts promote HCC development and progression3,5. Therefore, a better understanding of the role of HSC and their crosstalk with HCC cells may provide new therapeutic options for the treatment of HCC.
Fibroblast growth factor (FGF) signaling plays an important role in the regulation of many biological processes such as development, cell proliferation and differentiation, and its dysregulation is reported in different types of diseases including cancers6. The FGF family comprises 22 proteins that can be classified into intracrine, endocrine and paracrine factors. Paracrine FGFs can be further subclustered into five subfamilies (FGF1/2; FGF3/7/10/22; FGF4/5/6; FGF8/17/18 and FGF9/16/20). All FGFs interact with heparan sulfate proteoglycans. However, paracrine and endocrine FGFs show different affinity to these matrix components, which results in a predominantly local action of paracrine FGFs near the place of secretion6. The FGFs signal through four transmembrane tyrosine kinase FGF receptors (FGFR) namely FGFR1, FGFR2, FGFR3 and FGFR46.
Several alterations in FGF-signaling have been found to affect liver carcinogenesis7. Aberrant expression of the endocrine FGF19 and its high affinity FGFR4 contributes to HCC progression8. Furthermore, overexpression of FGFR2 and FGFR3 contributes to HCC development and metastasis9,10, further suggesting that FGF-signaling plays an important role in HCC.
While most previous studies had focused on the role of FGFRs in HCC, the aim of the present study was to get a deeper understanding of the expression and tumorigenic effects of different FGFR-ligands with a focus on paracrine FGFs and their role in the HSC-HCC crosstalk.
Results and Discussion
FGF9 expression in HCC
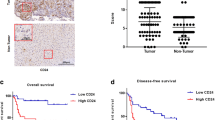
First, we systematically analyzed the expression of paracrine FGFs (subfamilies 1 (FGF1 and FGF2), 4 (FGF4, FGF5 and FGF6), 7 (FGF3, FGF7, FGF10 and FGF22), 8 (FGF8, FGF17, FGF18) and 9 (FGF9, FGF16, FGF20)) in activated human HSC from 3 different donors and 4 HCC cell lines (Hep3B, HepG2, PLC and Huh7) using qRT-PCR analysis. Notably, mRNA expression levels of FGF1, FGF2, FGF5, FGF7 and FGF9 were more than 100-fold higher in HSC compared with HCC cells (Fig. 1A). To get further insight into the role of these FGFs in HCC, we assessed the correlation between their tumorous expression levels and survival of HCC patients using the “SurvExpress” Biomarker validation for cancer gene expression database11. Computational stratification into “low-risk” and “high-risk” patient groups (based on prognostic index) revealed significant overexpression of FGF9 as well as reduced overall survival of high- compared to low-risk groups in the TCGA Liver Cancer dataset (n = 381) (Fig. 1B) as well as in the LIHC-TCGA HCC (n = 361) and Hoshida Golub Liver GSE10143 (n = 162) datasets (Suppl. Fig. 1A,B). In contrast, no correlation was found between tumorous FGF1-, FGF2-, FGF5-, and FGF7-expression levels and survival in HCC patients (data not shown).

Analysis of paracrine FGFs expression with focus on FGF9 in HCC. (A) Fold mRNA expression levels of paracrine FGFs (subfamilies 1 (FGF1 and FGF2), 4 (FGF4, FGF5 and FGF6), 7 (FGF3, FGF7, FGF10 and FGF22), 8 (FGF8, FGF17, FGF18) and 9 (FGF9, FGF16, FGF20)) in four human HCC cell lines (Hep3B, Huh7, PLC, HepG2) compared to expression in primary human hepatic stellate cells (HSC) from three different donors. (B) “SurvExpress-Biomarker validation for cancer gene expression” database analysis of FGF9 expression (right panel) and corresponding Kaplan-Meier curve for overall survival (left panel) in “TCGA liver cancer” dataset. Computational stratification of patients into “Low Risk” and “High Risk” groups was based on prognostic index and according to the “Maximized Risk Groups” algorithm. (C) Western blot analysis of FGF9 protein levels in primary human hepatic stellate cells (phHSC) and human HCC cell lines (Hep3B, HepG2, Huh7, PLC). (D) Immunofluorescence staining for FGF9 (green) and alpha-smooth muscle actin (alphaSMA; red) of primary human hepatic stellate cells. Nuclei were counterstained using DAPI (blue). (E) Correlation of FGF9 and alpha-sma mRNA expression in human HCC tissues (n = 14). (F) Representative image of immunofluorescence staining for FGF9 (green) and alphaSMA (red) in a human HCC tissue section. Nuclei were counterstained using DAPI (blue).
The high expression of FGF9 in HSC as compared with HCC cells and the strong correlation of high FGF9 expression in human HCC tissues with the survival of HCC patients prompted us to focus our further analysis on FGF9. Western blot analysis confirmed FGF9 expression in HSC while no FGF9 expression was detectable in the HCC cell lines (Fig. 1C). Immunofluorescence analysis showed a distinct FGF9-signal in cell-culture activated primary human HSC that colocalized with alpha-sma, a characteristic marker of activated HSC and stromal myofibroblasts3,4 (Fig. 1D). Analysis of tumor tissue samples from HCC patients revealed a significant correlation of the expression levels of FGF9 and alpha-sma (Fig. 1E). For comparison, FGF9 and alpha-sma mRNA expression levels were more than 10-fold higher in activated HSC compared with HCC-tissue levels (Suppl. Fig. 1C,D). Furthermore, immunofluorescence analysis showed co-localization of alpha-sma and FGF9 in human HCC tissues (Fig. 1F). In summary, these results indicate activated HSC/myofibroblasts as cellular source of FGF9 in HCC and suggest that enhanced FGF9 expression in HCC promotes tumor progression.
Effects of FGF9 on tumorigenicity of HCC cells
Therefore, we wanted to analyze the effects of recombinant FGF9 (rFGF9) on tumorigenicity of human HCC cells in functional in vitro assays. With up to 20 ng/ml we applied similar FGF9 doses as used in previous in vitro studies12,13,14,15,16. Stimulation with rFGF9 caused a dose-dependent induction of the proliferation in Hep3B and HepG2 cells but not in PLC cells (Fig. 2A). Since FGF9 has been identified to be a high affinity ligand for mainly FGFR2 and FGFR317, we compared the expression of these 2 FGF-receptors in HCC cell lines. While PLC cells showed comparable FGFR3 expression with HepG2 and Hep3B cells, FGFR2-expression was markedly reduced in PLC cells (Suppl. Fig. 1E,F). Thus, differences in FGFR2 expression might be a potential explanation for the varying FGF9 responsiveness regarding proliferation. In contrast, the very low FGF9 expression levels did not differ between the responsive (Hep3B and HepG2) and non-responsive (PLC) cells (Suppl. Fig. 1G) and further depletion of FGF9 with specific siRNA did not affect their proliferation (data not shown), further indicating that endogenous FGF9 expression in the different HCC cell lines has no significant impact on their proliferation.

Effect of FGF9 on tumorigenicity of HCC cells. (A) Proliferation of HCC cell lines Hep3B, HepG2 and PLC stimulated without (ctr.) or with recombinant FGF9 (rFGF9) for 72 h. (B) Quantification of colony number and size (left and middle panel) and representative images (right panels) in anchorage-dependent clonogenic assays with Hep3B cells treated without (ctr.) or with rFGF9. (C) Migratory activity of HCC cells following 4 h treatment with rFGF9 (20 ng/ml; left panel) and representative images of Boyden chamber filters (right panel). Arrowheads indicate migrated cells. (D) Western Blot analysis of phosphorylated ERK and JNK1/2 in rFGF9 (20 ng/ml) treated and control cells. (E) Effects of PD98059 (a selective inhibitor of the MEK/ERK pathway; 10 µM) and SP600125 (JNK inhibitor; 10 µM) on rFGF9 (20 ng/ml)-induced proliferation of Hep3B cells. (F) Effects of PD98059 and SP600125 on proliferation of FGF9 overexpressing (HCCFGF9) and control (HCCctr) Hep3B cells (G) FACS analysis of cell cycle fractions of HCC cells treated with conditioned media (CM) from HSC with siRNA-mediated FGF9 suppression (HSC-siFGF9) or HSC transfected with control siRNA (HSC-siCtr; left panel); representative images (right panel). (H) Volume of spheroids formed by Hep3B cells alone or mixed spheroids of Hep3B cells and control transfected HSC (HSCctr) or Hep3B cells and HSC transfected with an FGF9 expression plasmid (HSCFGF9); representative microscopic images. (Analysis have been performed in triplicates; *p < 0.05).
Next, we assessed the impact of FGF9 on HCC cells in clonogenicity assays, which reflects also stem cell properties and cell survival of tumors cells. Here, stimulation with FGF9 induced the colony number as well as the colony size of Hep3B cells (Fig. 2B). In HepG2 cells, FGF9 stimulation did not affect the colony size and only the highest FGF9 dose induced the colony number (Suppl. Fig. 2A). In contrast, FGF9 dose dependently induced the colony size of PLC cells but not the colony number (Suppl. Fig. 2B). Analysis of the impact of FGF9 on the migratory activity of HCC cells in transwell Boyden chamber assays revealed that FGF9 significantly induced the directed migration of Hep3B and HepG2 cells but had only a slight effect on PLC cells (Fig. 2C). Together these data indicate, that generally FGF9 induces tumorigenic characteristics of HCC cells but there are qualitative and quantitative differences between different tumor cells. It appears, that differences in FGF-receptor expression may only partly explain this phenomenon, which has also already been described in other tumor entities. For example, Sun et al. described FGF9 did not induce proliferation but had an anti-apoptotic effect in gastric cancer cells15. Activation of mitogen-activated protein kinases (MAPK) is known to be associated with HCC proliferation, migration and stem cell properties including enhanced clonogenicity1,18. Therefore, we next analyzed the effect of FGF9 stimulation on the phosphorylation of extracellular-signal regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in HCC cells (Fig. 2D). FGF9 induced ERK- and JNK-phosphorylation in Hep3B, HepG2 as well as PLC cells (Fig. 2D). Incubation with PD98059, a specific MEK/ERK-pathway-inhibitor, significantly but not completely reduced the rFGF9-induced growth promoting effect in Hep3B cells. SP600125, a specific JNK-inhibitor, significantly reduced both basal as well as FGF9 induced proliferation (Fig. 2E). Still, relative to basal levels, FGF9 induced proliferation also in the presence of SP600125 in Hep3B cells (Fig. 2E). In HepG2 cells, PD98059 had no significant impact on the FGF9 induced growth promoting effect while SP600125 significantly reduced basal cell growth and also blunted rFGF9-induced proliferation (Suppl. Fig. 2C).
In addition to the effect of the stimulation with recombinant FGF9, we assessed the impact of overexpression of FGF9 in HCC cells. HCC cells were transfected with an FGF9-expression plasmid or empty vector as control (Suppl. Fig. 2D). FGF9-overexpression significantly increased the proliferation of HCC cells and this FGF9-induced proliferation was efficiently blocked by the JNK inhibitor SP600125 while ERK-inhibition showed no significant effect (Fig. 2F). Together, these data indicate that FGF9 inducing effect of the ERK- and JNK-pathways is at least in part responsible for the observed effects on different tumorigenic characteristics of HCC cells.
Next, we wanted to simulate the interaction between HSC (derived FGF9) and HCC cells in vitro. Once, HCC cells were incubated with either conditioned media (CM) from HSC with siRNA mediated FGF9 suppression or HSC transfected with control siRNA (Suppl. Fig. 2E). FACS analysis revealed that the growth promoting effect of CM from control HSC was significantly higher as compared with CM from FGF9-depleted HSC (Fig. 2G). Furthermore, we used a spheroid formation assay to assess the interaction between HCC and HSC. Here, we used a complementary approach and compared the effects of FGF9-overexpressing HSC (transfected with an FGF9-expression plasmid) and control HSC (transfected with empty vector) (Suppl. Fig. 2F). After 11 days, mixed HSC-HCC spheroids were significantly larger than spheroids formed by pure HCC cells (Fig. 2H). Still, spheroids of HCC-cells and FGF9 overexpressing HSC were significantly larger than spheroids of HCC-cells and control HSC (Fig. 2H and Suppl. Fig. 2G). Together, these findings indicate that FGF9 is not the only but a significant factor by which hepatic stellate cells promote the tumorigenicity of HCC cells.
Effects of FGF9 on HCC cells in combination with sorafenib
The multi-kinase inhibitor sorafenib also inhibits Raf/MEK/MAPK signaling and is currently the only clinically established pharmacological therapy for HCC. Therefore, we next wanted to analyze the effect of FGF9 on HCC cells in combination with the multi-kinase inhibitor sorafenib. Stimulation with rFGF9 significantly decreased the efficacy of sorafenib to inhibit proliferation and to induce cell death in HCC cells (Fig. 3A,B). PI/Annexin V flow cytometric analysis confirmed that rFGF9 inhibited sorafenib-induced apoptosis of HCC cells (Fig. 3C). Together, these data indicate that FGF9 enhances sorafenib resistance of HCC cells. Previous studies have linked JNK activation19,20,21,22 or ERK activation23 to sorafenib resistance. To determine whether FGF9 induced activation of ERK or JNK in HCC cells is responsible for the observed effect on sorafenib resistance, we repeated the analysis in the presence of the specific inhibitors PD98059 (MEK/ERK-pathway) or SP600125 (JNK-pathway). Interestingly, only SP600125 had a pronounced effect on rFGF9-induced sorafenib resistance (Fig. 3D). This indicates that JNK-activation is, at least in part, responsible for FGF9-induced sorafenib resistance of HCC cells and thus further suggests FGF9 and FGF9-induced signaling, respectively, as promising strategy in HCC.

Effects of recombinant FGF9 on HCC cells in combination with sorafenib. (A) Proliferation of HCC cells treated with depicted doses of sorafenib (SF) and rFGF9 (20 ng/ml) for 48 h. (B) Corresponding microscopic images of HCC cells treated w/or w/o rFGF9 (20 ng/ml) and SF (2 µM). (C) Flow cytometric analysis of Annexin V-FITC and propidium iodide (PI) stained HCC cells following treatment with rFGF9 (20 ng/ml) and/or SF (2 µM). The right panel shows representative dot plots. (D) Effects of PD98059 (MEK/ERK inhibitor; 10 µM) or SP600125 (JNK inhibitor; 10 µM) on rFGF9 (20 ng/ml) induced sorafenib resistance of HCC cells. (Analyses have been performed in triplicates; *p < 0.05).
Modulation of FGF9 effects on HCC cells by FGFR-inhibitors
Therapeutic targeting of FGF-receptors (FGFR) with pharmacological inhibitors has shown promising results in preclinical HCC models24, and several FGFR inhibitors are in clinical trials to treat cancers harboring FGFR aberrations (e.g. ClinicalTrials.gov Identifier: NCT02965378). Therefore and to get further information on the receptors mediating the protumorigenic FGF9 effects in HCC cells, we compared the effects of the selective FGFR4 inhibitor BLU993125 and the FGFR1/2/3 inhibitor BGJ398 (infigratinib)25 on HCC cells in the presence or absence of rFGF9. BGJ398 completely blocked the rFGF9-induced ERK- and JNK-phosphorylation in HCC cells (Fig. 4A). In contrast, BLU9931 had no effect on FGF9 induced ERK-phsophorylation and only slightly reduced JNK phosphorylation in HCC cells (Fig. 4A). Furthermore, BGJ398 completely inhibited the growth inducing effect of FGF9 in Hep3B cells while BLU9931 exhibited no significant effect (Fig. 4B). Moreover, the increased number and size of colonies formed by FGF9 stimulated HCC cells was blunted by BGJ398 while BLU9931 did not significantly affect the FGF9 effects on HCC cells in clonogenicity assays (Fig. 4C,D). Thus, while most preclinical and clinical studies (e.g. ClinicalTrials.gov Identifier: NCT02834780 and NCT03144661) focus on selective FGFR4 inhibitors26, our data indicate that rather FGFR1/2/3 inhibition is suitable to inhibit the protumorigenic FGF9 effects on HCC cells.

Effect of FGFR inhibitors on HCC cells. Effects of BGJ398 (a FGF receptor 1-3 inhibitor, 100 nM) and BLU9931 (a FGF receptor 4 inhibitor, 100 nM) on (A) rFGF9 (20 ng/ml)-induced phosphorylation of ERK and JNK1/2, (B) rFGF9 (20 ng/ml)-induced proliferation, and (C) colony number and (D) colony size anchorage-dependent clonogenic assays with Hep3B cells treated without (ctr.) or with rFGF9 (20 ng/ml). (Analysis have been performed in triplicates; *p < 0.05).
Future studies are needed to assess which of FGFR 1, 2 or 3 majorly contributes to the pro-tumorigenic effects of FGF9 on HCC cells and whether there may be individual differences, which could be used for targeted therapy with selective FGFR inhibition, which are in development27. Furthermore, FGF traps or anti-FGF antibodies27 could be exploited to specifically target FGF9 alone or in combination with other drugs, such as sorafenib. It is further tempting to speculate whether FGF9 expression levels could serve as biomarker for such therapeutic strategies. In any case, FGF9 expression levels appear as novel prognostic marker for survival of HCC patients. A recent study identified miR-140-5p as tumor suppressor in HCC and suggested that part of its inhibitory effect on tumor growth is mediated via suppressing FGF9, which these authors identified as miR-140-5p target in HCC cells28. Although our study indicates HSC/stromal myofibroblasts as major source of FGF9 in HCC, it may be that in a subset of HCCs also the tumor cells contribute to local FGF9 levels.
In this study, we focused on FGF9 effects on HCC cells and its impact on resistance to sorafenib. In addition, FGF/FGFR signaling has been implicated in angiogenesis and immune surveillance25. Therefore, it is possible that FGF9 exhibits further tumor promoting effects and leads to resistance to immune checkpoint or VEGF/VEGF receptor (VEGFR)-targeted agents, which needs to be addressed in future studies.
In summary, our study indicates that FGF9 derived from activated HSC enhances the tumorigenicity and therapy resistance of HCC cells embedded in a fibrotic microenvironment. These data may form the basis for future studies assessing the potential of FGF9 as prognostic biomarker and targeting of FGF9-FGFR signaling as a new treatment approach for patients with HCC.
Materials and Methods
Cells and cell culture
Primary human hepatic stellate cells (HSC) were isolated and cultured as described29. In vitro activation of HSC was achieved by cell culture on uncoated tissue culture dishes29. Tissue samples for cell isolation were obtained from patients undergoing partial hepatectomy for metastatic liver tumors. All experimental procedures were performed according to the guidelines of the non-profit state-controlled HTCR (Human Tissue and Cell Research) with informed patients’ consent30. Only those liver tissues judged as noncancerous by local pathologists were used for cell preparation. Further exclusion criteria were known liver disease or histologic evidence for liver fibrosis or inflammation in surrounding nontumorous liver tissue. Human HCC cell lines PLC (ATCC CRL-8024), Hep3B (ATCC HB-8064), HepG2 (ATCC HB-8065) and Huh7 (ATCC PTA-4583) were cultured as described31. Spheroid co-culture experiments of HCC cells and HSC were performed as described31.
For stimulation experiments, cells were treated with recombinant human FGF9 (R&D Systems, Minneapolis, MN, USA), sorafenib (Cayman Chemicals, Ann Arbor, MI, USA), PD98059 (inhibitor of the MEK/ERK pathway; Calbiochem, La Jolla, CA, USA), SP600125 (JNK inhibitor; Calbiochem), the FGF receptor 1-3 inhibitor BGJ398 (Selleckchem, Munich, Germany) or the FGF receptor 4 inhibitor BLU9931 (Cayman Chemicals).
SiRNA-induced knockdown of FGF9 was used applying an si-RNA-pool-FGF9 (siTOOLs Biotech GmbH, Planegg, Germany) and Lipofectamine RNAiMax transfection reagent (Life Technologies, Darmstadt, Germany) as described32. Overexpression of FGF9 protein was induced by transfection of a human FGF9 open reading frame (ORF) pcDNA3.1+/C-(K)DYK vector from GenScript (Piscataway, NJ, USA; CAT#: OHu27298) using LipofectAMINE plus method (Life Technologies, Darmstadt, Germany) as described33. An according empty control vector without the FGF9 ORF was used as control.
Human tissue samples
Human HCC tissues and corresponding non-tumorous liver tissues were obtained from patients that underwent partial hepatectomy. All experimental procedures were performed according to the guidelines of the non-profit state-controlled HTCR (Human Tissue and Cell Research) with informed patients’ consent30. Sampling and handling of patient material were carried out according to the ethical principles of the Declaration of Helsinki.
Immunofluorescence staining
Formalin-fixed primary human HSC and sections of formalin-fixed and paraffin-embedded HCC tissues were used for immunofluorescence staining applying anti-FGF9 antibodies (AF-273-NA, 1:25; R&D Systems, Minneapolis, MN, USA) and anti-alpha smooth muscle actin antibodies (ab32575, 1:500; Abcam, Cambridge, MA, USA) and standard protocols as described34. The following secondary antibodies were used: Alexa Fluor 488-conjugated donkey anti-goat IgG (A11055, 1:1,000; Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and Cy3-conjugated donkey anti-rabbit IgG (711-165-152, 1:1,000; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Nuclei were counterstained using DAPI. For control of specificity, antibody diluent was applied instead of the primary antibody and rabbit and goat IgG (Sigma, Munich, Germany) were used as isotype controls. These control stainings showed no background signal (Suppl. Fig. 3).
In silico analysis
The “SurvExpress-Biomarker validation for cancer gene expression” database (http://bioinformatica.mty.itesm.mx:8080/Biomatec/SurvivaX.jsp) was used for analysis of liver cancer datasets11.
Analysis of mRNA expression
Total RNA was extracted using the PureLink RNA Mini kit (Ambion, Thermo Fisher Scientific, Waltham, MA, USA) following the manual’s instructions. Reverse transcription was performed using the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). Quantitative real-time polymerase chain reaction was performed on a LightCycler 480 System (Roche Diagnostics, Mannheim, Germany) using specific sets of primers. Amplification of cDNA derived from β-actin or 18S rRNA was used for normalization of data.
Protein analysis
Protein extraction and Western blotting were performed as described31 using the following primary antibodies: goat anti-FGF9 (AF-273-NA, 1:2,000; R&D Systems), rabbit anti-phospho-ERK (#9101, 1:1,000; Cell Signaling Technology, Danvers, MA, USA), rabbit anti-phospho-JNK (#9251, 1:1000; Cell Signaling Technology) and mouse anti-actin (MAB1501, 1:10,000; Merck Millipore, Billerica, MA, USA). Donkey anti-goat (sc-2020; 1:1,000; Santa Cruz Biotechnology, Heidelberg, Germany), chicken anti-rabbit (sc-2955; 1:10,000; Santa Cruz Biotechnology) and horse anti-mouse (Santa Cruz Biotechnology, sc-2005, 1:3,000) were used as secondary antibodies.
Analysis of cell death, cell proliferation and cell cycle
For quantification of apoptosis, cells were simultaneously stained with FITC-conjugated Annexin V and propidium iodide using the Annexin V-FITC Detection Kit (PromoKine, PromoCell GmbH, Heidelberg, Germany) as described35. Proliferation of cells was determined using a colorimetric XTT assay (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s protocol31. Cell cycle fractions were analyzed by flow cytometry as described36.
Clonogenic assay
Clonogenic assays were used to analyze anchorage-dependent colony formation and proliferation of cancer cells. The assay is based on the capability of single cells to grow into colonies and was described previously32.
Analysis of cell migration
Migratory activity of HCC cells following treatment with FGF9 for 4 h was quantified using Boyden chamber assay as described31, with DMEM supplemented with 20% FCS attached to the bottom chamber.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism Software version 6.01 (GraphPad Software, San Diego, CA, USA). Data are shown as the mean ± standard error of the mean (SEM). Data sets were compared with analysis of unpaired Student’s t-test. A p-value <0.05 was considered statistically significant.
References
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 2, 16018 (2016).
Gerbes, A. et al. Gut roundtable meeting paper: selected recent advances in hepatocellular carcinoma. Gut. 67, 380–388 (2018).
Baglieri, J., Brenner, D. A. & Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int J Mol Sci. 20 (2019.
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Filliol, A. & Schwabe, R. F. Contributions of Fibroblasts, Extracellular Matrix, Stiffness, and Mechanosensing to Hepatocarcinogenesis. Semin. Liver Dis. 39, 315–333 (2019).
Ornitz, D. M. & Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 (2015).
Sandhu, D. S., Baichoo, E. & Roberts, L. R. Fibroblast growth factor signaling in liver carcinogenesis. Hepatology. 59, 1166–1173 (2014).
Raja, A, Park, I, Haq, F. & Ahn, S.-M. FGF19-FGFR4 Signaling in Hepatocellular Carcinoma. Cells., 8 (2019).
Harimoto, N. et al. The significance of fibroblast growth factor receptor 2 expression in differentiation of hepatocellular carcinoma. Oncology. 78, 361–368 (2010).
Paur, J. et al. Fibroblast growth factor receptor 3 isoforms: Novel therapeutic targets for hepatocellular carcinoma? Hepatology. 62, 1767–1778 (2015).
Aguirre-Gamboa, R. et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 8, e74250 (2013).
Antoine, M. et al. Expression and function of fibroblast growth factor (FGF) 9 in hepatic stellate cells and its role in toxic liver injury. Biochem. Biophys. Res. Commun. 361, 335–341 (2007).
Hendrix, N. D. et al. Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer Res. 66, 1354–1362 (2006).
Mulder, D. J. et al. FGF9-induced proliferative response to eosinophilic inflammation in oesophagitis. Gut. 58, 166–173 (2009).
Sun, C. et al. FGF9 from cancer-associated fibroblasts is a possible mediator of invasion and anti-apoptosis of gastric cancer cells. BMC Cancer. 15, 333 (2015).
Wang, S. et al. Expression and purification of an FGF9 fusion protein in E. coli, and the effects of the FGF9 subfamily on human hepatocellular carcinoma cell proliferation and migration. Appl. Microbiol. Biotechnol. 101, 7823–7835 (2017).
Hecht, D., Zimmerman, N., Bedford, M., Avivi, A. & Yayon, A. Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Factors. 12, 223–233 (1995).
Min, L., He, B. & Hui, L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin. Cancer Biol. 21, 10–20 (2011).
Chen, W. et al. Activation of c-Jun predicts a poor response to sorafenib in hepatocellular carcinoma: Preliminary Clinical Evidence. Sci. Rep. 6, 22976 (2016).
Haga, Y. et al. Overexpression of c-Jun contributes to sorafenib resistance in human hepatoma cell lines. PLoS One. 12, e0174153 (2017).
Hagiwara, S. et al. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br. J. Cancer. 106, 1997–2003 (2012).
Nguyen, T. V., Sleiman, M., Moriarty, T., Herrick, W. G. & Peyton, S. R. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials. 35, 5749–5759 (2014).
Chen, W. et al. Regorafenib reverses HGF-induced sorafenib resistance by inhibiting epithelial-mesenchymal transition in hepatocellular carcinoma. FEBS Open. Bio. 9, 335–347 (2019).
Huynh, H. et al. Infigratinib Mediates Vascular Normalization, Impairs Metastasis, and Improves Chemotherapy in Hepatocellular Carcinoma. Hepatology. 69, 943–958 (2019).
Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 38, 3–15 (2016).
Lu, X., Chen, H., Patterson, A. V., Smaill, J. B. & Ding, K. Fibroblast Growth Factor Receptor 4 (FGFR4) Selective Inhibitors as Hepatocellular Carcinoma Therapy: Advances and Prospects. J. Med. Chem. 62, 2905–2915 (2019).
Ghedini, G. C., Ronca, R., Presta, M. & Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert. Rev. Anticancer. Ther. 18, 861–872 (2018).
Yang, H., Fang, F., Chang, R. & Yang, L. MicroRNA-140-5p suppresses tumor growth and metastasis by targeting transforming growth factor β receptor 1 and fibroblast growth factor 9 in hepatocellular carcinoma. Hepatology. 58, 205–217 (2013).
Mühlbauer, M. et al. A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease. Gastroenterology. 125, 1085–1093 (2003).
Thasler, W. E. et al. Charitable State-Controlled Foundation Human Tissue and Cell Research: Ethic and Legal Aspects in the Supply of Surgically Removed Human Tissue For Research in the Academic and Commercial Sector in Germany. Cell Tissue Bank. 4, 49–56 (2003).
Amann, T. et al. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 100, 646–653 (2009).
Dietrich, P. et al. Wild type Kirsten rat sarcoma is a novel microRNA-622-regulated therapeutic target for hepatocellular carcinoma and contributes to sorafenib resistance. Gut. 67, 1328–1341 (2018).
Schuierer, M. M., Graf, E., Takemaru, K.-I., Dietmaier, W. & Bosserhoff, A.-K. Reduced expression of beta-catenin inhibitor Chibby in colon carcinoma cell lines. World J. Gastroenterol. 12, 1529–1535 (2006).
Schiffner, S. et al. Tg(Grm1) transgenic mice: a murine model that mimics spontaneous uveal melanoma in humans? Exp. Eye Res. 127, 59–68 (2014).
Wobser, H. et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 19, 996–1005 (2009).
Dietrich, P. et al. Combined effects of PLK1 and RAS in hepatocellular carcinoma reveal rigosertib as promising novel therapeutic “dual-hit” option. Oncotarget. 9, 3605–3618 (2018).
Acknowledgements
This work was supported by grants from the German Research Association (DFG) (HE2458/15 and HE2458/18) and by the Human Tissue and Cell Research (HTCR) Foundation, a non-profit foundation regulated by German civil law, which facilitates research with human tissues and cells through the provision of an ethical and legal framework for sample collection. The authors are indebted to Jennifer Czekalla and Rudolf Jung for excellent technical assistance.
Author information
Authors and Affiliations
Contributions
T.S. and K.F. performed the experiments. T.S. and C.H. analyzed the data. P.D., W.E.T. and A.B. provided material and methods. T.S. and C.H. designed the project. T.S. and C.H. wrote the manuscript; all authors edited and reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seitz, T., Freese, K., Dietrich, P. et al. Fibroblast Growth Factor 9 is expressed by activated hepatic stellate cells and promotes progression of hepatocellular carcinoma. Sci Rep 10, 4546 (2020). https://doi.org/10.1038/s41598-020-61510-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61510-4
- Springer Nature Limited
This article is cited by
-
HepG2 exosomes coated luteolin nanoparticles remodeling hepatic stellate cells and combination with sorafenib for the treatment of hepatocellular carcinoma
Cancer Nanotechnology (2024)
-
Fibroblast growth factors induce hepatic tumorigenesis post radiofrequency ablation
Scientific Reports (2023)
-
Src is essential for the endosomal delivery of the FGFR4 signaling complex in hepatocellular carcinoma
Journal of Translational Medicine (2021)