
Abstract
We observed an increase in methicillin-susceptible Staphylococcus aureus (MSSA) infections at a Dutch neonatal intensive care unit. Weekly neonatal MSSA carriage surveillance and cross-sectional screenings of health care workers (HCWs) were available for outbreak tracing. Traditional clustering of MSSA isolates by spa typing and Multiple-Locus Variable number tandem repeat Analysis (MLVA) suggested that nosocomial transmission had contributed to the infections. We investigated whether whole-genome sequencing (WGS) of MSSA surveillance would provide additional evidence for transmission. MSSA isolates from neonatal infections, carriage surveillance, and HCWs were subjected to WGS and bioinformatic analysis for identification and localization of high-quality single nucleotide polymorphisms, and in-depth analysis of subsets of isolates. By measuring the genetic diversity in background surveillance, we defined transmission-level relatedness and identified isolates that had been unjustly assigned to clusters based on MLVA, while spa typing was concordant but of insufficient resolution. Detailing particular subsets of isolates provided evidence that HCWs were involved in multiple outbreaks, yet it alleviated concerns about one particular HCW. The improved resolution and accuracy of genomic outbreak analyses substantially altered the view on outbreaks, along with apposite measures. Therefore, inclusion of the circulating background population has the potential to overcome current issues in genomic outbreak inference.
Similar content being viewed by others


Introduction
In the third level neonatal intensive care unit (NICU) of our hospital, we observed a rise in methicillin-susceptible S. aureus (MSSA) infections in two consecutive years. Traditional MSSA typing methods brought the suspicion of health care workers (HCWs) being repeatedly involved in the outbreak transmission chains. However, as these traditional typing methods fell short in providing definitive answers, we set out to improve the resolution of our outbreak analysis by bacterial whole-genome sequencing (WGS).
Neonates have an immature immune system and are susceptible to opportunistic bacterial infections, especially in case of premature birth. Important bacteria involved in outbreaks at NICUs causing neonatal infections are Enterobacteriaceae and S. aureus. Although many studies have emphasized transmission of MRSA (methicillin-resistant S. aureus), its susceptible variant MSSA is, depending on local epidemiology, generally a more frequent cause of neonatal infections1,2,3. Neonatal bacterial infections are preceded by bacterial spread which involves both direct transmission by close interaction with visiting parents and HCWs and indirect transmission through medical equipment1,4,5,6. Proper hand hygiene, wearing disposable gloves and aprons, and cleaning and disinfection of surfaces and medical devices have shown to be effective measures to reduce the incidence of infections in NICUs by 33–60%7,8,9,10. To monitor the dynamics of potential pathogens in a NICU, carriage surveillance should be performed, as implemented at our NICU for Enterobacteriaceae and (both methicillin-susceptible and resistant) S. aureus. These advanced surveillance practices have become pivotal to the outbreak analysis performed in this study.
Outbreak analyses traditionally involve epidemiological analysis combined with microbial typing. Currently, traditional molecular methods for typing S. aureus include determination of the Multi-locus Sequence Type (MLST), the Variable Number Tandem Repeat (VNTR) signature of the staphylococcal protein A gene (spa typing), and the Multiple-Locus Variable number tandem repeat Analysis (MLVA)11. Although MLVA has the largest discriminatory power12, it covers only a fraction of the bacterial genetic material, and it is sensitive to influential sequencing errors.
With the advent of microbial whole-genome sequencing (WGS) information from the entire bacterial genome becomes available for typing and outbreak analyses13. To facilitate such analyses, numerous new outbreak detection methods and applications have become available14. Current examples of the application of WGS for outbreak analysis of S. aureus in neonates are mostly restricted to MRSA. Koser et al. first described WGS to confirm the epidemiological and phenotypic clustering of neonatal MRSA infections15. Next, a suspected persistent outbreak of MRSA carriage among neonates was confirmed by WGS, possibly maintained by ongoing carriage in a HCW involved16. In addition, WGS was demonstrated to enable differentiation between three contemporaneous MRSA outbreaks in a NICU17, and also pointed towards international transmission of MRSA from NICUs18. The sole example investigating MSSA, proposed that the application of WGS on neonatal infection isolates can also distinguish between patient-to-patient transmission and re-seeding from a separate source during an outbreak spanning multiple years19.
Despite these appealing examples, uncertainty in WGS based outbreak analysis lies within the interpretation of sequence similarity, and the establishment of a threshold for transmission-level relatedness between bacterial isolates. Here, we performed WGS on MSSA isolates from both carriage surveillance and infections on a NICU, to investigate whether this approach could better specify transmission-level relatedness and improve the reconstruction of a period of presumed transmission events. To our knowledge, we are the first to deduce outbreak clusters being deviations from the background level of genomic diversity in the bacterial population as observed in surveillance of that particular clinical setting.
Methods
Setting and surveillance policies
This study was performed at a third level 18-bed neonatal intensive care unit (NICU) of the Radboud university medical center, Nijmegen, the Netherlands during 2014 and 2015. As part of bacterial surveillance purposes, once weekly a throat and rectal swab were taken from each neonate present at the NICU and cultured for 2 days on BD Columbia CNA agar with 5% sheep blood Improved II (Becton Dickinson) at 36 °C. Colonies suspect for S. aureus that were positive in the slide coagulase test were tested for susceptibility to cefoxitin and mupirocin by agar diffusion. The first MSSA isolate cultured from a neonate was stored in glycerol 10% bouillon at −80 °C for typing purposes. Surveillance culture results were discussed at a monthly meeting with representatives of the NICU and the department of Infection Prevention and Control (IPC) to monitor colonization patterns.
The procedures described in this report were part of standard clinical care. Ethical approval was not required according to the Dutch CCMO guidelines. Human data were handled in accordance with the Declaration of Helsinki.
MSSA carriage and infection
MSSA carriage at a particular time point was defined as MSSA detected in at least one culture from a throat and rectal swab set. MSSA infection was defined as MSSA cultured from cerebrospinal fluid, blood, pleural fluid, wound swab, ocular swab or corneal scrape. Under certain conditions MSSA cultured from respiratory specimens was considered to represent possible or probable S. aureus infection (Supplementary Methods 1). MSSA isolates cultured from infection sites were stored. Clinical characteristics of neonatal MSSA infections were manually extracted from corresponding patient records.
Traditional outbreak investigations
During the study period, an outbreak was considered if closely related MSSA isolates were cultured from a throat and rectal swab set (carriage surveillance) or infection site in two or more patients within a month. Relatedness between isolates was assessed by agreement in traditional molecular S. aureus typing methods. First, the Variable Number Tandem Repeat signature of the spa gene (spa typing) was determined. Subsequently, if isolates’ spa type and temporal relationship made them suspected members of an outbreak, also Multiple-Locus Variable number tandem repeat Analysis (MLVA) was performed to determine the MLVA complex (MC) and underlying MLVA type (MT). If the time criterion was met, only isolates of equal MLVA types were considered to be sufficiently related to represent an outbreak. In retrospect, typing by whole-genome sequencing (WGS) was performed on a selection of the study isolates.
On occasion, when MSSA infections seemed to originate from carriage outbreaks, all involved health care workers (HCWs) were tested for MSSA carriage by a combined throat-nose swab and swabs from high frequency touched surfaces at the NICU were cultured. Those HCWs who carried outbreak spa/MLVA types were offered a five-day decolonization treatment and their corresponding isolates were subjected to WGS.
Genomic analyses
The second MSSA outbreak period in 2015, together with uncertainties about transmission and intervention, prompted for WGS. The isolates selected for WGS differed per year. To estimate the level of genetic variation in background surveillance, for 2014 we included any typed MSSA isolate from neonatal carriage or infection, plus the HCW carriage isolates of the outbreak MLVA types for WGS. Because for 2015 the MLVA types were not specified yet, it was decided for this year to forward all isolates with outbreak spa types for WGS. A detailed description of the applied WGS methods is provided in Supplementary Methods 2. In short, after DNA was extracted from cultured isolates by a cetrimonium bromide-based method, sequencing libraries were prepared for Illumina’s NextSeq 500 platform using the NexteraXT kit (Illumina, San Diego, CA, USA). Reads were filtered and subsequently assembled into contigs by SPAdes (version 3.10.1)20. Quality of the assembly was assessed by mapping the quality filtered reads back to the assembled contigs using bowtie2 (version 2.2.9)21. Pairwise single nucleotide polymorphism (SNP) detection across all shared sequence regions between any pair of isolates was performed using kSNP (version 3.021)22, followed by a custom script to obtain core high quality SNPs (hqSNPs) based on a specified sequence consistency and coverage. A phylogeny based on core hqSNPs was calculated using PhylML23 and visualized using iTOL (version 4.1.1)24. Minimum spanning trees (MSTs) were generated using Python library networkx 1.11 and visualized using Cytoscape (version 3.5.1)25. Because the Illumina NextSeq 500 platform yields paired-end reads of 150 bp, the draft genomes were unfit to reliably span and verify the repeat-based typings that were previously assigned to the study isolates. The pairwise SNP analysis allowed us to valorize information from the “core” genomic regions shared by any subset of isolates from the cohort. In a subset of closely related isolates, the core genome is likely to be larger than in the total cohort, providing a substantially higher resolution for estimating relatedness. Because of this varying magnitude of the core genome, we measured relatedness between two isolates as the fraction of SNP differences across the shared genomic regions of a given subset that was present in the particular pair, denominated as SNP difference ratio. To account for possible technical SNP errors, introduced throughout the genomic analyses, we included several duplicates, including culture from frozen stocks as well as sequencing from libraries onwards.
Study period from genomics perspective
Using the available genomic information, outbreaks during the study period were reconstructed. Outbreaks were now defined as groups of isolates with pairwise core hqSNP difference ratios between neighbors in the MST of the entire cohort below a particular threshold. This threshold could be no less than our technological SNP error rate (in the duplicates), yet was primarily based on the distribution of the number of pairwise core hqSNPs between MST-neighbors in the total cohort, including background surveillance. Through this approach, isolates were assigned to an outbreak cluster if they were genetically more similar than was expected based on the measured variation in background surveillance. While the threshold was selected arbitrarily, its appropriateness for the current cohort was subsequently assessed using epidemiological data. Epidemiological linkage was expected between the isolate pairs below the SNP threshold, while pairs above it ought to be unrelated.
Results
Surveillance
No methicillin-resistant Staphylococcus aureus (MRSA) was detected throughout the study period, which is expected with < 1% carriage in the Dutch population26. From 510 neonates, in total 2296 surveillance swabs were collected, of which 313 (13.6%) were cultured positive for MSSA. Among the surveillance cultures 18.5% (214 out of 1154) of throat swabs were positive for MSSA, compared to only 8.7% (99 out of 1142) of rectal swabs. Nineteen sole rectal carriers were identified. The monthly number of throat swabs collected ranged from 37 to 61, while the proportion of positive MSSA cultures ranged from 2 to 41% (Supplementary Table 1). The majority of the 510 neonates involved in surveillance was screened only once before discharge from the NICU, and MSSA carriage was identified in 19.6% of them.
Traditional outbreak analysis
Based on identification of equal MLVA types within one month time distance, five MSSA outbreaks were identified during the study period. The outbreak-MLVA types were observed simultaneously in both carriage as well as infection (Fig. 1). The forty neonates who developed an infection with MSSA are further detailed in Supplementary Table 2. On two occasions screening of surfaces and HCWs was performed, in May 2014 and November 2015. None of the 42 cultured high frequency touched surface samples were positive for MSSA. Of the 377 screened HCWs 24% carried MSSA of whom 6 carried an outbreak MLVA type (Supplementary Fig. 1). The large variety of spa types identified in the entire cohort is displayed in Fig. 2. We identified 68 unique spa types among 210 MSSA carriage isolates (121 first isolates from neonates and 89 from HCWs). The 93 isolates that underwent subsequent MLVA (49 neonates, 44 HCWs) showed 9 different MLVA clones, and 42 MLVA types. While most 2014 MSSA isolates were subjected to WGS, in 2015 isolates only outbreak spa types were selected for.

Traditional MSSA surveillance and outbreak view during the study period. The bars in the top panel indicate the monthly percentage of throat swabs positive for MSSA, collected in weekly neonatal carriage surveillance at the NICU over a 2 year period. The dots in the bottom panel indicate the absolute number of neonatal infections, according to specimen positive for MSSA. Members of the five presumed outbreaks are indicated by a given color. Vertical arrows indicate the two occasions on which cross-sectional screening of MSSA carriage in HCWs was performed. Abbreviations: MSSA: methicillin-susceptible S. aureus; MT: MLVA type; HCW: healthcare worker.

Population diversity and selection for whole-genome sequencing. Pie charts indicating the percentage of typed MSSA isolates assigned to a particular spa type or MLVA type, categorized by study year (rows) and origin (columns). Raised sections indicate the isolates that were eligible for whole-genome sequencing, which includes all neonatal isolates in 2014. Abbreviations: MSSA: methicillin-susceptible S. aureus; MT: MLVA type; HCW: healthcare worker.
Genomic analysis
A total of 84 different MSSA isolates were sequenced, of which 78 from neonates (27 with MSSA infection) and 6 from HCWs. Six other eligible isolates were not stored and no longer available for WGS. The 84 selected WGS isolates represented 23 different spa types, and 58 of these isolates that had qualified for MVLA had been assigned to 5 different MLVA clones and 15 MLVA types. Quality thresholds for the genomic outbreak analyses were reached by 98% of the isolates. Sequenced isolates had a median sequence coverage of 88 × , a median cumulative contig length of 2.75 × 106 base pairs (bps) and a median paired-end read alignment to the contigs of 98%. Pairwise analysis of isolates identified 29,292 SNP-affected base positions present in the core genome of the total cohort, of which 8,575 (29%) were of high quality. The distance between the culture duplicates was 1 core hqSNP out of 8,575 hqSNP-affected base positions compared, and 0 core hqSNPs between the sequencing duplicates.
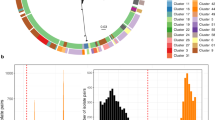
The concordance between spa typing, MLVA and genomic relatedness based on core hqSNPs in the total cohort is demonstrated in Fig. 3. Stratification of isolates according to spa type (inner lane) was in perfect agreement with genomic relatedness. However, intense admixture of MLVA types (outer lane) is observed in the branch corresponding to spa type t091.

Concordance between traditional typing methods and WGS based phylogeny. The circular phylogenetic tree is based on 8,575 core hqSNPs among the sequenced isolates. Each branch represents an MSSA isolate. The colored lanes indicate spa type (inner lane) and MLVA type (outer lane). RUMC labels represent identifiers of deposited sequencing data. The labels (A–D) anticipate representation of the isolate in the MST panels of Fig. 4. Abbreviations: MT: MLVA type; MC: MLVA clone; NA: not assigned; HCW: healthcare worker; MST: minimum spanning tree.
The extraction of subset specific core genomic regions significantly increased the resolution (the number of base positions screened for SNP differences) of our genomic outbreak analyses. For the 13 isolates of MLVA type 388 that were all regarded as members of a single outbreak by traditional analysis, a 6-fold increase in resolution by subset-analysis did not particularly change this perspective. Multiple HCWs were involved in this transmission chain (Fig. 4A). However, a genomic close-up of the subset with disagreement between stratification according to MLVA type and genomic relatedness, displays SNP difference ratios of up to 0.32% between neighbors central to the MST (Fig. 4B). Such large genomic differences make it highly unlikely that any of these MLVA types had been involved in long transmission chains. Moreover, it seems that the MLVA perspective has also induced erroneous separation of isolates that should have been attributed to a common outbreak. Examples are the extremely low SNP difference ratios between neighboring isolates of MT0371 and MT0368, or MT0007 and MT0366 in the MST. The re-assortment of isolates of these MLVA types by WGS clusters is supported by the common time periods in which they have been isolated. Figure 4C demonstrates how our subset-analysis alleviated concerns about a particular HCW. Contrary to what was concluded from MLVA, this HCW (isolate May_14 in Fig. 4C) involved in a first outbreak (represented by isolate Apr_14) had re-acquired a similar MSSA strain (isolate Oct_15), which was however unrelated to the second outbreak (represented by isolate Sep_15) with this MLVA type. Finally, knowledge of the genomic location of identified SNP differences between RUMC_0017 and RUMC_0071 (indicated by the arrow in Fig. 4D) confirmed that these were not introduced by a single event involving concentrated footprints, but by multiple sparse events probably accumulated over time (Supplementary Table 3).

Minimum spanning trees of core hqSNP subset analyses. Nodes represent MSSA isolates which are labeled by month of isolation and colored according to traditional MLVA type assigned. Nodes of a subset are connected to their closest neighbors based on relative distances expressed in SNP difference ratios. Edges are not scaled. The arrow in panel D indicates the core SNP differences whose genomic locations are further specified. Abbreviations: hqSNP: high quality single nucleotide polymorphism; MT: MLVA type; MC: MLVA clone; NA: not assigned; HCW: healthcare worker.
Study period from genomics perspective
The MSSA outbreaks were reconstructed using the available genomic information. Figure 5 demonstrates how, based on genomic distances in background surveillance, the threshold for transmission-level relatedness between neighbors in the MST was placed at a pairwise core hqSNP difference ratio of ≤0.03% (here equal to ≤3 hqSNP differences out of 8,575 SNP-affected base positions compared). Outbreaks were defined as groups of isolates that still met this criterion. To assess the appropriateness of this threshold for the current cohort we investigated the pairs of isolates around the cut-off for epidemiological linkage (Table 1).

Threshold for transmission-level genomic relatedness. Dots indicate pairwise core hqSNP difference ratios between neighbors in the minimum spanning tree of the entire WGS cohort, ordered by increasing value. The threshold for transmission-level genomic relatedness was placed at ≤0.03% (y-axis), separating the MSSA neighbors into two groups as indicated by the vertical dashed line. Abbreviations: hqSNP: high quality single nucleotide polymorphism; MST: minimum spanning tree; WGS: whole-genome sequencing; MSSA: methicillin-susceptible S. aureus.
Figure 6 displays the outbreaks during the study period reconstructed according to our genomics approach. Compared to the traditional outbreak analysis (MLVA-typing plus time relation), the genomic analysis demonstrated ten more separate outbreaks, yet with shorter transmission chains, at least five more infections attributed to an outbreak (f.e. those in November 2014), and three infections attributed to a different outbreak (f.e. those in August 2015). The number of outbreaks identified increased from 4 to 10 in 2014 and from 2 to 4 in 2015, alongside an additional 2 outbreaks spanning both years (indicated in yellow and orange). On the other hand, the average chain length of outbreaks decreased from 5.5 to 3.9 neonates in 2014 and from 6.0 to 3.3 neonates in 2015. Both alterations support a better containment of transmission in 2015 compared to 2014 than was presumed by traditional outbreak analysis.

Neonatal MSSA outbreaks from a genomics perspective. Although panels display the same events as in Fig. 1, members of outbreaks are now assigned according to our genomics approach. Colors are randomly assigned and not related to the previous figures. Abbreviations: MSSA: methicillin-susceptible S. aureus.
Discussion
Traditional outbreak tracing by spa typing and MLVA suggested that nosocomial transmission had contributed to neonatal MSSA infections in our patient cohort. Comparative genomics provided a large increase in resolution, especially in subset analyses, and yielded more accurate outbreak tracing than traditional typing methods. We based the genomic outbreak threshold on the background diversity in carriage surveillance, and acknowledged a substantially altered picture of outbreaks, which would have led to different outbreak measures.
One of the major issues that fueled this study, was the uncertainty about involvement of a particular HCW as a MSSA carrier in multiple outbreaks that included severe neonatal infections. Our genomic view alleviated these pressing concerns, and waived the working restrictions considered for this HCW. Furthermore, because transmission chains were shorter than indicated by traditional typing methods, IPC efforts shifted from vertical measures directed at putative persistent sources of transmission, towards horizontal measures like hand hygiene and supportive infrastructure.
Because we observed concurrent patterns of particular MSSA variants in carriage and infection, we believe that weekly neonatal surveillance and cross-sectional sampling of HCWs provided a good representation of the circulating MSSA population. However, one should keep in mind that such assessments are not designed to, and will not provide a complete capture of all transmission, and are likely to be affected by carriage density and chances of detection by screening technique used.
The comparison between 2014 and 2015 may have been affected by bias from distinct selections for WGS. If all 2015 isolates had been sequenced, potentially more outbreak members would have been revealed, yet unlikely up to the same number as detected in 2014. Moreover, in 2015 the most important risk of missing outbreak members due to MLVA misclassification, was excluded as WGS selection was based on outbreak spa type in 2015 (instead of MLVA type in 2014).
A relative limitation of our genomic method is that Illumina sequencing followed by de novo assembly does not allow for complete reconstruction of bacterial genomes, partly at the expense of repetitive elements. Although this issue can be overcome by long-read sequencing technologies such as Oxford Nanopore [10] or Pacific Biosciences single-molecule real-time sequencing [10, 30], it is unsure whether genetic information from such complex regions would improve outbreak tracing. Although the genomic analyses as performed in this initial study are computationally demanding, they served well for sorting unresolved transmission chains. Especially if the surveillance-based concept presented in this study would be proven applicable to other outbreak settings as well, it would be worthwhile to further automate the genomic analyses, including outbreak threshold selection and outbreak subset analysis.
The strength and novelty of the current study lies in the combination of surveillance practices and particular genomics methods. Although surveillance of MSSA carriage is no common practice, its initiation at our NICU quickly paid off when a rise in MSSA infections was observed and could partly be attributed to nosocomial transmission. In addition, the availability of isolates from the circulating MSSA carriage population provided us the unique opportunity to investigate whether knowledge of background genetic diversity could tackle the issue of determining thresholds for genomic outbreak calling. Two aspects of our genomic methods were designed to maximize the proportion of the genomes that could be utilized for comparison. First, because genomes were reconstructed de novo from the sequencing reads, identification of genetic regions was not restricted to those present on a reference genome. Second, in addition to the SNP analysis on the core genome of the entire cohort, the applied k-mer based pairwise SNP analysis allowed us to substantially expand the genomic regions for comparative subset analysis. Compared to recently applied core genome and whole genome MLST methods that rely on alleles27,28, our method is more dynamic as it includes any genomic region in common between a pair of isolates, including non-coding regions. This “zooming in” subset analyses appeared not only advantageous because of an absolute increase in base positions compared, but there was also more discriminatory signal in the accessory regions. We observed that for the same pair of isolates, an expansion of the core genome generally increased the percentage of hqSNPs detected, probably because genomic regions that are not universally essential to S. aureus survival allow for more variability. With this increased resolution the neighbors in the MST stayed put, yet their degree of relatedness became better specified. Finally, to account for the possibility that multiple SNP differences were in fact introduced by a single event like horizontal gene transfer, we designed a method to determine the distribution of SNP locations across multiple bacterial genomes without using a reference genome.
For S. aureus an MLVA type is determined by the pattern of 24 bp repeats within eight genetic regions (V09_01-V61_01-V61_02-V67_01-V21_01-V24_01-V63_01-V81_01). An explanation for the excessive admixture of four MLVA types compared to the WGS-based stratification in part of our MSSA population could be that these MLVA types were defined by highly similar repeat patterns (MT0368: 14-1-1-5-1-11-8-6; MT0007: 16-1-1-5-1-11-8-6; MT0366: 18-1-1-5-1-11-8-6; MT0371: 16-1-1-5-1-11-7-6). This suggests that either the MLVA typing method is susceptible to errors, or that MLVA repeat regions are simply not always representative for the remainder of the genome. The knowledge of epidemiological links between isolates of our study cohort made us confident that the genomic outbreak view was more plausible and therefore more reliable than MLVA type clustering. While in line with previous reports spa typing concurred with WGS-based phylogeny29,30, it lacked the required resolution as exemplified by the five separate outbreaks that were identified under spa type t091 using comparative genomics.
Because our specified genomic methods including particular SNP confidence requirements have not been applied before, the selected SNP threshold cannot be directly compared to that in other genomic outbreak studies. However, previous WGS outbreak studies on S. aureus infections among neonates accepted isolates to belong to an outbreak cluster if they differed dozens of core genomic sites, compared to our restriction at 3 hqSNPs. The admixture of isolates from multiple spa types31 did not occur in our study. In most studies, a relatively devoid group of isolates was arbitrarily considered as separate outbreak15,16,17,18,19. Sometimes WGS analysis resulted in a further separation, but never in re-assortment of outbreak isolates like we observed. While several studies referred to clonal mutation rates reported in unrelated cohorts to infer transmission-level relatedness, Coll et al. based their outbreak threshold of ≤50 core SNP differences in a monophyletic group of isolates on those observed within multiple isolates from single patients, and the maximum number of SNP differences between isolates within the largest epidemiologically trusted outbreak in their cohort32. Although accuracy and resolution may further benefit from knowledge on the local circulating carriage population, WGS benchmark initiatives could provide an appropriate platform for comparing the effects from different genomic outbreak methodologies on the outbreak assessment for a particular cohort33. While our threshold for genomic outbreak calling was supported by the epidemiological linkage between isolates, this threshold was put rather low, demanding tight genomic relatedness to meet clustering. These settings resulted in a genomic view of more and shorter outbreaks compared to traditional typing methods. Although we cannot exclude that the selected threshold was too strict, the re-assortment of outbreaks did not lead to indistinct singletons but rather separations into more homogeneous sub-clusters, still suggestive of a credible genomic outbreak reconstruction.
The genomic view revealed pairs of isolates that were collected months apart but displayed identical core genomes. Given the vast genetic diversity observed in the circulating MSSA population, we considered that these isolates were very likely to be closely related. Likewise, previous studies identified many unsuspected transmission clusters upon re-assessment of S. aureus cohorts using WGS31,32. Accordingly, in our genomic view time-linkage was not regarded as a prerequisite for outbreak clustering. This contributed to the increased number of outbreaks detected and the larger number of infections being attributed to nosocomial transmission.
Despite the non-exhaustive typing strategy applied in this study, MSSA isolates from 19 out of 40 neonatal MSSA infections were demonstrated likely to be members of transmission chains involving other neonates. Although prevention of nosocomial transmission of micro-organisms does not necessarily ban all neonatal infections, the attention for MSSA transmission seemed to have resulted in less spread and infections in the second year of the study. By our genomic view, infection prevention measures sorted an even larger reduction of outbreaks than presumed by traditional outbreak analysis. In addition, the outbreaks in 2015 appeared more modest, which could have waived the incentive to perform a second screening of MSSA carriage among HCWs.
Future studies are required to assess whether the current approach can again be successfully applied in similar as well as different outbreak settings involving other pathogens. Particularly because the rate at which mutations accumulate varies between bacterial species and lineages34. In line, within our cohort we noticed that the degree of SNP variation seemed to differ between lineages (Supplementary Fig. 2). It is unsure at which level such variation would violate the representativeness of a single outbreak threshold for an entire cohort. A related question is how extensive (in terms of density, but also proximity in place and time) background surveillance needs to be in order to identify an adequate threshold for transmission-level genomic relatedness.
Conclusions
In this study, WGS-based reconstruction of MSSA outbreaks was clearly superior to that based on traditional typing methods. As accurate insight in transmission is crucial for proper selection and evaluation of interventions for containment, we recommend WGS as the preferred method for assessment of bacterial outbreaks. Consensus needs to be reached on what would be needed for real-time genomic surveillance to become the leading method for identification of nosocomial outbreaks35,36.
Data availability
The dataset supporting the conclusions of this article is available in the Sequence Read Archive repository, BioProject accession number PRJNA523569, https://www.ncbi.nlm.nih.gov/sra/PRJNA523569.
Preprint
This article was submitted to an online preprint archive37.
References
Romano-Bertrand, S. et al. Staphylococcus aureus in a neonatal care center: methicillin-susceptible strains should be a main concern. Antimicrobial resistance Infect. Control. 3, 21 (2014).
Carey, A. J., Duchon, J., Della-Latta, P. & Saiman, L. The epidemiology of methicillin-susceptible and methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit, 2000–2007. J Perinatol 30, 135–139, https://doi.org/10.1038/jp.2009.119 (2010).
Gastmeier, P., Schwab, F., Behnke, M. & Geffers, C. Decreasing healthcare-associated infections (HAI) is an efficient method to decrease healthcare-associated Methicillin-resistant S.aureus (MRSA) infections Antimicrobial resistance data from the German national nosocomial surveillance system KISS. Antimicrobial resistance and infection control 1, 3-3, https://doi.org/10.1186/2047-2994-1-3 (2012).
Adams-Chapman, I. & Stoll, B. J. Prevention of nosocomial infections in the neonatal intensive care unit. Curr. Opin. Pediatr. 14, 157–164 (2002).
Sax, H. et al. Control of a cluster of community-associated, methicillin-resistant Staphylococcus aureus in neonatology. J. hospital Infect. 63, 93–100 (2006).
Achermann, Y. et al. Epidemiology of Methicillin-Susceptible Staphylococcus aureus in a Neonatology Ward. Infect. Control. hospital Epidemiol. 36, 1305–1312 (2015).
Won, S. P. et al. Handwashing program for the prevention of nosocomial infections in a neonatal intensive care unit. Infect. Control. hospital Epidemiol. 25, 742–746 (2004).
Pessoa-Silva, C. L. et al. Reduction of health care associated infection risk in neonates by successful hand hygiene promotion. Pediatrics 120, e382–390 (2007).
Svenningsen, N. W., Bekassy, A. N., Christensen, P. & Kamme, C. Nosocomial Klebsiella pneumoniae infection: clinical and hygienic measures in a neonatal intensive care unit. Scand. J. Infect. Dis. 16, 29–35 (1984).
Lenglet, A., Faniyan, O. & Hopman, J. A Nosocomial Outbreak of Clinical Sepsis in a Neonatal Care Unit (NCU) in Port-Au-Prince Haiti, July 2014–September 2015. PLoS Curr 10 (2018).
Harmsen, D. et al. Typing of methicillin-resistant Staphylococcus aureus in a university hospital setting by using novel software for spa repeat determination and database management. J. Clin. Microbiol. 41, 5442–5448 (2003).
Bosch, T. et al. Multiple-locus variable number tandem repeat analysis is superior to spa typing and sufficient to characterize MRSA for surveillance purposes. Future microbiology 10, 1155–1162 (2015).
Joseph, S. J. & Read, T. D. Bacterial population genomics and infectious disease diagnostics. Trends Biotechnol. 28, 611–618 (2010).
Quainoo, S. et al. Whole-Genome Sequencing of Bacterial Pathogens: the Future of Nosocomial Outbreak Analysis. Clin. microbiology Rev. 30, 1015–1063 (2017).
Koser, C. U. et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 366, 2267–2275 (2012).
Harris, S. R. et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect. Dis. 13, 130–136 (2013).
Madigan, T. et al. Whole-genome sequencing for methicillin-resistant Staphylococcus aureus (MRSA) outbreak investigation in a neonatal intensive care unit. Infection control and hospital epidemiology, 1–7 (2018).
Earls, M. R. et al. Intra-Hospital, Inter-Hospital and Intercontinental Spread of ST78 MRSA From Two Neonatal Intensive Care Unit Outbreaks Established Using Whole-Genome Sequencing. Front. Microbiol. 9, 1485 (2018).
Roisin, S. et al. Pan-genome multilocus sequence typing and outbreak-specific reference-based single nucleotide polymorphism analysis to resolve two concurrent Staphylococcus aureus outbreaks in neonatal services. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases (2016).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Gardner, S. N., Slezak, T. & Hall, B. G. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinforma. 31, 2877–2878 (2015).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–245 (2016).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Paget, J. et al. MRSA Carriage in Community Outpatients: A Cross-Sectional Prevalence Study in a High-Density Livestock Farming Area along the Dutch-German Border. PLoS One 10, e0139589 (2015).
Leopold, S. R., Goering, R. V., Witten, A., Harmsen, D. & Mellmann, A. Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J. Clin. Microbiol. 52, 2365–2370 (2014).
Maiden, M. C. et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736 (2013).
Park, K.-H. et al. Molecular epidemiology of Staphylococcus aureus bacteremia in a single large Minnesota medical center in 2015 as assessed using MLST, core genome MLST and spa typing. PLoS ONE 12, e0179003 (2017).
Hamed, M. et al. Whole genome sequence typing and microarray profiling of nasal and blood stream methicillin-resistant Staphylococcus aureus isolates: Clues to phylogeny and invasiveness. Infection, Genet. evolution: J. Mol. Epidemiol. Evolut. Genet. Infect. Dis. 36, 475–482 (2015).
Durand, G. et al. Routine Whole-Genome Sequencing for Outbreak Investigations of Staphylococcus aureus in a National Reference Center. Front. Microbiol. 9, 511 (2018).
Coll, F. et al. Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Sci Transl Med 9 (2017).
Ahrenfeldt, J. et al. Bacterial whole genome-based phylogeny: construction of a new benchmarking dataset and assessment of some existing methods. BMC Genomics 18, 19 (2017).
Didelot, X. & Maiden, M. C. Impact of recombination on bacterial evolution. Trends Microbiol. 18, 315–322 (2010).
Peacock, S. J., Parkhill, J. & Brown, N. M. Changing the paradigm for hospital outbreak detection by leading with genomic surveillance of nosocomial pathogens. Microbiology 164, 1213–1219 (2018).
Rossen, J. W. A., Friedrich, A. W., Moran-Gilad, J., Genomic, E. S. Gf & Molecular, D. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. microbiology infection: Off. Publ. Eur. Soc. Clin. Microbiology Infect. Dis. 24, 355–360 (2018).
Cremers, A. J. H et al. Surveillance-embedded genomic outbreak resolution of methicillin-susceptible Staphylococcus aureus in a neonatal intensive care unit. Preprint at, https://www.biorxiv.org/content/10.1101/584359v2 (2019).
Acknowledgements
This study was not supported by a particular funding body.
Author information
Authors and Affiliations
Contributions
A.C. and J.C. wrote the first version of the manuscript. A.C. and J.C. performed the data collection. J.Z. performed the laboratory assays. A.C. performed the epidemiological data analyses and J.C. performed the bioinformatics analyses. H.W. and J.H. initiated and supervised the study. All authors substantially contributed to the interpretation of data and reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cremers, A.J.H., Coolen, J.P.M., Bleeker-Rovers, C.P. et al. Surveillance-embedded genomic outbreak resolution of methicillin-susceptible Staphylococcus aureus in a neonatal intensive care unit. Sci Rep 10, 2619 (2020). https://doi.org/10.1038/s41598-020-59015-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59015-1
- Springer Nature Limited
This article is cited by
-
Comparison of fast Fourier transform infrared spectroscopy biotyping with whole genome sequencing-based genotyping in common nosocomial pathogens
Analytical and Bioanalytical Chemistry (2022)