
Abstract
Microhaplotypes are an emerging type of forensic genetic marker that are expected to support multiple forensic applications. Here, we developed a 124-plex panel for microhaplotype genotyping based on next-generation sequencing (NGS). The panel yielded intralocus and interlocus balanced sequencing data with a high percentage of effective reads. A full genotype was determined with as little as 0.1 ng of input DNA. Parallel mixture experiments and in-depth comparative analyses were performed with capillary-electrophoresis-based short tandem repeat (STR) and NGS-based microhaplotype genotyping, and demonstrated that microhaplotypes are far superior to STRs for mixture deconvolution. DNA from Han Chinese individuals (n = 256) was sequenced with the 124-plex panel. In total, 514 alleles were observed, and the forensic genetic parameters were calculated. A comparison of the forensic parameters for the 20 microhaplotypes with the top Ae values in the 124-plex panel and 20 commonly used forensic STRs showed that these microhaplotypes were as effective as STRs in identifying individuals. A linkage disequilibrium analysis showed that 106 of the 124 microhaplotypes were independently hereditary, and the combined match probability for these 106 microhaplotypes was 5.23 × 10−66. We conclude that this 124-plex microhaplotype panel is a powerful tool for forensic applications.
Similar content being viewed by others


Introduction
The microhaplotype is a powerful new type of forensic genetic marker1,2. It is the combination of two or more closely linked single-nucleotide polymorphisms (SNPs) within DNA segments of 200 base pairs (bp), and offers multiple forensic applications3,4,5,6,7. Short tandem repeat (STR) genotyping is currently the dominant technology in forensic DNA laboratories. Although it works well with single-sourced DNA samples, great challenges are encountered with DNA mixtures because stutters in the major donor DNA can be indistinguishable from alleles in the minor donor DNA3,8. Stutters are unavoidable during the replication of repetitive DNA, and they severely interfere with mixture deconvolution. SNPs are not repetitive sequences, but are typically biallelic, which restricts their utility in the analysis of mixtures. Microhaplotypes have the advantages of both STRs and SNPs because they are multiallelic and do not produce stutters during amplification. Therefore, microhaplotypes are perfect genetic markers for mixture deconvolution.
Although capillary electrophoresis (CE)-based genetic analyzers are widely used in forensic DNA laboratories, these machines are unsuitable for microhaplotype genotyping8. Several methods have been used for microhaplotype detection. TaqMan assays have been used to type each SNP that constitutes a microhaplotype8, followed by a PHASE software analysis to determine the cis/trans relationships between individual SNP alleles. Single-strand conformational polymorphisms9 and high-resolution melting curves4 have also been used for microhaplotype genotyping. These methods are simple and inexpensive, but they can pose problems when multiplexing different loci or dealing with mixed samples. MinION, a nanopore sequencing machine, has also been used for microhaplotype sequencing10, but the accuracy of sequencing for forensic applications must be improved.
Next-generation sequencing (NGS) is well accepted by the forensic community. Both the Illumina and Ion Torrent sequencers are high throughput, with appropriate read lengths for microhaplotypes11,12, and NGS can directly determine the phase between SNP alleles. Based on these characteristics, NGS is considered the optimal strategy for microhaplotype genotyping, and the development of NGS has made microhaplotypes a powerful new type of genetic marker for forensic analyses2. Zhu et al.13, Qu et al.14, Turchi et al.12, and Kidd et al.15 have studied microhaplotypes for forensic applications on the Miseq, HiSeq, Ion Personal Genome Machine (PGM), and Ion S5™ platforms, respectively. Attempts to develop NGS-based microhaplotype panels and microhaplotype population data have also been reported. In 2017, 89 microhaplotypes were sequenced with two primer pools in 73 Italian samples12, and this panel was later optimized to 87 loci by the same research group16. Another research team constructed a 74-plex microhaplotype assay and sequenced 278 samples from three different populations15. In the present study, we developed and evaluated a multiplex amplification system containing 124 microhaplotype loci. Parallel mixture experiments were performed with CE-based STR and NGS-based microhaplotype genotyping methods to compare their capacities for forensic mixture deconvolution. Microhaplotype allelic diversity and forensic estimations were determined for a Han Chinese population.
Results
The 124-plex microhaplotype panel
A total of 124 microhaplotype loci were multiplexed in a single primer pool. The number of SNPs contained at each locus ranged from 2 to 5, and 52 loci contained ≥ 3 SNPs (Supplementary Table S1). The molecular extent of the loci ranged from 13 to 210 nt, with an average of 108 nt. The primer sequences, primer concentrations, and amplicon sizes of the 124-plex panel are summarized in Table 1. The amplicons ranged from 63 to 298 bp, with an average size of 212 bp (Supplementary Fig. S1).
To evaluate the performance of this assay, we sequenced 10 reference samples. The numbers of total reads and reads representing microhaplotype alleles were calculated and are shown in Fig. 1. Around 100,000 total reads were obtained for each sample. The reads representing alleles accounted for over 90% of the total reads, and even 99% for some samples, indicating that the quality of the sequencing data was good.

Read counts and percentage of reads representing the alleles for 10 reference samples. Number of effective reads (those called as microhaplotype alleles) are shown in orange, and the total reads are shown in blue.
The allele coverage ratio (ACR) was used to evaluate the heterozygosity balance. The ACRs were calculated for the 10 reference samples by dividing the lower coverage allele by the higher coverage allele at each locus. All average ACRs were above 0.7, indicating that the heterozygosity balance of the 124-plex assay was good (Fig. 2). To examine the interlocus balance of this 124-plex panel, we calculated the average percentage depth of coverage (DoC) for each locus (Fig. 3). Each locus accounted for 0.2%–2% of the effective reads, 0.8% on average.

Average allele coverage ratio (ACR) for each locus. Horizontal black line, number of heterozygotes for each calculated ACR. Error bars represent standard deviations.

Average percentage (%) depth of coverage (DoC) for each locus. Error bars represent standard deviations.
To evaluate the sensitivity of the 124-plex assay, a dilution series of genomic DNA 9947 A (1.0, 0.5, 0.2, and 0.1 ng) was sequenced. All 124 microhaplotypes were successfully genotyped with a sequencing depth of ≥ 30 × when 1.0 ng, 0.5 ng, 0.2 ng, or 0.1 ng of input DNA was used (Supplementary Table S2 and Supplementary Figs. S2–S5), demonstrating the highly sensitive performance of the 124-plex assay.
Mixture study
To compare the effectiveness of microhaplotypes and STRs in the analysis of forensic mixtures, we prepared artificially mixed DNA samples with commercial genomic DNAs 9947 A and 2800 M, and performed parallel CE-based STR profiling and NGS-based microhaplotype genotyping experiments (Table 2 and Supplementary Figs. S5–S18). Representative data are summarized and compared in Fig. 4.

Representative STR profiles and representative microhaplotype genotyping histograms for the mixture experiments. Signal peaks for D16S539-12 and CSF1PO-11 in the 1:6 mixture are indicated as “Allele” and “Stutter”, respectively, for comparison. Numbers under each microhaplotype allele are the numeral allele names assigned to allow the microhaplotype data to be read conveniently.
Allele dropouts can severely interfere with a mixture analysis. Therefore, we examined the dropout alleles of the minor contributor (9947 A), and calculated the number of loci with fully called 9947 A alleles for each artificially mixed sample. In the STR profiles, no allele dropout was observed for the 1:1, 1:3, or 1:6 mixture (Table 2). Two alleles (D22S1045-11 and D2S1338-19) of the minor contributor dropped out in the analysis of the 1:9 mixture (Supplementary Fig. S12), and only 38% of the STR loci (8/21) reported full 9947 A alleles when the mixture ratio was 1:19 (Table 2, Supplementary Fig. S13). In contrast, 92% of the microhaplotypes (114/124) reported full 9947 A alleles for the 1:19 mixture (Table 2, Supplementary Fig. S18). No allele dropin was observed for the 1:3, 1:6, 1:9, or 1:19 mixture. Only two artefacts dropped in (mh02KK003-GTC and mh20kk059-AG) with low sequencing depths (40X and 30X, see Supplementary Fig. S19) when analyzing the 1:1 mixture. These data indicated that the NGS-based microhaplotypes were superior to the CE-based STRs in genotyping the alleles of the minor contributor.
We then investigated the effect of STR stutters on the analysis of these mixtures. When 9947 A and 2800 M were mixed at a 1:1 ratio, the alleles from both contributors were very similar in peak height or sequencing depth (Fig. 4, Supplementary Figs. S9 and S14). Neither STR nor microhaplotype was effective in mixture deconvolution. When the mixture ratio was 1:3, the peak heights of alleles from the minor contributor were significantly lower than those of the major contributor and significantly higher than the STR stutters (Fig. 4, Supplementary Figs. S10 and S15). Both STRs and microhaplotypes were effective in mixture deconvolution. However, at mixture ratios of 1:6, 1:9, and 1:19, the minor contributor STR alleles were indistinguishable from the stutters of the major contributor because their peak heights were similar (Fig. 4). For example, in the 1:6 mixture, D16S539-12 (an allele of the minor contributor 9947 A) and CSF1PO-11 (a stutter of the major contributor 2800 M) were both at the n-1 stutter position, with similar intensities. Their peak heights were 5%−10% of those of their possible parent alleles, which is typical for STR stutters. Incorrect allele/stutter interpretation can readily occur in such situations. However, with microhaplotype genotyping, the alleles from the major and minor contributors were easily distinguishable in the various mixture ratios based on their sequencing depths (Table 2, Supplementary Figs. S15–S18). Taken together, only 38.10% and 4.76% of the STR loci were effective in analyzing the 1:9 and 1:19 mixtures, respectively, whereas 99.19% and 91.94% of the microhaplotypes were effective in analyzing the same mixtures, respectively (Table 2). These data confirm that microhaplotypes are reliable genetic markers for the deconvolution of forensic mixtures.
Population data
A total of 256 Han Chinese individuals residing in Gansu Province were genotyped, and 514 alleles were observed (Table 3), with approximately four alleles per locus on average. Thirteen alleles were observed for locus mh01KK-117, which was the highest number in this dataset. Single alleles were observed for two loci, mh10KK-084 and mh17KK-076, indicating that there was no genetic diversity at these two loci in this Han Chinese population. Therefore, the forensic parameters were not calculated for these two loci. The forensic statistical parameters were calculated for the other 122 loci, and are summarized in Table 4.
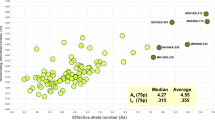
The PD values ranged from 0.0232 to 0.9623, with an average of 0.6799. The PD values for 90 loci were > 0.6, indicating that the individual identification capacity of the panel was high. The PEs for 66 loci were > 0.2, with 0.7855 (mh13KK-218) the highest PE value. Observed heterozygosity (Ho) was 0.0070–0.8952, and expected heterozygosity (He) was 0.0117–0.8656. The Ae values for 28 loci were > 3 (Fig. 5), and for another 23 loci, Ae was 2.5–3. Notably, the Ae values for mh13KK-218 and mh05KK-170 were even higher than 7.

Histogram of the Ae values for the 124 microhaplotypes.
To compare the individual identification capacities of the microhaplotypes and STRs, we summarized the PD and Ae values for the 20 microhaplotypes with the highest Ae values in the 124-plex panel and 20 commonly used forensic STRs (data under review in another manuscript) in Supplementary Table S3. The PD values for the microhaplotypes were 0.8691–0.9623 (0.9036 on average), which were very close to the PD range for STRs, 0.7794–0.9592 (0.9094 on average). The Ae values for the microhaplotypes and STRs were also similar. These data suggest that these 20 microhaplotypes are almost as effective as the commonly used forensic STRs for the identification of individuals.
To examine whether the microhaplotypes located on the same chromosome were linked to each other, we calculated LD. The p-values for pairwise linkage analyses are presented in Supplementary Table S4. Among the 124 microhaplotypes, 28 were linked in 10 pairs or groups (Supplementary Table S5) after correction for multiple testing (p < 0.0000065565). The locus with the highest Ae value within each linkage pair or group was used to calculate the combined forensic genetic parameters, whereas the other microhaplotypes within the linkage pairs or groups were not. Thus, based on 106 independent microhaplotypes, the combined match probability (CMP) and combined power of exclusion (CPE) were calculated to be 5.23 × 10−66 and (1–4.28 × 10−16), respectively.
Discussion
Since the concept of microhaplotypes was introduced, their unique advantages as novel genetic markers in the field of forensics have been gradually demonstrated. Various research groups have conducted extensive research into microhaplotypes and provided data for different populations. Hiroaki et al. studied 27 multiple-SNP haplotype blocks in a Japanese population4. Chen and coworkers presented a novel panel of 26 microhaplotypes, with relatively high Ae (>3.0) and small sequence lengths (<50 bp)17. Voskoboinik et al. reported a panel of 10 highly polymorphic haplotypes, each containing more than 10 SNPs10. However, fewer surveys have been conducted with highly multiplexed systems. In this study, we developed a single-tube 124-plex assay for forensic microhaplotypes for use with next-generation sequencing.
The sequencing data from the 124-plex panel showed good intralocus and interlocus balance (Figs. 2 and 3), with over 90% of the reads classified as effective (Fig. 1). Mixture deconvolution is one of the major forensic applications for which microhaplotypes are advantageous, and it is noteworthy that the excellent intralocus balance characteristic of this panel provides a reliable foundation for mixture analyses.
Microhaplotypes are expected to provide a better solution than STRs to forensic mixture analyses because they circumvent the inference by stutters3,18,19,20. However, the extent to which microhaplotypes can improve mixture deconvolution has been unclear. Therefore, we undertook parallel mixture experiments and in-depth comparative analyses of CE-based STR and NGS-based microhaplotype genotyping. Our results show that only 38.10% and 4.76% of STR loci effectively analyzed 1:9 and 1:19 mixtures, respectively, whereas 99.19% and 91.94% of the microhaplotypes effectively analyzed the same mixtures, respectively (Table 2). The microhaplotypes were also superior to STRs in the analysis of forensic mixture because they avoided not only inference by stutters, but also the dropout of minor contributor alleles. It should be noted that these results were obtained by single experiments at each mixture ratio and needed further verification.
Probabilistic genotyping software, including LRmix21, STRmix22, and EuroForMix23, have been developed. Using semicontinuous or fully continuous models, these programs provide optional solutions for mixed STR profile deconvolution. As noted by Bennett et al.24, similar probabilistic calculations could also be helpful in mixed microhaplotype data analyses.
To evaluate their capacities to identify individuals and family/clan relationships in a Han Chinese population, we sequenced the DNA of 256 unrelated individuals. A statistical analysis showed that the majority of microhaplotypes sequenced were highly polymorphic and informative in the Gansu Han population. The CMPs for most commercial forensic STR kits range from 10−17 to 10−26 25,26,27. In this study, the CMP for 106 microhaplotypes was 5.23 × 10−66, which is tens of orders of magnitude lower than those of STR multiplex systems. These data demonstrate that microhaplotypes are powerful genetic markers for the precise identification of individuals.
Some less polymorphic microhaplotypes in the Han Chinese population were kept in the 124-plex panel, including 2 markers which showed no genetic diversity. The ancestry inference capacity of these microhaplotypes has been extensively discussed by Kidd et al.8,15,28,29. Potential application of the 124-plex panel in ancestry inference awaits further studies.
Conclusions
We have developed an NGS-based 124-plex panel of microhaplotypes. Mixture experiments showed that the microhaplotypes are superior to STRs in forensic mixture analysis because they avoid not only interference by stutters, but also the dropout of minor contributor alleles. The DNA of 256 Chinese Han individuals was sequenced with the 124-plex panel. The estimated forensic parameters showed that the 20 microhaplotype loci with the highest Ae values in the 124-plex panel were as efficient as STRs in the identification of individuals, and that CMP for 106 microhaplotypes was 5.23 × 10−66. These data demonstrate that the 124-plex microhaplotype panel provides an additional tool for forensic applications.
Materials and Methods
DNA samples
Blood samples were collected from unrelated Han Chinese individuals. Written informed consent was given by the blood donors and this work was approved by the Ethical Review Board of the Institute of Forensic Science, Ministry of Public Security of China (Beijing, China). All methods were performed in accordance with the relevant guidelines and regulations. DNA was extracted with the MagAttract M48 DNA Manual Kit (Qiagen, Limburg, Germany), according to the manufacturer’s guidelines. The extracted DNA samples were quantified with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) on a Qubit fluorometer (Thermo Fisher Scientific).
The female genomic DNA standard 9947 A (Promega, Madison, WI, USA) was used in the sensitivity assays. Massive parallel sequencing was performed on a dilution series of genomic samples (1.0, 0.5, 0.2, or 0.1 ng). For the mixture experiments, standard genomic DNAs 9947 A and 2800 M (Promega) were mixed in ratios of 1:1, 1:3, 1:6, 1:9 and 1:19, to a total amount of 1.0 ng.
Multiplex amplification
Primers were designed for the 130 microhaplotype loci reported by Kidd et al.8 with the Primer Premier 5.0 software30. After repeated optimization of the primer sequences and the PCR conditions, 124 microhaplotypes were successfully multiplexed in a single reaction system (Table 1). The PCRs were performed in a total volume of 20 μL containing 20 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.6 mM MgCl2, 0.8 mg/ml bovine serum albumin, 0.2% (v/v) Tween 20, 3.2% (v/v) glycerol, 0.02% (w/v) NaN3, 200 mM each dNTP, 2 U of Taq DNA polymerase (Roche, Basel, Swiss), primer pairs (concentrations indicated in Table 1), and 1 ng of template DNA. The PCR conditions were 95 °C for 11 min, followed by 28 cycles of 30 s at 94 °C, 2 min at 60 °C, and 1 min at 72 °C, with a final elongation step at 60 °C for 60 min.
Library preparation and sequencing
The PCR products were purified with the QIAquick 96 PCR Purification Kit (Qiagen) and the TruSeq DNA PCR-Free HT Kit (Illumina, San Diego, CA, USA) and used for library preparation, according to the manufacturer’s guidelines. The libraries were sequenced on a MiSeq FGx platform (Illumina) using the Miseq Reagent Kit v2 (Illumina), with a read length of 250 bases.
Data analysis
FASTQ data were generated with the Miseq FGx Control Software 1.0.15.0 (Illumina). The MHTyper software31 was employed for microhaplotype allele calling, with the sequencing depth threshold set at 30 reads. The Hg19 human genome was used as the reference sequence. The allele frequencies and forensic statistical parameters (match probability, MP; power of discrimination, PD; power of exclusion, PE; typical paternity index, TPI) were calculated with Modified-PowerStat spreadsheet 1.232. Arlequin 3.533 was used to calculate the observed heterozygosity, expected heterozygosity, Hardy–Weinberg equilibrium, and linkage disequilibrium (LD). The effective number of alleles (Ae) was calculated with the formula described in a previous publication3.
CE-based STR genotyping
The GlobalFiler® Kit (Thermo Fisher Scientific) was used for CE-based STR genotyping, according to the manufacturer’s recommendations. An aliquot of PCR product (1 µL) was added to 10 µL of deionized formamide (Thermo Fisher Scientific) containing the internal size standards. All samples were separated on a 3500XL Genetic Analyzer (Thermo Fisher Scientific) using POP™-4 Polymer (Thermo Fisher Scientific) and a 36 cm capillary array (Thermo Fisher Scientific). The GeneMapper® ID-X software v4.0 (Thermo Fisher Scientific) was used for fragment sizing and allele calling.
References
Kidd, K. K. et al. Microhaplotype loci are a powerful new type of forensic marker. Forensic Sci. Int: Genet. Suppl. Ser. 4, e123–e124 (2013).
Kidd, K. K. et al. Current sequencing technology makes microhaplotypes a powerful new type of genetic marker for forensics. Forensic Sci. Int. Genet. 12, 215–224 (2014).
Kidd, K. K. & Speed, W. C. Criteria for selecting microhaplotypes: mixture detection and deconvolution. Investig. Genet. 6, 1 (2015).
Hiroaki, N. et al. Approaches for identifying multiple-SNP haplotype blocks for use in human identification. Leg. Med. 17, 415–420 (2015).
Kidd, K. K. et al. Genetic markers for massively parallel sequencing in forensics. Forensic Sci. Int: Genet. Suppl. Ser. 5, e677–e679 (2015).
Pu, Y. et al. Microhaplotype: Ability of personal identification and being ancestry informative marker. Forensic Sci. Int: Genet. Suppl. Ser. 6, e442–e444 (2017).
Zhu, J. et al. Genotyping polymorphic microhaplotype markers through the Illumina® MiSeq platform for forensics. Forensic Sci. Int. Genet. 39, 1–7 (2019).
Kidd, K. K. et al. Evaluating 130 microhaplotypes across a global set of 83 populations. Forensic Sci. Int. Genet. 29, 29–37 (2017).
Chen, P. et al. Microhaplotype identified and performed in genetic investigation using PCR-SSCP. Forensic Sci. Int. Genet. 28, e1–e7 (2017).
Voskoboinik, L., Motro, U. & Darvasi, A. Facilitating complex DNA mixture interpretation by sequencing highly polymorphic haplotypes. Forensic Sci. Int. Genet. 35, 136–140 (2018).
Wang, H. et al. NGS technology makes microhaplotype a potential forensic marker. Forensic Sci. Int: Genet. Suppl. Ser. 5, e233–e234 (2015).
Turchi, C., Pesaresi, M. & Tagliabracci, A. A microhaplotypes panel for forensic genetics using massive parallel sequencing. Forensic Sci. Int: Genet. Suppl. Ser. 6, e117–e118 (2017).
Zhu, J. et al. FLfinder: A novel software for the microhaplotype marker. Forensic Sci. Int: Genet. Suppl. Ser. 5, e622–e624 (2015).
Qu, S. et al. Estimate the heterozygote balance of microhaplotype marker with massively parallel sequencing. Forensic Sci. Int: Genet. Suppl. Ser. 6, e375–e376 (2017).
Oldoni, F. et al. Microhaplotypes for ancestry prediction. Forensic Sci. Int: Genet. Suppl. Ser. 6, e513–e515 (2017).
Turchi, C., Melchionda, F., Pesaresi, M. & Tagliabracci, A. Evaluation of a microhaplotypes panel for forensic genetics using massive parallel sequencing technology. Forensic Sci. Int. Genet. 41, 120–127 (2019).
Chen, P. et al. Evaluation of the Microhaplotypes panel for DNA mixture analyses. Forensic Sci. Int. Genet. 35, 149–155 (2018).
Zhu, J. et al. Genotyping microhaplotype markers through massively parallel sequencing. Forensic Sci. Int: Genet. Suppl. Ser. 6, e314–e316 (2017).
Oldoni, F., Kidd, K. K. & Podini, D. Microhaplotypes in forensic genetics. Forensic Sci. Int. Genet. 38, 54–69 (2019).
Chen, P. et al. A microhaplotypes panel for massively parallel sequencing analysis of DNA mixtures. Forensic Sci. Int. Genet. 40, 140–149 (2019).
Prieto, L. et al. Euroforgen-NoE collaborative exercise on LRmix to demonstrate standardization of the interpretation of complex DNA profiles. Forensic Sci. Int. Genet. 9, 47–54 (2014).
Bright, J. A. et al. Developmental validation of STRmix, expert software for the interpretation of forensic DNA profiles. Forensic Sci. Int. Genet. 23, 226–239 (2016).
Bleka, O., Storvik, G. & Gill, P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic Sci. Int. Genet. 21, 35–44 (2016).
Bennett, L. et al. Mixture deconvolution by massively parallel sequencing of microhaplotypes. Int. J. Leg. Med. 133, 719–729 (2019).
Adnan, A., Zhan, X., Kasim, K., Rakha, A. & Xin, X. Population data and phylogenetic structure of Han population from Jiangsu province of China on GlobalFiler STR loci. Int. J. Leg. Med. 132, 1301–1304 (2018).
Besecker, J., Peri, G., Davis, M., Zubizarreta, J. & Hampikian, G. Allele frequencies of 15 STR loci (Identifiler™ kit) in Basque-Americans. Leg. Med. 31, 17–19 (2018).
Srivastava, A. et al. Genetic data for PowerPlex 21™ autosomal and PowerPlex 23 Y-STR™ loci from population of the state of Uttar Pradesh, India. Int. J. Leg. Med. 133, 1381–1383 (2019).
Bulbul, O. et al. Ancestry inference of 96 population samples using microhaplotypes. Int. J. Leg. Med. 132, 703–711 (2018).
Kidd, K. K. et al. Selecting microhaplotypes optimized for different purposes. Electrophoresis 39, 2815–2823 (2018).
Qin, P., Niu, C., Ruan, M., Zeng, G. & Wang, X. A novel bifunctional europium complex as a potential fluorescent label for DNA detection. Analyst 135, 2144–2149 (2010).
Zhang, C. et al. MHTyper: a microhaplotype allele-calling pipeline for use with next generation sequencing data. Aust J Forensic Sci, published online, https://doi.org/ 10.1080/00450618.2019.1699956 (2019).
Guo, J. et al. Genetic polymorphism of 21 non-CODIS STR loci for Han population in Hunan Province, China. Forensic Sci. Int. Genet. 17, 81–82 (2015).
Excoffier, L. & Lischer, H. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Acknowledgements
The authors thank Mr. Yan-Dong Cao for technical assistance. This work was supported by grants from the National Natural Science Foundation of China (81971797), the National Key R&D Program of China (2017YFC0803503), the Ministry of Public Security of China (2017JSYJA05), and the Institute of Forensic Science, Ministry of Public Security of China (2018FGKFKT03).
Author information
Authors and Affiliations
Contributions
L.W., S.J.N. and J.Y. conceived and designed the experiments. J.B.P., M.R., Q.F.C. and H.W. performed most of the experiments. A.Q.J., C.Z. and K.L.K. analyzed the data. L.W. wrote the manuscript. All authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pang, JB., Rao, M., Chen, QF. et al. A 124-plex Microhaplotype Panel Based on Next-generation Sequencing Developed for Forensic Applications. Sci Rep 10, 1945 (2020). https://doi.org/10.1038/s41598-020-58980-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58980-x
- Springer Nature Limited
This article is cited by
-
Forensic genetic analysis of single-nucleotide polymorphisms and microhaplotypes in Koreans through next-generation sequencing using precision ID identity panel
Genes & Genomics (2023)
-
An overview of SNP-SNP microhaplotypes in the 26 populations of the 1000 Genomes Project
International Journal of Legal Medicine (2022)
-
Development and validation of a novel 133-plex forensic STR panel (52 STRs and 81 Y-STRs) using single-end 400 bp massive parallel sequencing
International Journal of Legal Medicine (2022)
-
Identification and sequencing of 59 highly polymorphic microhaplotypes for analysis of DNA mixtures
International Journal of Legal Medicine (2021)
-
Noninvasive prenatal paternity determination using microhaplotypes: a pilot study
BMC Medical Genomics (2020)