
Abstract
Vertebral body bone marrow aspirate (V-BMA), easily accessible simultaneously with the preparation of the site for pedicle screw insertion during spinal procedures, is becoming an increasingly used cell therapy approach in spinal surgery. However, the main drawbacks for V-BMA use are the lack of a standardized procedure and of a structural texture with the possibility of diffusion away from the implant site. The aim of this study was to evaluate, characterize and compare the biological characteristics of MSCs from clotted V-BMA and MSCs from whole and concentrate V-BMAs. MSCs from clotted V-BMA showed the highest cell viability and growth factors expression (TGF-β, VEGF-A, FGF2), the greatest colony forming unit (CFU) potency, cellular homogeneity, ability to differentiate towards the osteogenic (COL1AI, TNFRSF11B, BGLAP) and chondrogenic phenotype (SOX9) and the lowest ability to differentiate toward the adipogenic lineage (ADIPOQ) in comparison to all the other culture conditions. Additionally, results revealed that MSCs, differently isolated, expressed different level of HOX and TALE signatures and that PBX1 and MEIS3 were down-regulated in MSCs from clotted V-BMA in comparison to concentrated one. The study demonstrated for the first time that the cellular source inside the clotted V-BMA showed the best biological properties, representing an alternative and advanced cell therapy approach for patients undergoing spinal surgery.
Similar content being viewed by others
Introduction
Bone marrow aspiration is an easy, safe and inexpensive method that makes possible a direct transplantation of mesenchymal stem cells (MSCs), endothelial progenitor cells, hematopoietic stem cells, other progenitor cells, growth factors, e.g. bone morphogenetic proteins (BMPs), platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), and several interleukins, into the defect site1,2,3. Despite only a very small percentage (0.01–0.001%) of MSCs is found among the totality of mononuclear cells in bone marrow aspirate (BMA), the attendance of non-adherent osteogenic cells and the potential collaboration among BMA cell types in tissue repair suggest that the use of whole BMA, instead of expanded, concentrated and purified MSCs, is preferable for bone cell therapy4. Recently the use of iliac crest BMA is becoming increasingly trendy also in spinal surgery in order to overcome the problems linked to the harvest and use of iliac crest autograft and/or local autograft (spinous processes, laminae)5,6. However, several issues of the process need specific attention in order to optimize the efficacy and reduce potential side effects7. In addition, although bone marrow is commonly aspirated by the iliac crest, during spinal surgery its harvest leads to an increase in operative and rehabilitation time and to further morbidity in the donor site8. These complications are particularly critical in adolescent idiopathic scoliosis patients who have no pain before surgery and in osteoporotic patients that have a greatest risk of bleeding complications, involuntary penetration of the inner cortex of the ilium and nerve injury9,10,11. To overcome these limitations, several studies explored different BMA harvest sites, finding that the vertebral body, accessible by transpedicular aspiration during spinal procedures, is a robust site for aspiration12,13,14. Additionally we recently demonstrated that human MSCs from whole vertebral body BMA (V-BMA) (not concentrated and/or purified) were successfully isolated and showed best biological properties14. It was also shown that MSCs from whole V-BMA expressed definite level of HOX genes, genes able to control characteristic morphologies along regions of the vertebral column, and that HOXB8, a gene with key role in the regulation of adult MSCs, was up-regulated with greater efficiency in comparison to MSCs obtained from concentrated V-BMA14. Given the easy availability, safety and biological performances offered by V- BMA use for spinal fusion procedures, we have thought to evaluate clotted V-BMA. This choice started from a previous systematic literature review where the potential use of clotted BMA emerged as a novel cell therapy strategy for orthopedic procedures15. This review highlighted that, although there are very few studies about clotted BMA, and most of them are on cartilage regeneration (i.e. microfractures), BMA clot seems to be an effective, reliable, and easy strategy favoring tissue regeneration. A recent preclinical in vivo study carried out by Lim et al. demonstrated that clotted BMA from iliac crest alone can be considered as an alternative option to autograft for long bone healing16. Thus, theoretically, these data demonstrated that clotted BMA represent an attractive approach for bone regeneration. These promising results were probably due to the intricate cascade of chemical reactions during the process of coagulation that comprise platelets degranulation, which allows the release of several osteotropic cytokines and growth factors17,18. Additionally, it is necessary to emphasize that the fibrinolytic activity that takes place during the first days within a clot could offer a further source of angiogenic factors (fibrin split products)17. However, to date, no preclinical study assessed the in vitro biological properties and characteristics of MSCs derived from clotted BMA and evaluated the potential advantages of this alternative and little known approach to the current standard techniques.
By using human V-BMA, the main objective of this study was to evaluate, characterize and compare the biologics characteristics of MSCs from clotted V-BMA to MSCs from whole and concentrate V-BMAs. Using different qualitative and quantitative analyses, we want to prove if clotted V-BMA could be effectively used to enhance spinal fusion procedures offering significant advantages over whole and concentrate V-BMA, providing a novel, advanced and alternative strategy favoring tissue regeneration.
Results
Cell morphology and viability
In MSCs derived from whole and clotted V-BMAs red blood cells, platelets, and leucocytes were still observable within the first week of culture. However, after 10 days of culture most of the impure cells were discarded and at 14 days a homogenous population of spindle-shaped and plastic-adherent cells was observable in all culture conditions (Fig. 1).

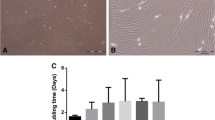
Cells morphology after 10 days of culture of MSCs from (a) whole BMA, (b) concentrated BMA and (c) clotted BMA. Magnification 4×.
Three, 7 and 14 days after cells culture, MSCs from whole and clotted V-BMA showed significantly higher viability in comparison to MSCs from concentrated V-BMA (***p < 0.0005) (Fig. 2). Significantly higher values of cells viability were found at all experimental time in MSCs from clotted V-BMA also in comparison to MSCs from whole V-BMA (***p < 0.0005) (Fig. 2).

Cell viability by Alamar blue dye performed on MSCs derived from whole BMA (dark bar), concentrated BMA (grey bar) and clotted BMA (light bar) after 3, 7 and 14 days of culture (***p < 0.0005) in basal medium. 3, 7 and 14 days: ***Whole and clotted BMAs vs. concentrated BMA; ***clotted BMA vs. whole BMA.
Expression of surface markers
In all culture conditions MSCs were almost completely negative for CD45 (leukocyte common antigen) and CD34 (gp105–120), haematopoietic surface markers, and for CD31 (platelet endothelial cell adhesion molecule) which indicated that they were not of endothelial origin. MSCs in all culture conditions expressed ecto-5′-nucleotidase (CD73), CD90 (thymocyte differentiation antigen-1, Thy-1) and matrix receptor CD105 (endoglin, SH2). However, the isolated cells highlighted a distinct phenotypic population (>90% homogeneous) only in clotted V-BMA. In fact, a percentage <90 were found in MSCs from whole V-BMA for CD73 and in MSCs from concentrated V-BMA for CD44, CD73 and CD90 (Table 1).
Colony-forming units assay
CFUs were significantly increased in MSCs from clotted V-BMA (25.83 ± 1.60) in comparison to MSCs from whole (19.33 ± 3.79: p < 0.05) and concentrated V-BMAs (13.33 ± 3.79; p < 0.0005) (Fig. 3).

Colony forming units (CFUs) count of MSCs derived from whole, concentrated and clotted BMAs after 10 days of culture in basal medium. The graphs indicate the number of positive colonies/well by toluidine blue staining. *Clotted BMA vs. whole BMA; ***Clotted BMA vs. concentrated BMA. *p<0.05; ***p < 0.0005. Representative images of CFUs observed by toluidine blue staining after10 days of culture, magnification 4×.
In vitro osteogenic, adipogenic and chondrogenic differentiation
In all culture conditions, LIVE/DEAD staining confirmed the viability of MSCs exposed to osteogenic and adipogenic medium (Fig. 4). Osteogenic induced-MSCs derived from whole, concentrated and clotted V-BMAs showed a change of cells shape and presence of mineralized matrix (red staining). However, in MSCs from whole and clotted V-BMAs a greater amount of calcium deposits were seen and this aspect was more marked in clotted V-BMA (Fig. 4). A decrease in cell density and a change of cells shape, that become more elongated and flat, were observed in all culture conditions when MSCs were exposed to adipogenic medium. The reduction in cell density was more evident in particular for MSCs from whole and clotted V-BMAs (Fig. 4). Concerning the chondrogenic differentiation of MSCs, in all culture conditions the presence of chondrocytes inside the lacunae separated by extracellular matrix was seen (Fig. 4). Although in all culture conditions MSCs differentiated towards the chondrogenic phenotype, MSCs from clotted V-BMA highlighted a greater amount of chondrocytes that were also organized in isogenous group (Fig. 4). Immunostaining showed the presence of Collagen Type II in all MSCs micromasses with more prominent staining in MSCs derived from whole and clotted V-BMAs, while the staining was less evident in MSCs derived from concentrated V-BMA (Fig. 4).

Representative images of in vitro osteogenic (Left: Alizarin Red S staining, magnification 4×; Right: LIVE/DEAD fluorescent staining, magnification 10×), adipogenic (Left: Oil Red O, magnification 40×; Right: LIVE/DEAD fluorescent staining, magnification 10×) and chondrogenic (Left: Alcian Blue/Nuclear Fast Red staining, magnification 80×; Right: Collagen Type II magnification 80×) differentiation of MSCs from (a) whole, (b) concentrated and (c) clotted BMAs. Black arrows: chodrocytes; Red arrows: extracellular matrix.
Gene expression analysis
TGF-β and VEGF-A gene expression showed significant higher values in MSCs from clotted V-BMA in comparison to MSCs from concentrated V-BMA (TGF-β: p < 0.05; VEGF: p < 0.05) (Fig. 5). Additionally, MSCs from clotted V-BMA revealed significantly higher values of TGF-β, VEGF-A and also FGF2 in comparison to MSCs from whole V-BMA (p < 0.05) (Fig. 5). Finally, significantly higher values were also seen for VEGF-A and FGF2 in MSCs from concentrated V-BMA in comparison to MSCs from whole V-BMA (p < 0.05) (Fig. 5).

Gene expression measured by quantitative reverse transcriptase polymerase chain reaction of TGF-β, VEGF and EGF in MSCs from whole, concentrated and clotted BMAs. TGF-β, VEGF-A: *MSCs from clotted V-BMA vs. MSCs from concentrated V-BMA and MSCs from whole V-BMA; EGF2: *MSCs from clotted V-BMA vs. MSCs from whole V-BMA; VEGF-A, FGF2: *MSCs from concentrated V-BMA vs. MSCs from whole V-BMA. *p < 0.05.
After 21 days of osteogenic induction, RT-PCR demonstrated that MSCs from clotted V-BMA showed significantly higher expression of COL1A1, TNFRSF11B and BGLAP in comparison to MSCs from concentrated (COL1A1: p < 0.0005; TNFRSF11B: p < 0.0005; BGLAP: p < 0.05) and whole V-BMAs (COL1A1: p < 0.005; TNFRSF11B: p < 0.005; BGLAP: p < 0.05) (Fig. 6a). Likewise, significantly higher values of RUNX2 and ALP were seen for clotted V-BMA in comparison to concentrate ones (RUNX2 and ALP: p < 0.0005) (Fig. 6b). Finally, COL1A1 (p < 0.05) also had significantly higher values in whole V-BMA when compared to concentrated V-BMA (Fig. 6a).

Gene expression measured by quantitative reverse transcriptase polymerase chain reaction of osteogenic markers comparing the differentiation potential of MSCs from whole BMA, concentrated BMA, clotted BMA after 21 days of culture. (a) COL1A1: ***clotted BMA vs. concentrated BMA; **clotted BMA vs. whole BMA; *whole BMA vs. concentrated BMA. TNFRSF11B: ***clotted BMA vs. concentrated BMA; **clotted BMA vs. whole BMA. BGLAP: *clotted BMA vs. concentrated BMA; *clotted BMA vs. whole BMA. RUNX2: ***clotted BMA vs. concentrated BMA. ALP: ***clotted BMA vs. concentrated BMA. *p < 0.05; **p < 0.005; ***p < 0.0005.
Adipogenic differentiation of MSCs from whole and clotted V-BMAs was significantly reduced (p < 0.05) in comparison to MSCs from concentrated V-BMA, as shown by ADIPOQ expression (Fig. 7). Additionally, MSCs from whole V-BMA had a significant lower value of ADIPOQ (p < 0.05) in comparison to MSCs from clotted B V-MA (Fig. 7).

Gene expression measured by quantitative reverse transcriptase polymerase chain reaction of adipogenic markers comparing the differentiation potential of MSCs from whole BMA, concentrated BMA, clotted BMA after 21 days of culture. ADIPOQ: *concentrated BMA vs. whole BMA and clotted BMA; *whole BMA vs. clotted BMA.*p < 0.05.
Chondrogenic differentiation capability revealed a significant higher value of SOX9 expression (p < 0.05) in clotted V-BMA in comparison to whole and concentrated V-BMAs (Fig. 8). Significant differences were also seen for MSCs derived from whole V-BMA that highlighted a higher value of SOX9 in comparison to MSCs from concentrated V-BMA (p < 0.05) (Fig. 8).

Gene expression measured by quantitative reverse transcriptase polymerase chain reaction of chondrogenic markers comparing the differentiation potential of MSCs from whole BMA, concentrated BMA, clotted BMA after 30 days of culture. SOX9: *clotted BMA vs. whole and concentrated BMA; *whole BMA vs. concentrated BMA. *p < 0.05.
After 21 days of osteogenic induction PBX1 and MEIS3 (p < 0.0005) showed significantly higher values in MSCs from concentrated V-BMA in comparison to MSCs from clotted V-BMA (Fig. 9). Additionally, significantly higher values (p < 0.05) were found also for HOXB8 in MSCs from whole V-BMA in comparison to MSCs isolated from concentrated and clotted V-BMAs (Fig. 9).

Gene expression measured by quantitative reverse transcriptase polymerase chain reaction of HOX and TALE signatures for MSCs from whole BMA, concentrated BMA, clotted BMA after 21 days of osteogenic induction. HOXB8: *whole BMA vs. concentrated BMA and clotted BMA; PBX1 and MEIS3: ***concentrated BMA vs. clotted BMA. *p < 0.05; ***p < 0.0005.
Discussion
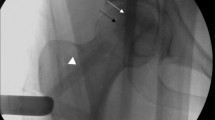
Spine fusion success depends on different factors, several linked to the patient (age, smoking, previous spine surgery, osteoporosis, osteoarthritis) and other linked to the surgery, such as use of instrumentation, surgical approach, use of bone graft/substitutes19,20. The increase of aging population and the need for multilevel spinal fusion surgery, request techniques capable to increase success rate of fusion and reduce pseudarthrosis rate21. A promising approach is the use of BMA from iliac crest6,21. Despite the clinical favorable outcome, its use is still limited due to several complications, such as increase in operative and rehabilitation time and to additional morbidity in the donor site22. To overcome these limitations V-BMA, accessible by transpedicular aspiration during spinal procedures, represent a promising cell source for fusion and its minimally invasive application may represent an efficacious biological cell source for spinal surgery outcomes improvement12,14. In this study we propose to evaluate and compare MSCs from clotted V-BMA as advanced and alternative cell therapy approach, with best biological properties and characteristics than MSCs derived from whole and concentrated V-BMA, for spinal fusion surgery. We hypothesized that despite V-BMA clot formation is usually considered a complication hampering its use during spinal surgery its biological and physical properties could make it a good candidate as autologous cell therapy in spinal surgery. The V-BMA clot with its complex environment of many cell types, growth factors and osteotropic cytokines is supposed to perform the necessary physiological functions to accelerate spinal fusion. Thanks to its structural texture V-BMA clot could be also placed where necessary at the implant site maximizing efficacy and minimizing clinical complications. However, main drawbacks in the use of clotted V-BMA could be the real surgical feasibility of the procedure in the clinical theatre and the need to have high amounts of V-BMA in presence of multilevel (long) spinal fusion. To confirm the surgical feasibility of this procedure we tested, directly in the clinical theatre, for the first time, the technical feasibility (Fig. 10) demonstrating its practicability. Concerning the need of high amounts of V-BMA in presence of multilevel (long) spinal fusion we assume that begin able to harvest V-BMA from each site for pedicle screw insertion there will be no problem to obtain the required amount of V-BMA. Finally, this easier procedure could totally avoid the burden of the bench to bedside translation of the use of this strategy also increasing the cost-effectiveness of the intervention.

Surgical feasibility of V-BMA clot procedure in the clinical theatre.
Our results showed that at all experimental time in vitro cells viability and CFU potency of MSCs from clotted V-BMA were increased compared to all the other culture conditions. These results may be due to the culture methods, i.e. to the clot by itself, that initially presents erythrocytes and platelets but also to the presence of a greater amount of growth factors, i.e. TGF-β, VEGF-A and FGF2, issued by the α-granules of platelets during the coagulation of bone marrow23,24,25. This clinically relevant source of growth factors present in greater amount in the clotted V-BMA may be more effective in the clinical scenario where increased vascularity and healing are mandatory. Additionally, TGF-β is reported to have anabolic and anti-inflammatory effects and it is known for its regulation of MSC proliferation and colonies formation26. VEGF, which is part of a subset of the PGDF family, is as a potent promoter of angiogenesis and has a powerful role in tissue healing27. Concerning FGF2, it was seen that it enhances MSCs proliferation and cooperates in maintaining MSCs three lineage differentiation potential during in vitro expansion28.
With reference to cellular homogeneity, it was seen that in V-BMA clot about 10% of cells were positive for CD31 and CD34 markers, thus suggesting that these cells may contain several populations with different phenotypic and biological properties. However, despite the currently adopted MSC definition relies on the International Society for Cellular Therapy criteria (adherence to plastic under standard culture conditions, expression or lack of expression specific surface markers, in-vitro three-lineage differentiation potential), functional and phenotypic differences may exist across tissue sources, culture conditions, and extent of ex-vivo expansion29,30. In fact, it is important to emphasize that although prevailing opinion states that MSC are CD34−, in vitro characterization mainly occurs following some passages of culture. This aspect not represents the in vivo or originally extracted cell phenotype. In fact, freshly extracted stem cells, from different anatomical sites, have shown to hold CD34+ cells31,32,33. CD34+ MSC are there immediately following withdrawal but quickly diminish after a short time in culture34,35. In addition, CD34 expression was linked with long-term proliferative capability and elevated colony forming efficiency34,36. CD34 has also been found on embryonic stem cell derived MSC, thus suggesting to be a marker of premature human MSC33,37. Also concerning CD31, greatly expressed on endothelial cells and to diverse degrees also on some hematopoietic cells, including monocytes, granulocytes and platelet38, it was reported the existence of large multinuclear CD31+ cells in the beginning of human bone marrow MSCs culture39. These observations imply that MSCs from V-BMA clot ‘heterogeneity’ is not an isolated case; however, this aspect must be further investigated to determine whether co-expression of specific MSC markers confers to V-BMA clot cells specific and peculiar properties. Additionally, differently, for the other culture condition, and in particular for concentrated V-BMA, a decrease in MSCs population, positive for CD44 and CD73, were also observed. This aspect is of critical importance because it can lead to different degrees of effectiveness after MSCs transplantation40. CD44 that was expressed by 93% of MSCs from whole V-BMA, 52% of MSCs from concentrated V-BMA and 100% of MSCs from clotted V-BMA regulates and operates in cell adhesion, migration, homing, proliferation and apoptosis and have also a role in stemness properties and maintenance41,42. CD73+ positive populations that in our study were expressed only by 64% of MSCs from concentrated V-BMA and by high percentage in the other culture conditions are significantly enriched in clonogenic cells, able to generate CFUs43. In our study, this data was confirmed also by the lower CFUs number that was evaluated in MSCs from concentrated V-BMA. Concerning CD90, that was expressed by high percentage in MSCs from whole and clotted V-BMAs and by a lower percentage in MSCs from concentrate V-BMA, it has a critical role in stem cell growth and differentiation potential44. In fact, even if all culture conditions produce mature cells of all mesenchymal lineages when differentiating into osteocytes, adipocytes and chondrocytes, RT-PCR revealed different regulation of genes marker for the three-lineage differentiation between the examined methods of isolation. A significant increase of genes marker of osteogenic differentiation, i.e. COL1AI, TNFRSF11B and BGLAP, were observed in MSCs from clotted V-BMA in comparison to all culture conditions. The COL1AI down-regulation in MSCs from whole and concentrate V-BMAs underlines a reduction in their final osteogenic potential45. In our study a down-regulation was also observed for TNFRSF11B (osteoprotegerin) expression, a member of tumor necrosis factor receptor super family, principally produced by osteoblasts and osteocytes, able to reduce osteoclasts activities46. Similarly, gene expression levels of osteocalcin (BGLAP), a protein released by adult osteoblasts, was down-regulated in MSCs from whole and concentrate V-BMA47. MSCs from clotted V-BMA showed also higher expression level of RUNX2, the key inducer of ALPL, COL1A1 and BGLAP genes, and ALPL in comparison to MSCs from concentrated V-BMA. These results additionally confirmed the data obtained by Alizarin Red S staining that showed a greater presence of calcium phosphate deposits in MSCs from clotted V-BMA, with a more intense red-orange staining of mineralized bone matrix. The higher incidence of calcium phosphate deposits and the higher expression of osteogenic markers in MSCs from clotted V-BMA proved the greater bone formation ability of these cells. Similarly to the osteogenic differentiation also the chondrogenic differentiation ability was higher in MSCs from clotted V-BMA, as confirmed by Collagen Type II immunostaining and SOX9 gene expression, a member of the family of Sox (Sry-type HMG box) genes. SOX9, defined as a “master regulator” of the chondrocyte phenotype, is critical for the chondrogenic differentiation ability and for the production of extracellular matrix48. Unlike osteogenic and chondrogenic differentiation ability, adipogenic one showed few lipid vacuoles (Oil Red staining) and lower ADIPOQ gene expression in MSCs from clotted V-BMA. These results indicated that MSCs from clotted V-BMA were able to differentiate onto the adipogenic lineage when exposed to definite inducing factors but with a lower ability in comparison with all the other culture condition. This greater ability to differentiate towards the osteogenic and chondrogenic phenotype associated to the lower ability to differentiate toward the adipogenic lineage, provide to the MSCs isolated from clotted V-BMA the best biological properties and characteristics.
We recently demonstrated that vertebral MSCs, isolated through different methods, expressed diverse level of HOX and TALE signatures, genes that control specific vertebral elements morphology. Thus, also in this study we analyzed whether MSCs from whole, concentrated and clotted vertebral V-BMAs might, after osteogenic induction, modulate the expression of these genes14,49. Our results revealed that MSCs from whole, concentrated and clotted V-BMAs express different level of HOX and TALE genes with significant differences for PBX1 and MEIS3 between MSCs from concentrated and clotted V-BMAs. This last showed reduced level of these genes. Although there are no reports regarding the screening of TALE groups that are involved in the osteogenesis of vertebral MSCs, it was reported that PBX1 is down-regulated during osteoblast maturation since its presence in osteoprogenitor cells and its reduction in mature osteoblasts50. These data were also confirmed by the fact that PBX1 depletion by shRNA increases osteoblast differentiation50. Additionally, in accordance with our results it was also reported that increased PBX1 level in MSCs reduced the expression of early (RUNX2 and ALPL) and late osteoblast genes (BGLAP), whereas decreased levels PBX1 lead to increased osteoblast gene expression50. Concerning MEIS3 expression no specific data were reported concerning its role during MSCs osteogenic differentiation ability. However, Roson-Burgo at al. supposed MEIS3 as interactor of Kruppel-like factor 4 (KLF4) in human bone marrow MSCs51. KLF4 showed a possible control over mesenchymal differentiation and it was seen that KLF4 DNA-binding protein presents a critical role in the regulation of MSC transcriptional activity, thus acts in maintaining cells in an undifferentiated state52,53. Thus, as for KLF4, we hypothesized also for MEIS3 a down-regulation when vertebral MSCs were submitted to differentiation pressure. Finally, concerning the highest expression level of HOXB8 in MSCs from whole V-BMA this data confirmed our previous study where this gene, involved in the hematopoietic stem and early progenitor cells expansion54, was strongly transcribed in whole V-BMA14.
Until now, the real biological properties and characteristics of clotted V-BMA are not yet evaluated. Our study demonstrated for the first time that the cellular source inside the vertebral clotted BMA showed the best biological properties, representing an alternative strategy for patients undergoing spinal surgery. The use of vertebral clotted BMA not only eliminates the need to concentrated and/or purified BMA but also gives an attractive tool for cellular therapy able to provide higher biomechanical stability to the graft site in comparison to the existing approaches (i.e. whole and concentrated V-BMAs). Vertebral BMA can be harvested at the same time of the preparation of the site for pedicle screw insertion and reintroduced after clotting (15–30 minutes) in the fusion site alone and/or in combination with a graft, without extra surgical time or involvement of other donor site. We hypothesize that the easier clinical access to vertebral BMA in the patient, the need of cellular in vitro expansion/purification and the ability to offer a greater stability to the graft site will totally avoid the burden of the ‘bench to bedside translation’ of the use of this procedure also decreasing the cost-effectiveness of the intervention.
Materials and Methods
Bone marrow aspirate collection and processing
The study was approved by the Ethics Committee of IRCCS Istituto Ortopedico Rizzoli (Protocol n. 9499_v-MSC) and was carried out in accordance with relevant guidelines and regulations (IRCCS Istituto Ortopedico Rizzoli has kept the ISO 9001 Quality certification since 2008, with special reference to the Research area). Written informed consent was obtained from all subjects involved in the study. Exclusion criteria were human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), diabetes, pregnancy, bone diseases, drugs active on bone metabolism, primary bone tumors, metastases, minors and/or patients incapable of giving consent personally. Four milliliters of human vertebral aspirate was harvested into a 10 ml syringe from each vertebral pedicle, during the preparation of the pilot hole for pedicle screw fixation, of six female patients (mean age: 57.33± 10.19; body mass index and bone mineral index matched) undergoing spinal surgical procedures involving posterolateral arthrodesis. This process was repeated until a total volume of 16 ml bone marrow was obtained from each patient. Vertebral aspirate was divided in 3 equal parts of 5.33 ml each. Two parts have been placed into test tubes containing heparin as an anticoagulant for MSCs isolation from 1) whole V-BMA and 2) concentrated V-BMA. The other part has been placed in test tube without any anticoagulant for MSCs isolation from 3) clotted V-BMA. Subsequently, V-BMAs were transferred to the laboratory and they were processed as described below (Fig. 11).

Study experimental set-up. Human BMA harvest from vertebrae divided in 3 equal parts: two parts placed into test tubes containing heparin as anticoagulant for MSCs isolation from whole and concentrated BMAs, one part placed in test tube without any anticoagulant for MSCs isolation from clotted BMA.
MSCs from whole V-BMA
As previously described14, whole V-BMA was immediately put in the culture flask and cultured with Dulbecco’s Modified Eagles Medium (DMEM, Sigma–Aldrich, St. Louis, MO), containing 10% Fetal Bovine Serum (FBS, Lonza), 100 U/ml penicillin, 100 mg/ml streptomycin (Gibco, Life Technologies, Carlsbad, CA) and 5 mg/ml plasmocin (Invivogen, San Diego, CA).
MSCs from concentrated V-BMA
V-BMA mononuclear cell fraction was harvested for MSC isolation using conventional density gradient centrifugation (Histopaque, Sigma, Aldrich, MO, USA), as reported in our previous works14. Briefly, the entire aspirate volume was mixed 1:1 with phosphate buffer solution (PBS). The diluted bone marrow was subsequently layered on top of Histopaque and centrifuged at 2.200 rpm for 30 minutes. The mononuclear cells in the interface were extracted and washed twice with PBS by centrifuging at 1.800 rpm for 10 minutes. The cells pellet was then suspended and cultured in DMEM containing 10% FBS (Sigma–Aldrich, St. Louis, MO), 100 U/ml penicillin, 100 mg/ml streptomycin (Gibco, Life Technologies, Carlsbad, CA) and 5 mg/ml plasmocin (Invivogen, San Diego, CA).
MSCs from clotted V-BMA
V-BMA in the test tube without anticoagulant clotted in about 15–30 minutes and subsequently it was entirely, like an explants, put in a culture flask with Dulbecco’s Modified Eagles Medium (DMEM, Sigma–Aldrich, St. Louis, MO), containing 10% Fetal Bovine Serum (FBS, Lonza), 100 U/ml penicillin, 100 mg/ml streptomycin (Gibco, Life Technologies, Carlsbad, CA) and 5 mg/ml plasmocin (Invivogen, San Diego, CA).
All cultures (whole, concentrated and clotted) were incubated at 37 °C in 5% CO2 and under hypoxia (2% O2), in order to better mimic the microenvironment of the stem cells niche. Non-adherent cells were washed and culture medium changed after 72 h. Subsequently, culture medium was replaced twice a week, up to 85–90% of cell confluence.
Cell viability
After removing non-adherent cells (after 3 days), MSCs were observed twice a week up to 85–90% of cell confluence and the images acquired by a standard light microscope (Nikon Eclipse, Ti-U, Nikon Italia Srl, Italy) equipped with a digital camera at 4× and 10× magnification.
At 3, 7 and 14 days of cell culture, Alamar blue dye test (Serotec, Oxford, UK) was used, as previously described14, to evaluate cell viability. Briefly, the reagent is a dye, which incorporates an oxidation-reduction indicator that changes color in response to the chemical reduction of growth medium, resulting from cell growth. It was added to each culture well (1:10 v/v) for 4 h at 37 °C. After transferring the supernatants to 96-well plates, the absorbance was read spectrophotometrically at 570- and 600-nm wave lengths (for the fully oxidized and reduced forms of reagent) by MicroPlate reader (BioRad, CA, USA). The results, obtained as optical density (OD), were processed following the manufacturer’s instructions and expressed as reduction percentage.
Flow cytometry analyses
The characteristics of MSCs from whole, concentrated and clotted V-BMAs were investigated by characterizing cell-surface markers of isolated cells using flow cytometry analysis12. To test surface antigen expression, 0.5–1 × 105 MSCs for each antigen were washed with PBS, centrifuged at 260 g for 5 min, and incubated at 4 °C for 30 min in flow cytometry buffer (FCB, 2% FBS in PBS) adding 0.5 μg/ml of fluorescein isothiocyanate (FITC)-conjugated antibody against CD31, CD45, CD34, CD44, CD73, CD90 and CD105. FITC-conjugated nonspecific IgG was used as isotype control (BioLegend, San Diego, CA, USA). Cell fluorescence was evaluated with FACSCanto II instrument (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed by FACSDiva software (Becton Dickinson).
Colony-forming unit-fibroblasts (CFU-F) Assay
The number of colony-forming units (CFUs) was evaluated for MSCs from whole, concentrated and clotted V-BMAs. Two-hundred MSCs/cm2 were plated onto six-well plates and cultured for 10 days. At the endpoint, cells were fixed with 10% formalin for 20 min and stained with 0.1% toluidine blue in 1% paraformaldehyde (PFA) for 1 h12,14. The aggregates with ≥20 cells were visually scored as colonies and counted with a light microscope (Olympus BX51).
In vitro osteogenic, adipogenic and chondrogenic differentiation
Osteogenic, adipogenic, and chondrogenic differentiations were induced for MSCs from whole, concentrated and clotted V-BMAs. To induce osteogenic and adipogenic differentiation, MSCs were plated at a density of 7.0 × 103 cells per cm2 onto six-well plates and incubated in culture medium (DMEM with 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin and 5 mg/ml plasmocin) for 3 days. After 3 days by initial seeding, the medium was replaced with osteogenic and adipogenic medium and cells were cultured for 21 days. Osteogenic medium consisted of culture medium supplemented with dexamethasone 10−8 M (sol.1:100), ascorbic acid 50 μg/ml and β-glycerophospate 10 mM. Adipogenic medium consisted of culture medium supplemented with isobutylmethylxanthine 500 μM, indomethacin 100 μM, dexamethasone 1 × 10−6 and insulin 2.5 mg. Osteogenic or adipogenic induction medium was changed every three days for 21 days. LIVE/DEAD staining was used to assess cell viability after 21 days of culture in osteogenic and adipogenic medium. Briefly, LIVE/DEAD staining was performed by incubating the cells with a working solution of calcein AM (4 μm) and ethidium homodimer-1 (1 μm) for 45 minutes at 37 °C. Cells were washed with PBS and images acquired by a fluorescence microscopy (Nikon Eclipse, Ti-U, Nikon Italia Srl, Italy) equipped with a digital camera at 10× and 20× magnification.
To induce chondrogenesis, 2.5 × 105 cells per tube were pelleted (micromasses) at 1.500 rpm for 10 minutes. Pellet cultures were incubated for 3 days in culture medium before being switched to chondrogenic medium and grown for further 30 days. Chondrogenic medium consisted of culture medium supplemented with 5 μg/ml insulin, 5 μg/ml transferrin and 5 μg/ml selenous acid, 0.1 μM dexamethasone, 0.17 mM ascorbic acid–2-phosphate, 1 mM sodium pyruvate, 0.35 mM proline and 10 μg/ml transforming growth factor-β3 (TGF-β3). Chondrogenic induction medium was changed every three days for 30 days12,14.
Staining of differentiated cells
For evaluating differentiation potentials, after 21 days osteogenic cultures were fixed in 10% formaldehyde for 15 minutes and stained with 2% Alizarin Red S (Sigma-Aldrich) for 30 minutes at room temperature to detect calcium depositis. To detect lipid accumulation after 21 days, adipogenic cultures were fixed in 4% paraformaldehyde for 10 minutes at room temperature and stained with 1.8% Oil Red O (Sigma-Aldrich) for 15 minutes at room temperature. After 30 days, chondrogenic pellets (micromasses) were fixed in 10% formaldehyde for 30 min, washed in distilled water, and dehydrated in increasing ethanol series. Finally, they were clarified in xylene (VWR International, Milan, Italy) and embedded in paraffin (Thermo Fisher Scientific, Waltham, MA) blocks. Blocks were sectioned along a transversal plane and cut into 5 μm sections; 3 consecutive sections for each sample were stained with Alcian Blue/Nuclear Fast Red and 2 additional sections were immunostained for Collagen Type II. Briefly, after fixation, sections were rinsed in PBS and permeabilized by incubation in 0.3% hydrogen peroxide in PBS solution for 15 min. Slides were pre-treated for antigen unmasking with 0.2% Pronase (Sigma–Aldrich, US-MO) solution in PBS for 30 minutes at 37 °C. After washing, the slides were incubated at room temperature for 1 hours with Blocking Serum (Vectastain Universal-Quick-Kit, Vectors Laboratories, US-CA) to prevent nonspecific bindings, followed by incubation with specific rabbit polyclonal antibodies against Collagen Type II (NSJ Bioreagents, CA) overnight at 4 °C. After rinsing in PBS, the slides were incubated with an anti-rabbit HRP-conjugated secondary antibody (Bethyl Laboratories, US-TX) and Streptavidin/Peroxidase complex (Vectastain Universal-Quick-Kit). Finally, reactions were developed using Vector NovaRed Substrate Kit for Peroxidase (Vectors Laboratories, US-CA). Negative controls, by omitting the primary antibody, were included to check proper specificity and performance of the applied reagents.
Images were taken using a standard light microscope (Nikon Eclipse, Ti-U, Nikon Italia Srl, Italy) equipped with a digital camera at 4x, 10x, 20x and 80x of magnification.
RNA isolation and cDNA synthesis
Before three lineage differentiation and at 21 days of osteogenic and adipogenic differentiation, total RNA was isolated by PureLink RNA Mini Kit (Life Technologies, Carlsbad, CA, USA) according to manufacturer’s instructions. For condrogenic differentiation, after 30 days, micromasses were homogenized and total RNA extraction was performed by phenol/chlorophorm method using TRIzol reagent (Invitrogen, Life Technologies, Carlsbad, CA) and chloroform 99% (Sigma-Aldrich); samples were vortexed thoroughly, incubated for 2 min at RT and centrifuged at 12,000 g for 20 min at 4 °C. The resulting aqueous phase was then purified with PureLink RNA mini kit columns (Life Technologies) following manufacturer’s direction. For all samples total RNA was eluted with RNase-free water and quantified by NanoDrop 2000 (Thermo Scientific, Waltham, MA, USA). Purified RNA was reverse transcribed with Superscript VILO cDNA Synthesis kit (Invitrogen, Life Technologies, Carlsbad, CA, USA), reaction was carried out in a 2720 Thermal Cycler (Applied Biosystems, Foster City, CA) at 25 °C for 10 min, 42 °C for 60 min and 85 °C for 5 min. cDNA was diluted for gene expression analysis a concentration of 5 ng/ul with nuclease-free H2O14.
Quantitative real-time PCR (qPCR)
Quantitative real-time PCR (qPCR) analysis was performed in a LightCycler Instrument (Roche Diagnostics) using QuantiTect SYBR Green PCR kit (Qiagen, Hilden, Germany). The following protocol was pursued: initial denaturation at 94 °C for 15 min and 40 cycles of amplification (15 s at 94 °C, 20 s at the appropriate annealing temperature for each target and 20 s at 72 °C). The protocol was concluded by melting curve analysis to check for amplicon specificity. The threshold cycles (Ct) were determined for each sample and these values were used for comparative gene expression analysis. For TGF-β, VEGF-A and FGF2 expression it was employed the 2−ΔCt method while for all the others genes we employed 2−ΔΔCt method and using for all genes Actin beta (ACTB) expression as ref. 55. The 2−ΔΔCt results were expressed as fold variation relative to the value of undifferentiated cells control for each condition14.
Gene expression analysis
TGF-β, VEGF-A and FGF2 genes expression were evaluated before osteogenic, adipogenic and chondrogenic induction. ALPL, BGLAP, COL1A1, OPG and RUNX2 genes were evaluated for osteogenic induction, ADIPOQ and PPRG genes for adipogenic induction, and ACAN and SOX9 genes for chondrogenic induction. cDNA of cells in osteogenic differentiation was also analyzed for the expression profile of HOX and TALE gene. In particular, 11 genes were evaluated: HOXA1, HOXA6, HOXA7, HOXA11, HOXB8, HOXB9, HOXD9, HOXD10, MEIS2, MEIS3 and PBX1. All these genes were analyzed by specific QuantiTect Primer Assay (Qiagen, Hilden, Germany)14. All primers are listed in Table 2.
Statistical analysis
Statistical analysis was performed using R v.3.5.3 software [R: The R Project for Statistical Computing, R Foundation for Statistical Computing. (2008). https://www.r-project.org/ (accessed August 15, 2018)]; package ‘WRS2’56 was used to analyse data. Data are reported as mean ± standard deviation (SD) at a significant level of p < 0.05. Cell viability for each experimental time and CFUs data were analyzed with a one-way ANOVA followed by Scheffé post hoc test. After having verified normal distribution (Shapiro-Wilk test) and homogeneity of variance (Levene test), gene expression data were analyzed with robust one-way (FM) and a post hoc comparison test based on trimmed means was used to estimate the effects or the interactions of factors between groups (\(\hat{\psi }\) pairwise trimmed mean differences) through percentile bootstrap57,58.
Data availability
The data reported in this study are available from the corresponding author.
References
Bain, B. J. Bone marrow biopsy morbidity and mortality. Br. J. Haematol. 121(6), 949–51 (2003).
Kitchel, S. H., Wang, M. Y. & Lauryssen, C. L. Techniques for aspirating bone marrow for use in spinal surgery. Neurosurg. 57(4 Suppl), 286–9 (2005).
Imam, M. A. et al. A systematic review of the clinical applications and complications of bone marrow aspirate concentrate in management of bone defects and nonunions. Int. Orthop. 41(11), 2213–2220 (2017).
Hernigou, P., Poignard, A., Manicom, O., Mathieu, G. & Rouard, H. The use of percutaneous autologous bone marrow transplantation in nonunion and avascular necrosis of bone. J. Bone Jt. Surg. Br. 87(7), 896–902 (2005).
Hernigou, P. et al. Allografts supercharged with bone-marrow-derived mesenchymal stem cells possess equivalent osteogenic capacity to that of autograft: a study with long-term follow-ups of human biopsies. Int. Orthop. 41(1), 127–132 (2017).
Salamanna, F. et al. Mesenchymal Stem Cells for the Treatment of Spinal Arthrodesis: From Preclinical Research to Clinical Scenario. Stem Cell Int. 2017, 3537094 (2017).
Schottel, P. C. & Warner, S. J. Role of Bone Marrow Aspirate in Orthopedic Trauma. Orthop. Clin. North. Am. 48(3), 311–321 (2017).
Vaz, K. et al. Bone grafting options for lumbar spine surgery: a review examining clinical efficacy and complications. SAS J. 4(3), 75–86 (2010).
Pesenti, S. et al. Bone substitutes in adolescent idiopathic scoliosis surgery using sublaminar bands: is it useful? A case-control study. Int. Orthop. 41(10), 2083–2090 (2017).
Riley, R. S. et al. A pathologist’s perspective on bone marrow aspiration and biopsy: I. Performing a bone marrow examination. J. Clin. Lab. Anal. 18(2), 70–90 (2004).
Konda, B. et al. Safe and successful bone marrow biopsy: an anatomical and CT-based cadaver study. Am. J. Hematol. 89(10), 943–6 (2014).
Barbanti Brodano, G. et al. Mesenchymal stem cells derived from vertebrae (vMSCs) show best biological properties. Eur. Spine J. 22(Suppl 6), S979–84 (2013).
Badrinath, R., Bohl, D. D., Hustedt, J. W., Webb, M. L. & Grauer, J. N. Only prolonged time from abstraction found to affect viable nucleated cell concentrations in vertebral body bone marrow aspirate. Spine J. 14(6), 990–5 (2014).
Salamanna, F. et al. Biological Rationale for the Use of Vertebral Whole Bone Marrow in Spinal Surgery. Spine . 43(20), 1401–1410 (2018).
Salamanna, F. et al. Bone marrow aspirate clot: A technical complication or a smart approach for musculoskeletal tissue regeneration? J. Cell Physiol. 233(4), 2723–2732 (2018).
Lim, Z. X. H. et al. Autologous bone marrow clot as an alternative to autograft for bone defect healing. Bone Jt. Res. 8(3), 107–117 (2019).
Muschler, G. F. et al. Spine fusion using cell matrix composites enriched in bone marrow-derived cells. Clin. Orthop. Relat. Res. 407, 102–18 (2003).
Palta, S., Saroa, R. & Palta, A. Overview of the coagulation system. Indian. J. Anaesth. 58(5), 515–23 (2014).
Kim, Y. J., Bridwell, K. H., Lenke, L. G., Rhim, S. & Cheh, G. Pseudarthrosis in long adult spinal deformity instrumentation and fusion to the sacrum: prevalence and risk factor analysis of 144 cases. Spine 31(20), 2329–36 (2006).
Barbanti Bròdano, G. et al. Hydroxyapatite-Based Biomaterials Versus Autologous Bone Graft in Spinal Fusion: An In Vivo Animal Study. Spine 39(11), 661–668 (2014).
Schroeder, J., Kueper, J., Leon, K. & Liebergall, M. Stem cells for spine surgery. World J. Stem Cell 7(1), 186–94 (2015).
Tural-Kara, T., Özdemir, H., Fitöz, S., Çiftçi, E. & Yalçınkaya, F. Bone marrow aspiration complications: Iliopsoas abscess and sacroiliac osteomyelitis. Turk. J. Pediatr. 58(5), 562–565 (2016).
Horn, P. et al. Isolation of human mesenchymal stromal cells is more efficient by red blood cell lysis. Cytotherapy 10(7), 676–85 (2008).
Italiano, J. E. Jr et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 111(3), 1227–33 (2008).
Schallmoser, K. et al. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfus. 47(8), 1436–46 (2007).
Grafe, I., Alexander, S. & Peterson, J. R. TGF-beta family signaling in mesenchymal differentiation. Cold Spring Harb. Perspect. Biol. 10(5), A022202 (2018).
Ferrara, N. & Gerber, H. P. The role of vascular endothelial growth factor in angiogenesis. Acta Haematol. 106(4), 148–156 (2001).
Tsutsumi, A. et al. Retention of multilineage differentiation potential of mesenchymal cells during proliferation in response to FGF. Biochem. Biophys. Res. Commun. 288, 413–419 (2001).
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8(4), 315–7 (2006).
Reinisch, A. et al. Epigenetic and in vivo comparison of diverse MSC sources reveals an endochondral signature for human hematopoietic niche formation. Blood 125(2), 249–60 (2015).
Yoshimura, K. et al. Characterization of freshly isolated and cultured cells derived from the fatty and fluid portions of liposuction aspirates. J. Cell Physiol. 208(1), 64–76 (2006).
Ferraro, G. A. et al. Human adipose CD34+ CD90+ stem cells and collagen scaffold constructs grafted in vivo fabricate loose connective and adipose tissues. J. Cell Biochem. 114(5), 1039–49 (2013).
Sidney, L. E., Branch, M. J., Dunphy, S. E., Dua, H. S. & Hopkinson, A. Concise review: evidence for CD34 as a common marker for diverse progenitors. Stem Cell 32(6), 1380–9 (2014).
Quirici, N. et al. Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Exp. Hematol. 30(7), 783–91 (2002).
Kuçi, S. et al. CD271 antigen defines a subset of multipotent stromal cells with immunosuppressive and lymphohematopoietic engraftment-promoting properties. Haematologica 95(4), 651–9 (2010).
Simmons, P. J. & Torok-Storb, B. CD34 expression by stromal precursors in normal human adult bone marrow. Blood 78(11), 2848–53 (1991).
Kopher, R. A. et al. Human embryonic stem cell-derived CD34+ cells function as MSC progenitor cells. Bone 47(4), 718–28 (2010).
Woodfin, A., Voisin, M. B. & Nourshargh, S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 27(12), 2514–23 (2007).
Gheisari, Y. & Ahmadbeigi, N. Mesenchymal Stem Cells and Endothelial Cells: A Common Ancestor? Arch. Iran. Med. 19(8), 584–7 (2016).
De Girolamo, L. et al. Mesenchymal stem/stromal cells: a new “cells as drugs” paradigm. Efficacy and critical aspects in cell therapy. Curr. Pharm. Des. 19(13), 2459–73 (2013).
Schieker, M. et al. The use of four-colour immunofluorescence techniques to identify mesenchymal stem cells. J. Anat. 204(2), 133–9 (2004).
Zhu, H. et al. The role of the hyaluronan receptor CD44 in mesenchymal stem cell migration in the extracellular matrix. Stem Cell 24(4), 928–35 (2006).
Suto, E. G. et al. Prospectively isolated mesenchymal stem/stromal cells are enriched in the CD73+ population and exhibit efficacy after transplantation. Sci. Rep. 7(1), 4838 (2017).
Rege, T. A. & Hagood, J. S. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochim. Biophys. Acta 1763(10), 991–9 (2006).
Caplan, A. I. Review: mesenchymal stem cells: cell-based reconstructive therapy in orthopedics. Tissue Eng. 11(7-8), 1198–211 (2005).
Bielby, R., Jones, E. & McGonagle, D. The role of mesenchymal stem cells in maintenance and repair of bone. Injury 38(Suppl 1), 26–32 (2007).
Hanna, H., Mir, L. M. & Andre, F. M. In vitro osteoblastic differentiation of mesenchymal stem cells generates cell layers with distinct properties. Stem Cell Res. Ther. 9(1), 203 (2018).
Wright, E. M., Snopek, B. & Koopman, P. Seven new members of the Sox gene family expressed during mouse development. Nucleic Acids Res. 21(3), 744 (1993).
Rux, D. R. & Wellik, D. M. Hox genes in the adult skeleton: Novel functions beyond embryonic development. Dev. Dyn. 246(4), 310–317 (2017).
Gordon, J. A. et al. Pbx1 represses osteoblastogenesis by blocking Hoxa10-mediated recruitment of chromatin remodeling factors. Mol. Cell. Biol. 30(14), 3531–41 (2010).
Roson-Burgo, B., Sanchez-Guijo, F., Del Cañizo, C. & De Las Rivas, J. Transcriptomic portrait of human Mesenchymal Stromal/Stem Cells isolated from bone marrow and placenta. BMC Genomics 15, 910 (2014).
Piek, E. et al. Osteo-transcriptomics of human mesenchymal stem cells: accelerated gene expression and osteoblast differentiation induced by vitamin D reveals c-MYC as an enhancer of BMP2-induced osteogenesis. Bone 46(3), 613–27 (2010).
Saulnier, N. et al. Gene profiling of bone marrow- and adipose tissue-derived stromal cells: a key role of Kruppel-like factor 4 in cell fate regulation. Cytotherapy 13(3), 329–40 (2011).
Davidson, A. J. et al. cdx4 mutants fail to specify blood progenitors and can be rescued by multiple hox genes. Nat. 425(6955), 300–6 (2003).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gen e expression data using real-time quantitative PCR and the 2(-Delta Delta C(T))Method. Methods 25, 402–408 (2001).
Mair P., Wilcox R. Robust Statistical Methods Using WRS2 package. Behav Res Methods (2019).
Wilcox, R. R. & Tian, S. T. Measuring effect size: a robust heteroscedastic approach for two or more groups. J. Appl. Stat. 38(7), 1359–1368 (2011).
Wilcox, R. R. Introduction to Robust Estimation and Hypothesis Testing - 4th Edition, Academic Press, Elsevier Inc., Amsterdam, The Netherlands (2017).
Acknowledgements
This work was supported by grants from IRCCS Istituto Ortopedico Rizzoli (Ricerca Corrente), 5 × 1000 – year 2017 and by ‘Fondazione del Monte di Bologna e Ravenna’ for the project “Cellule mesenchimali staminali da corpo vertebrale come prospettiva biologica innovativa per la chirurgia vertebrale” (Code: PRWEB-6564). The authors thank the Dr. Annapaola Parrilli for the three-dimensional reconstruction of the vertebra shown in Figure 11.
Author information
Authors and Affiliations
Contributions
Study conception and design: F.S. and M.F.; statistical analysis: G.G.; interpretation of the data: F.S., M.F., B.B.G., A.G.; acquisition of data: F.S., D.C. and L.S.; drafted the manuscript: F.S. and M.F. All authors were involved in revising it critically for important intellectual content and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salamanna, F., Contartese, D., Giavaresi, G. et al. A Rationale for the Use of Clotted Vertebral Bone Marrow to Aid Tissue Regeneration Following Spinal Surgery. Sci Rep 10, 4115 (2020). https://doi.org/10.1038/s41598-020-60934-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60934-2
- Springer Nature Limited
This article is cited by
-
A cautionary tale about the use of colony-forming efficiency as a proxy for the survival of mesenchymal stem cells
Stem Cell Research & Therapy (2020)