
Abstract
Advanced glycation end-products (AGEs) have been reported as results of increased oxidative stress. Consequently, the search for new antioxidant and anti-glycating agents is under intense investigation. Plant-derived procyanidins have previously demonstrated anti-glycation properties. Thus, this study aimed to isolate procyanidins from Annona crassiflora fruit peel, a species from the Brazilian Savanna, and investigate their antioxidant and anti-glycation effects. Free radical scavenging and quenching properties, formation of reactive oxygen species (ROS), AGEs, protein carbonyl and thiol groups, lipid peroxidation, crosslinked AGEs, as well as glycated catalase activity, were analyzed. In addition, in silico assessment of absorption, distribution, metabolism, excretion and toxicity was carried out. The procyanidins-enriched fraction, named here as F7, showed high antioxidant and anti-glycation capacities, with inhibitory activities against lipid peroxidation, and AGEs and ROS formation. In addition, there were reductions in AGEs-induced crosslinks and protein carbonyls and protective effects against oxidation of thiol groups and glycated-catalase. ADMET predictions of F7 showed favorable absorption and distribution, with no hepatotoxicity or mutagenicity. Together, our results support the anti-glycation activities of the procyanidins-enriched fraction from A. crassiflora, and suggest that these effects are triggered, at least in part, by scavenging free radical and dicarbonyls intermediates.
Similar content being viewed by others


Introduction
Glycation is a non-enzymatic reaction involving reactive carbonyls of sugars with amino groups of a protein, lipid or nucleic acid generating Schiff bases, which rearrange to Amadori products1. Due to instability of Amadori products, further rearrangement reactions occur that lead to the formation of advanced glycation end-products (AGEs)2. The damaging potential of AGEs results from direct alterations on protein structures and functions due to AGEs per se or the cross-linking effect of some AGEs. Recently, AGEs have been reported as results of increased oxidative stress, since many of them are generated by a combination of oxidation and glycation processes3,4,5. Oxidative stress is a result of an imbalance between free radical production and antioxidant defense mechanisms6. Moreover, it is well known that AGEs are increasingly formed under hyperglycemic conditions2. Thus, oxidative stress, reactive oxygen species (ROS) and AGEs are involved in the pathogenesis of diabetes mellitus and its complications7,8.
Natural products are considered important sources of investigation of potential new drugs, and studies of bioactive molecules effective in the control of oxidative stress and non-enzymatic glycation are promising. Plants with known antioxidant and anti-glycation properties are receiving a lot of attention as they are believed to have less adverse effects9. Proanthocyanidins, a class of flavonoids, are the most studied secondary metabolites found in plants due to their antioxidant properties10,11,12. Recent studies have shown the anti-glycation potential of procyanidins, oligomeric natural compounds belonging to the class of proanthocyanidins formed from catechin and epicatechin molecules13,14.
The fruit peel of Annona crassiflora Mart., a native species of the Brazilian Savanna biome belonging to the Annonaceae family, has been recently shown by our group as a source of proanthocyanidins15. Different parts of this species are popularly used for treating wounds, rheumatism, venereal diseases, diarrhea, snakebites and microbial infections16,17. Previously, we conducted a pre-purification of the crude ethanol extract of A. crassiflora fruit peel, obtaining non-cytotoxic polyphenols fractions (EtOAc and n-BuOH)15. Additionally, we related the inhibition of glycation by EtOAc fraction. However, only the preliminary BSA-fructose model was tested without focusing on specific aspect of AGE inhibition and its mechanism.
Thus, the present study aimed to purify procyanidins from A. crassiflora fruit peel with antioxidant and anti-glycation effects by examining free radical scavenging and quenching properties, ROS, AGEs, protein carbonyl and thiol groups formation, lipid peroxidation, crosslinked AGEs, as well as protective effect on glycated catalase. In addition, in silico assessment of the absorption, distribution, metabolism, excretion and toxicity (ADMET) profile of the main procyanidins was carried out.
Results
Extraction yield
The ethanolic extraction of A. crassiflora fruit peel had a total yield of about 5%. Among the organic fractions from ethanol extract, EtOAc fraction presented a yield of approximately 5.5%. Proanthocyanidins were purified from EtOAc fraction by column chromatography, which resulted in the grouping of 12 fractions (F1-F12) according to the Rf values. F6 and F7 showed higher yields, 24.6 and 14.0%, respectively, followed by F4, F5, F8 and F3 (5.2, 3.7, 1.6 and 1.4%, respectively). The other fractions had yields lower than 1%.
Phytochemical prospection
Total phenolics and proanthocyanidins contents of the purified proanthocyanidins fractions from A. crassiflora fruit peel are shown in Table 1. In general, all fractions presented amount of total phenolics and proanthocyanidins higher than 150 mg GAE g−1 and 250 mg CE g−1, respectively. As expected, EtOAc fraction also contained a substantial amount of phenolic compounds. Among the proanthocyanidins fractions, F7 had the highest values of total phenolics (660.6 mg GAE g−1). In addition, proanthocyanidins were concentrated mainly in F7 fraction (1295.7 mg CE g−1).
Because F7 had the highest value of proanthocyanidins content, this fraction was subjected to a HPLC-ESI-MS/MS analysis in order to identify its main proanthocyanidins. According to the high-resolution spectra of the molecular ions, error values (ppm), retention times, sequential mass spectra and comparisons with data from the literature15,18 and Metlin library, we identified oligomers of B-type procyanidin, including dimer, trimer, tetramer and pentamer (Table 2). The chromatogram and sequential mass spectra can be found as Supplementary material (Figs. S1–S6).
Antioxidant capacity
In all antioxidant capacity assays, F7 had the highest values (data not shown). Because of this, the biological assays were performed with F7 and EtOAc fractions, and quercetin as control. In the ORAC assay, F7 had higher antioxidant capacity than EtOAc (9691 µmol trolox eq g−1, p < 0.01), with a similar value to quercetin (12283 µmol trolox eq g−1) (Fig. 1a). In relation to the FRAP method, F7 showed higher antioxidant activity than EtOAc and quercetin (2438 µmol trolox eq g−1, p < 0.01) (Fig. 1b). In addition, the analysis using DPPH antiradical test showed that F7 and EtOAc had a high antioxidant capacity (IC50 5.4 and 6.1 µg mL−1, respectively). It is worth emphasizing that these fractions showed similar results to quercetin in the DPPH method (IC50 4.1 µg mL−1) (Fig. 1c).

Antioxidant capacity analysis of proanthocyanidins-enriched and EtOAc fractions from A. crassiflora fruit peel and quercetin by ORAC (a), FRAP (b) and DPPH (c) methods. Values (mean ± standard deviation) are expressed as µmol trolox equivalents g−1 (a,b), percentage of antioxidant activity and IC50 values. (c) Note: F7: procyanidins-enriched fraction, EtOAc: ethyl acetate fraction. **p < 0.01 versus EtOAc; ##p < 0.01 and ###p < 0.001 versus quercetin (control).
The cell viability and reduced of ROS production using the F7 and EtOAc fractions are shown in Fig. 2. As expected, F7, EtOAc and quercetin did not stimulate ROS production. F7 was able to reduce ROS production at the concentrations of 0.1, 1 ad 10 µg mL−1 (p < 0.05) (Fig. 2a,b), whereas that EtOAc reduced ROS production only at 1 and 10 µg mL−1 (p < 0.05) (Fig. 2c,d). Quercetin also reduced ROS production at all concentrations tested (p < 0.001) (Fig. 2e,f). The results also showed that F7 and EtOAc fractions were not cytotoxic in the concentration of 10 μg mL−1 (Fig. 2g).

ROS production and cell viability analyzes using the proanthocyanidins-enriched and ethyl acetate fractions from A. crassiflora fruit peel and quercetin in macrophages. Values (mean ± standard deviation) are expressed as relative luminescent units (RLU) for F7 (a), EtOAc (c) and quercetin (e), and area under the curve (AUC) for F7 (b), EtOAc (d) and quercetin (f). (g) Shows cellular viability of macrophages treated with F7, EtOAc and quercetin (10 µg mL−1). Note: F7: proanthocyanidins-enriched fraction, EtOAc: ethyl acetate fraction, Q: quercetin, Med: medium, Zym: zymosan. *p < 0.05, **p < 0.01 and ***p < 0.001 versus Zym; ###p < 0.001 versus Med.
Inhibition of lipid peroxidation
The addition of F7, EtOAc and quercetin to the liver lipid peroxide induced by FeSO4-ascorbate significantly reduced TBARS formation. All concentrations of F7, EtOAc and quercetin (0.5, 5 and 50 µg mL−1) reduced lipid peroxidation (p < 0.001) (Fig. 3a). Furthermore, F7 and EtOAc at 5 and 50 µg mL−1 increased total antioxidant capacity in the liver (p < 0.01) (Fig. 3b).

Analysis of lipid peroxidation (a) and total antioxidant capacity (b) in the livers of healthy rats treated for 60 minutes with 0.5, 5 and 50 µg mL−1 of proanthocyanidins-enriched and EtOAc fractions from A. crassiflora fruit peel and quercetin. Values expressed as mean ± standard deviation. Note: F7: proanthocyanidins-enriched fraction, EtOAc: ethyl acetate fraction. *p < 0.05, **p < 0.01 and ***p < 0.001 versus control.
Inhibition of the formation of advanced glycation end products (AGEs)
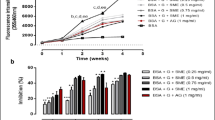
In the BSA-fructose model, F7 was able to inhibit glycation over 90% at 20 μg mL−1, as well as quercetin; EtOAc at 20 µg mL−1 reduced glycation over 80% (Fig. 4a). It is worth mentioning that their IC50 values did not present significant differences. In addition, F7 inhibited fluorescent AGE production by over 80% at 345 µg mL−1 in BSA-methylglyoxal assay, with an IC50 value lower than EtOAc (128.9 µg mL−1, p < 0.01) (Fig. 4b). Anti-glycation assay in arginine–methylglyoxal model showed that F7 and EtOAc inhibited fluorescent AGE generation over 50% at 345 µg mL−1, presenting no difference between their IC50 values (249.3 and 280.3 µg mL−1, respectively) (Fig. 4c). Quercetin showed the lowest IC50 values in BSA-methylglyoxal and arginine-methylglyoxal assays (24.4 µg mL−1). The anti-glycation results obtained for F7 and EtOAc fractions showed significant high correlations between BSA-fructose and BSA-methylglyoxal (r = 0.66, p < 0.05), BSA-fructose and arginine-methylglyoxal (r = 0.75, p < 0.05) and BSA-methylglyoxal and arginine-methylglyoxal (r = 0.96, p < 0.05) methods. A high correlation was also observed for F7 and EtOAc between non-enzymatic glycation and lipid peroxidation assays (r ~ 0.72, p < 0.05).

Glycation inhibitory activity analysis of the proanthocyanidins-enriched and EtOAc fractions from A. crassiflora fruit peel and quercetin in BSA-fructose (BSA-FRU) (a), BSA-methylglyoxal (BSA-MGO) (b) and arginine-methylglyoxal (ARG-MGO) (c) models. Values (mean ± standard deviation) are expressed as a percentage of glycation inhibition and IC50 values. Note: F7: proanthocyanidins-enriched fraction, EtOAc: ethyl acetate fraction. **p < 0.01 versus EtOAc; ##p < 0.01 and ###p < 0.001 versus quercetin (control).
Glycation-induced protein carbonyl and thiol group formation
BSA glycated for 4 days showed an increase in protein carbonyl groups compared to BSA alone (p < 0.001) (Fig. 5). Compared to glycated BSA, both F7 and EtOAc inhibited formation of protein-bound carbonyls groups at all concentrations tested (6.25, 62,5 and 625 µg mL−1, p < 0.001) (Fig. 5a,b). Quercetin reduced carbonyls groups in BSA only at 62.5 and 625 µg mL−1) (Fig. 5c). Glycated BSA had a lower thiol content compared to BSA alone (p < 0.05) (Fig. 5). F7 and EtOAc had a protective effect against oxidation of thiol groups (p < 0.05), as well as quercetin, when compared to glycated BSA (Fig. 5d–f). Also, the anti-glycation properties of F7 and EtOAc fractions were correlated with their capacity to inhibit protein carbonylation (r ~ 0.75, p < 0.05).

Effects of proanthocyanidins-enriched and EtOAc fractions from A. crassiflora fruit peel and quercetin on protein carbonyl (a–c, respectively) and thiol (d–f, respectively) groups of fructose-modified BSA. Data are presented as mean ± standard deviation. Note: F7: proanthocyanidins-enriched fraction, EtOAc: ethyl acetate fraction. *p < 0.05, **p < 0.01 and ***p < 0.001 versus glycated-BSA (control).
Crosslinked advanced glycation end products (AGEs)
SDS-PAGE gels showing the effects of F7 on formation of crosslinked AGEs formed by glycation of BSA and lysozyme by fructose or methylglyoxal are shown in Fig. 6. A major band around 66 kDa with high density was observed for native BSA (Fig. 6a,c, lane a), but nevertheless this band density was low in glycated BSA (Fig. 6a,c, lane b). F7 was able to slightly increase density of the 66 kDa band of BSA glycated by fructose (Fig. 1, lanes c–h). However, F7 seems to have no effect on BSA glycated by methylglyoxal (Fig. 1c). Furthermore, glycation of lysozyme by fructose or methylglyoxal produces crosslinked AGEs that present as dimers, with approximate molecular weights of 28 and 36 kDa, as shown in Fig. 6b,d (lane b), when compared to native lysozyme (Fig. 6b,d, lane a). F7 reduced AGE-induced dimerization of lysozyme in a dose-dependent manner (Fig. 6b,d, lanes c–h). These results were similar to the quercetin (Supplementary Material, Fig. S7).

SDS-PAGE gels showing the effects of the proanthocyanidins-enriched fraction from A. crassiflora fruit peel (F7) on formation of crosslinked AGEs formed by glycation of BSA (10 mg mL−1) and lysozyme (10 mg mL−1) by fructose (a,b, respectively) or methylglyoxal (c,d, respectively) for 4 weeks at 37 °C. Unmodified BSA and lysozyme (lane a), glycated BSA and lysozyme (lane b) or BSA and lysozyme glycated in the presence of 625 (lane c), 345 (lane d), 187.5 (lane e), 62.5 (lane f), 34.5 (lane g) and 18.75 µg mL−1 (lane h) of F7.
Glycated catalase activity and fluorescence
The effect of F7 and EtOAc on activity and fluorescence of glycated catalase is shown in Fig. 7. Effect of 16 mmol L−1 fructose as glycating agent on the catalytic activity of catalase was evaluated for 96 h (Fig. 7a). As expected, high concentration of fructose caused a decrease in catalase activity compared with the control, representing catalytic activity of catalase without fructose before incubation (day 0). Catalytic activity of glycated catalase decreased to approximately 46% on the 2nd day, 16% on the 3rd day, and about 6% on the 4th day (Fig. 7a). This decrease in catalase activity was attenuated by EtOAc on the 2nd day (57%, p < 0.001), and by F7 on the 3rd and 4th days (26 and 13%, respectively, p < 0.05) (Fig. 7a). These results are in agreement with the fluorescent intensity values observed for glycated catalase and glycated catalase incubated with F7 and EtOAc for 96 h (Fig. 7b). An increase of fluorescence intensity (approximately 115%, p < 0.001) was observed for glycated catalase when compared with catalase alone. When catalase is incubated for 96 h at 37 °C with fructose and F7 or EtOAc, a decrease in AGE-specific fluorescence was observed (p < 0.001) (Fig. 7b). Quercetin was not able to increase the activity and reduce the fluorescent intensity of glycated catalase. To ensure F7 and EtOAc fractions do not have any effect on reducing hydrogen peroxide absorbance, hydrogen peroxide was placed in the presence of these fractions and no reduction in the absorbance value was observed.

Activity (a) and fluorescence intensity (b) of catalase in the presence of 16 mmol L−1 fructose and 3 µg mL−1 proanthocyanidins-enriched and EtOAc fractions from A. crassiflora fruit peel and quercetin, for 96 h. H2O2 was used as substrate for catalase activity assay. Note: CAT: catalase, FRU: fructose, F7: proanthocyanidins-enriched fraction, EtOAc: ethyl acetate fraction and Q: quercetin. *p < 0.05, **p < 0.01 and ***p < 0.001 versus CAT + FRU.
In Silico pharmacokinetic properties prediction
The prediction of pharmacokinetic properties of procyanidins of F7 is shown in Table 3. All compounds showed intestinal absorption above 40%, with the exception of B procyanidin pentamer (33%), and had no interference of metabolism by binding families of cytochrome P enzymes. The predicted intestinal absorption was based on the original compounds. The steady-state volume of distribution (VDss) values of F7 procyanidins range from −0.046 to 0.011, indicating uniform distribution. With regard to the total clearance, the larger oligomers showed the lowest values. Also, all procyanidins presented no hepatotoxicity or mutagenicity.
Discussion
In this study, we performed an ethanolic extraction of A. crassiflora fruit peel, a liquid-liquid partitioning of the ethanol extract and a column chromatography of EtOAc in order to obtain purified proanthocyanidins. Previously, we identified various phenolic compounds in EtOAc, such as caffeoyl-glucoside, (epi)catechin, feruloyl-galactoside, quercetin-glucoside, kaempferol and procyanidin B215. The column chromatography concentrated the proanthocyanidins, with F7 having the highest values of total phenolics and proanthocyanidins (Table 1). These results are in accordance with HPLC-ESI-MS/MS analysis, which indicated the presence of B-type procyanidins oligomers in F7, such as dimer, trimer, tetramer and pentamer (Table 2).
The high content of proanthocyanidins in F7 might explain its high antioxidant capacity. F7 had higher antioxidant capacities by the oxygen radical absorption capacity (ORAC), 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging and ferric reducing antioxidant power (FRAP) methods (Fig. 1). These methods used together reinforce its antioxidant properties, since they are based on different antioxidative reaction mechanisms involving hydrogen atom and electron transfer19,20. FRAP method evaluates the ability to transfer electrons, ORAC analyzes the hydrogen transfer potential, while DPPH assay detects both hydrogen and electrons transfer capacities20,21. In the FRAP assay, F7 and EtOAc had higher antioxidant activity than quercetin, which may indicate that these fractions have greater ability to reduce oxidants by electrons transfer than quercetin. However, in the ORAC assay, both quercetin and F7 showed similar antioxidant activity, suggesting similar hydrogen transfer capacity between them. The antiradical activity of proanthocyanidins is principally based on the redox properties of their hydroxyl groups22. Through the availability of their phenolic hydrogens, proanthocyanidins act as hydrogen donating radical scavengers and singlet oxygen quenchers and electron-donating agents, with properties to form stable antioxidant-derived radicals11,23,24. In addition, F7 was able to reduce zymosan-induced ROS production in macrophages with no cytotoxicity (Fig. 2). It is worth mentioning that cellular-based assays provide a more realistic model of the in vivo antioxidant capacity of phytochemicals.
It is well known that oxidative stress and increased free radical production induce lipid peroxidation, since lipids are most susceptible to the attack of reactive oxygen and nitrogen species25. This may compromise the functions of lipoproteins due to the decrease in membrane fluidity25. Also, lipid peroxidation leads to complex products, such as hydroperoxides, aldehydes and polymeric materials, which may exert cytotoxic and genotoxic effects26. The antioxidant potential of F7 contributed to the reduction of lipid peroxidation in the liver by Fe2+-ascorbate-induced lipid peroxidation assay (Fig. 3). This can be explained by the fact that F7 was able to control reactive oxygen species production. Previous studies have also demonstrated the potential of proanthocyanidins in reducing lipid peroxidation in diabetic animals27,28.
In addition to overproduction of ROS, increased AGEs formation are also usually manifested in oxidative stress conditions4. Free radicals could mediate the conversion of Amadori products to AGEs29. Previously, we demonstrated the antioxidant and anti-glycation potential of ethanol extract of A. crassiflora fruit peel and its EtOAc fraction15. However, we tested only on the BSA-fructose model. Here, we focused on some specific aspect of AGEs inhibition and its mechanism. Interestingly, F7 exhibited an inhibitory potential against AGEs formation in the BSA-fructose, BSA-methylglyoxal and arginine-methylglyoxal models, with IC50 values lower than EtOAc (Fig. 4). It is noteworthy that these models together evaluate all stages of protein glycation30. According to Wang, et al.30, the arginine model evaluates a major and specific source of AGE generate by reactive carbonyls, such as methylglyoxal, that in reaction with the guanidine group of arginine forms a fluorescent AGE called argpyrimidine. The argpyrimidine can accumulate in various tissues causing systemic inflammatory processes and cataracts in lens31. Thus, F7 may act on the middle and all stages of protein glycation, as well as on a major and specific source of AGEs production.
Fructose can be generated from sorbitol oxidation and its accumulation on ocular lens and peripheral nerves is due to a high blood glucose concentration32. Methylglyoxal is primarily generated as an intermediate in the glycolysis pathway, being an reactive carbonyl33. Both fructose and oxidation products such as methylglyoxal may promote glycation of proteins and lipids, with the consequent generation of AGEs and ROS30, which can lead to diabetic retinopathy and peripheral neuropathy34,35. Therefore, inhibition of non-enzymatic glycation mediated by fructose and its oxidant products is a viable strategy to prevent AGEs formation and the complications related to diabetes.
Protein oxidation has a key role in AGEs formation and together with glycation can form protein-bound carbonyl groups36. In this study, carbonyl groups were generated by incubating BSA with a high concentration of fructose. F7, as well as EtOAc, inhibited protein-bound carbonyl groups with the results comparable to the quercetin (Fig. 5). In addition, the glycation mediated accumulation of carbonyl groups in proteins may lead to protein cross-linking14,37, which was observed for BSA and lysozyme treated with fructose and methylglyoxal (Fig. 6). F7 was able to reduce AGE-induced dimerization of lysozyme in a dose-dependent manner, especially when incubated with fructose. Our results corroborate a study conducted by Muthenna, et al.14, which showed that a procyanidin-B2 fraction of cinnamon was able to inhibit glycation and protein cross-links, and to reduce carbonyl content of eyes lens. Furthermore, the loss of protein thiol groups also occurs in glycation, which may potentially lead to severe structural changes in various biological structures38. The oxidation of thiol groups was observed following glycation of BSA and this was inhibited by F7 and EtOAc fractions (Fig. 5). The carbonyl and thiol assays provide mechanistic information concerning the effects of the procyanidin-enriched fraction of A. crassiflora on AGEs.
In general, many antioxidant compounds, especially proanthocyanidins, show anti-glycation properties39,40, which are associated with the presence of hydroxyl groups41. Oligomeric procyanidins present higher antioxidant and glycation inhibitory activities than monomers42,43. They can reduce AGEs formation by different mechanisms, such as inhibiting ROS formation during non-enzymatic glycation, Schiff base and Amadori products, as well as blocking AGE-RAGE receptors, capturing precursors such as 1,2-dicarbonyls and/or interacting with glucose and preventing it from binding to proteins14,44,45,46. Our results suggest that the proanthocyanidins-enriched fraction might exert its anti-glycation activity by scavenging free radical and dicarbonyls intermediates.
Antioxidant enzymes have important roles in the defense system for controlling ROS production and repairing damage resulting from oxidative stress47. Catalase is one of the main enzymes involved in the elimination of ROS, particularly hydrogen peroxide, being highly glycated48,49. Glycation of catalase can lead to a decrease in its activity and, consequently, to an imbalance in oxidants and antioxidants homeostasis50. A decrease in catalase activity was observed in the presence of fructose. Our results showed that the catalase samples which contained F7 in addition to fructose had more enzyme activity compared with the same samples without the proanthocyanidins-enriched fraction after 72 and 96 h of incubation; this was observed for EtOAc at 48 h of incubation (Fig. 7). In addition, an increase of fluorescence intensity was observed when catalase was incubated with fructose for 96 h, indicating the presence of fluorescent compounds being formed during glycation49,51. F7 and EtOAc fractions reduced AGE-specific fluorescence in catalase incubated with fructose. The results of our study showed that the proanthocyanidins-enriched fraction protected the enzyme to some extent against AGEs formation induced by fructose and can, at least in part, negate the effects of glycation on catalase and act as anti-glycation agent.
The in silico predicted pharmacokinetic properties showed favorable intestinal absorption ( > 40%) for proanthocyanidins, however, the higher the oligomer the lower its absorption52. According to Spencer, et al.53, the higher the polymerization index of the monomer, the more readily the components are cleaved. Thus, the monomeric and dimeric forms may be the forms presented at highest concentration to the small and large intestine. Proanthocyanidin monomers and dimers, as well as their methylated and glucuronidated forms are the main metabolites of proanthocyanidins in plasma52. The results of steady state volume of distribution (VDss) indicate that all proanthocyanidins of F7 can be distributed evenly providing an equal level of blood plasma54. Also, the dimer and trimer proanthocyanidins showed the highest values of total clearance, indicating that they are excreted more rapidly than the higher oligomers54. Furthermore, there was no negative interference in metabolism, since no inhibition of cytochrome P enzymes such as CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 was detected. With regard to toxicity, all compounds showed no hepatotoxicity or mutagenicity, corroborating data from Yamakoshi, et al.55, Bak, et al.56 and Yu and Swaminathan57.
In summary, the present study describes the characterization of proanthocyanidins from A. crassiflora fruit peel as anti-glycating agent and their mechanism of action. F7 showed high antioxidant and anti-glycation capacities, with inhibitory activities against lipid peroxidation, and AGEs and ROS formation. In addition, there were reductions in AGEs-induced protein carbonyls and protective effects against oxidation of thiol groups and glycated-catalase. The proanthocyanidins were predicted to have good pharmacokinetics properties and no toxicity. The fruit peel of A. crassiflora, a species from the Brazilian Savanna, a global biodiversity hotspot, could be potential source of nutraceuticals that offer combined antioxidant and anti-glycation properties to combat the oxidative stress and non-enzymatic glycation, which are present in many pathologies including diabetes mellitus.
Methods
Drugs and reagents
Reagents and solvents of analytical grade were purchased from Sigma (Sigma, St Louis, MO, USA) or from local suppliers. Water used was Milli-Q obtained by deionized water that has filtered on a millipore filter.
Preparation of plant material and proanthocyanidins fractions
Fruits of A. crassiflora were collected in the north region of Minas Gerais State in March 2017. The plant was identified by André Vito Scatigna from the Biology Institute of the Federal University of Uberlandia, Uberlândia-MG, Brazil. A voucher specimen (HUFU68467) was deposited in the Herbarium Uberlandense. The peels of A. crassiflora fruit were separated from pulps, and the dried and powdered peels (7.0 kg) were extracted by the maceration method for three days with 98% ethanol (18 L) in room temperature (25 °C). This process was performed 6 times. After filtration, ethanol was removed using a rotary evaporator (Bunchi Rotavapor R-210, Switzerland) in a water bath at 40 °C under reduced pressure (344.7 g, 4.92%). The crude EtOH extract (100.0 g) was resuspended in MeOH:H2O solution (9:1, v v−1, 500 mL), filtered (insoluble residue 15.7 g), and submitted to a liquid-liquid extraction according to a protocol designed by Justino, et al.15 using n-hexane (5 × 200 mL, 4.7 g), CH2Cl2 (5 × 200 mL, 8.3 g), EtOAc (5 × 200 mL, 4.7 g), and n-BuOH (5 × 200 mL, 37.0 g). Additionally, an aqueous fraction (15.2 g) was obtained. The extractive solvents were removed using a rotary evaporator under reduced pressure at 40 °C and the fractions were frozen and lyophilized (L101, Liobras, SP, Brazil) to remove the remaining water. EtOAc fraction (2.8 g) was dissolved in MeOH:H2O (3.5:6.5) and applied to a size exclusion column (Sephadex LH-20, 500 × 22 mm, GE Health-care Bio-Sciences AB, Uppsala, Sweden) pre-equilibrated with MeOH:H2O (3.5:6.5) for 4 h. Initially, sugars and low-molecular-weight phenolic compounds (as flavonols) were eluted from the chromatographic column with MeOH:H2O (3.5:6.5, v v−1, 500 mL). Further, the proanthocyanidins were eluted with acetone:H2O (60:40, v v−1; 250 mL)58 and the fractions were analyzed by TLC and grouped according to the Rf values (12 fractions). The solvents were removed using rotary evaporated under reduced pressure at 40 °C and lyophilized to dryness.
Phytochemical prospection
Total phenolic content
In a 96-well microplate was inserted into each well 5 μL of fractions (500 µg mL−1, methanol), 25 μL of Folin-Ciocalteu (10%, water) and 195 μL of water. After 6 min of incubation at 25 °C, 75 μL of Na2CO3 (7%, water) was added and the mixture was incubated at 25 °C in the dark for 2 h. The absorbance was read at 760 nm (Molecular Devices, Menlo Park, CA, USA)59. The total phenolic content was obtained using an analytical curve constructed with gallic acid standards. The results were expressed as milligrams of gallic acid equivalents per gram of sample (mg GAE g−1).
Total proanthocyanidins content
In a 96-well microplate was inserted into each well 10 µL of each fraction (500 µg mL−1, methanol) was incubated with 200 µL of vanilin (4%, methanol) and 100 µL of HCl at 25 °C for 15 min. After incubation, the absorbance was read at 500 nm (Molecular Devices, Menlo Park, CA, USA)59. The total proanthocyanidins content was determined using an analytical curve constructed with catechin standards. The results were expressed as milligrams of catechin equivalents per gram of sample (mg CE g−1).
Liquid chromatography-mass spectrometry analysis
The most active procyanidins-enriched fraction in the biological assays were analyzed by high-performance liquid chromatography-electrospray ionization tandem mass spectrometry (HPLC–ESI–MS) to identify the chemical compounds, according to Justino, et al.60 and Justino, et al.15. HPLC Agilent Infinity model 1260 was coupled in Mass Spectrometer Agilent model 6520 B Quadrupole Time of Flight Q-TOF with ESI source. The chromatographic parameters were: 50 × 2.1 mm column, pore diameter of 110 Å and particles of 1.8 µm (Agilent Zorbax model) and a mobile phase containing water acidified with formic acid at 0.1% (v v−1) (A) and methanol (B). The gradient solvent system (B) was: 2% (0 min); 98% (0–15 min); 100% (15–17 min); 2% (17–18 min) and 2% (18–22 min), with a flow of 0.35 mL min−1 and detection at 280 and 360 nm. The ionization parameters were: 8 L min−1 secant gas at 220 °C, nebulizer pressure at 58 psi and 4.5 kVa energy in the capillary. Sequential mass spectrometry (MS/MS) analyzes were performed on different collision energies (5–30 eV) for the positive and negative modes for each molecular ion. The identified compounds were proposed according to the high-resolution spectra of the molecular ions, error values (ppm), retention times, sequential mass spectra and comparisons with data from the literature and Metlin library.
In vitro antioxidant capacity
DPPH antiradical activity test
The method using the stable free radical 2,2-diphenyl-1-picrylhydrazyl (DPPH) test was performed according to Justino, et al.60. Briefly, the fractions (diluted in methanol) were incubated with DPPH (0.06 mmol L−1, methanol) at 30 °C for 20 min. Reduction in the absorbance of the mixture was measured at 517 nm (Molecular Devices, Menlo Park, CA, USA). Quercetin was used as a positive control. Scavenging of DPPH radicals was calculated using the following formula: Antioxidant activity (%) = [(Abs DPPH – Abs sample)/Abs DPPH] x 100, where Abs DPPH is the absorbance of DPPH radical and Abs sample is the absorbance of the DPPH radical in presence of fraction/control. A blank was performed with the fraction/control to reduce the contribution of the color to the absorbance.
ORAC antioxidant activity test
The method of oxygen radical absorbance capacity (ORAC) was performed according to Justino, et al.60. The fractions (diluted in methanol) were incubated with fluorescein (0.085 nmol L−1, phosphate buffer, 75 mmol L−1, pH 7.4) at 25 °C for 15 min. 2,2′-Azobis-(2-amidino-propane)-dihydrochloride (153 mmol L−1, phosphate buffer, 75 mmol L−1, pH 7.4) was added to the mixture and the loss of fluorescence intensity was measured at 37 °C for 90 min, by calculating the area under the curve (485 nmex/528 nmem) (Perkin-Elmer LS 55, Massachusetts, USA). The antioxidant capacity was determined using an analytical curve constructed with trolox as standard (µmol TE/g). Quercetin was used as a positive control.
FRAP antioxidant activity test
The method of ferric reducing antioxidant power (FRAP) was performed according to Benzie and Strain61. Briefly, the fractions (diluted in methanol) were incubated at 37 °C for 6 min with FRAP reagent containing the following reagents in 10:1:1 proportion: sodium acetate buffer (300 mmol L−1, pH 3.6), 2,4,6-tris(2-pyridyl)-s-triazine (10 mmol L−1) and ferric chloride (20 mmol L−1). The absorbance was measured at 593 nm (Molecular Devices, Menlo Park, CA, USA) and the antioxidant activity was determined using an analytical curve constructed with trolox as standard (µmol TE g−1). Quercetin was used as a positive control. A blank was performed with the fractions/control to reduce the contribution of the color to the absorbance.
Generation of bone marrow-derived macrophages (BMDM)
This method was based on Larmonier, et al.62. Macrophages were obtained by differentiation of bone marrow cells from 6–8 week old male C57BL/6 mice. Bone marrow cells were extracted by flushing bone cavity with RPMI 1640 into a 15 mL sterile polypropylene tube. The cells were homogenized and cell suspension was centrifuged (400 xg for 8 min, 4 °C). The pellet was homogenized in RPMI-1640 supplemented with 20% of fetal bovine serum (Gibco), 2 mmol L−1 L-glutamine, 100 U mL−1 penicillin, 100 µg mL−1 streptomycin and 30% L929-cell conditioned medium (LCCM), which was used as a source of macrophage colony stimulating factor (M-CSF). Cells were kept at 37 °C and 5% CO2 atmosphere; 5.0 × 106 cells were cultured to generate BMDM. BMDM were collected after 4 days by removing the supernatant of culture dishes and detaching adherent cells using sterile ice-cold phosphate-saline buffer (PBS). The animal proceedings were all previously approved by institutional Animal Ethics Committee (protocol number 032/18).
Cell viability
This method was based on Lee and Park63. BMDM were suspended in RPMI-1640 supplemented in according with aforementioned, except LCCM which was not added. Cells were seeded in 96-well microplate at density of 0.2 × 106 cells/well and were treated for 24 h with the fractions (diluted in RPMI-1640). Next, the supernatant was removed and incubated with 100 µL of 5 mg mL−1 MTT (3-(4,5-dimethylthiazolyl-2)−2,5-diphenyltetrazolium bromide) solution at room temperature for 2 h. The purple formazan formed by viable cells were solubilized in dimethyl sulfoxide (DMSO) and the absorbance was measured at 570 nm (Molecular Devices, Menlo Park, CA, USA).
Inhibition of ROS production
This method was based on Shih, et al.64. Briefly, BMDM cells, previously diluted in Hank’s Balanced Salt Solution (HBSS) without phenol red (Sigma-Aldrich), were seeded to white opaque 96-well microplate at a density of 0.2 × 106 cells/well. Cells were pre-incubated with the fractions (diluted in HBSS) for 30 min at 37 °C and 5% CO2 atmosphere. Next, luminol (final concentration of 625 nmol L−1) was added to each well and the generation of ROS was induced by opsonized- zymosan from Saccharomyces cerevisiae (final concentration of 100 µg mL−1). Opsonized-zymosan was previously prepared by incubating zymosan with serum of mice for 30 min at 37 °C. Zymosan suspension was centrifuged at 400 xg for 8 min and the pellet was homogenized in HBSS. The generation of ROS by BMDM was monitored for 4 h through chemiluminescence emission resulting from luminol oxidation by using a microplate reader (Perkin-Elmer LS 55, Massachusetts, USA). Results were expressed as Relative Luminescence Units (RLU).
Inhibition of non-enzymatic lipid peroxidation
The inhibition of Fe2+-ascorbate-induced lipid peroxidation was assayed by a modified method of Justino, et al.60. Male Wistar rats (200–270 g) were housed in a controlled environment (22 ± 2 °C) with a 12-h light/dark cycle (lights on between 6:00 AM and 6:00 PM) and fed standard laboratory chow and tap water ad libitum. All animal experiments were complied with the ARRIVE guidelines and approved by the Ethics Committee in Animal Experimentation of the Federal University of Uberlândia (CEUA/UFU-015/17). The liver tissues were removed and homogenized in phosphate buffer (1:10 w v−1, pH 7.4), then centrifuged at 3000 × g for 10 min at 4 °C. Lipid peroxidation was determined according to the thiobarbituric acid reactive substances (TBARS) method. Liver homogenate (0.5 mL) was incubated with phosphate buffer (0.9 mL), 0.1 mmol L−1 FeSO4.6H2O (0.25 mL), 0.1 mmol L−1 ascorbic acid (0.25 mL) and fractions/control (0.1 mL, diluted in water) at 37 °C for 60 min. Then, the mixture was incubated with 1 mL of 10% TCA (w v−1) and 1 mL of 0.67% thiobarbituric acid (in 7.1% Na2SO4) at 100 °C for 15 min. The samples were centrifuged at 3000 × g for 15 min and the absorbance of the complex was measured at 532 nm (Molecular Devices, Menlo Park, CA, USA). The supernatant was also used for FRAP assay (see methodology above). Quercetin was used as a positive control.
Inhibition tests for the formation of advanced glycation end products (AGEs)
BSA-fructose assay
The anti-glycation assay using the BSA-fructose model was performed according to Franco, et al.65. The fractions (diluted in methanol) were incubated in the dark with 50 mg mL−1 BSA, 1.25 mol L−1 fructose and 200 mmol L−1 sodium phosphate buffer containing 0.02% sodium azide, pH 7.4, at 37 °C for 72 h. Control samples without fructose and/or fractions were incubated under the same conditions. After incubation, 20% TCA (w v−1) was added and the mixture was centrifuged at 10,000 × g for 10 min. The pellet was resuspended in phosphate buffer and the fluorescence intensity was measured at 350 nmex/420 nmem (Perkin-Elmer LS 55, Massachusetts, USA). Quercetin was used as a positive control and methanol as a negative control. The blank was carried substituting fructose by phosphate buffer. The results were presented as a percentage of glycation inhibition (GI), calculated according to the following equation:
where: F sample is fluorescence intensity in presence of fraction/control; F blank is fluorescence intensity in the absence of fructose and fraction/control; and F control is fluorescence intensity in the absence of fraction/control.
BSA-methylglyoxal assay
The anti-glycation assay using the BSA-methylglyoxal model was performed according to Justino, et al.60. The fractions (diluted in methanol) were incubated in the dark with 50 mg mL−1 BSA, 53.3 mmol L−1 methylglyoxal and 200 mmol L−1 sodium phosphate buffer containing 0.02% sodium azide, at 37 °C for 72 h. Control samples without fructose and/or fractions were incubated under the same conditions. After incubation, 20% TCA (w v−1) was added and the mixture was centrifuged at 10000 × g for 10 min. The pellet was resuspended in phosphate buffer and the fluorescence intensity was measured at 340 nmex/380 nmem (Perkin-Elmer LS 55, Massachusetts, USA)30. Quercetin was used as a positive control and methanol as a negative control. The blank was carried substituting methylglyoxal by phosphate buffer. The results were presented as a percentage of glycation inhibition, using the same equation in BSA-fructose model.
Arginine-methylglyoxal assay
The anti-glycation assay using the arginine-methylglyoxal model was performed according to Justino, et al.60. The fractions (diluted in methanol) were incubated in the dark with 106.6 mmol L−1 arginine, 53.3 mmol L−1 methylglyoxal and 200 mmol L−1 sodium phosphate buffer containing 0.02% sodium azide, at 37 °C for 72 h. Control samples without fructose and/or fractions were incubated under the same conditions. After incubation, the fluorescence intensity was measured at 340 nmex/380 nmem (Perkin-Elmer LS 55, Massachusetts, USA). Quercetin was used as a positive control and methanol as a negative control. The blank was carried substituting methylglyoxal by phosphate buffer. The results were presented as a percentage of glycation inhibition, using the same equation in BSA-fructose model.
Measurement of protein-bound carbonyl and thiol groups of glycated BSA
BSA at 50 mg mL−1 was incubated in the dark with 1.25 mol L−1 fructose with or without the fractions (diluted in methanol) in 200 mmol L−1 sodium phosphate buffer containing 0.02% sodium azide, pH 7.4, at 37 °C for 4 days. Control sample without fructose and fractions was incubated under the same conditions. Carbonyls in BSA and in glycated BSA in the presence or absence of fractions were identified by 2,4-dinitrophenylhydrazine (DNPH), precipitated with 20% TCA, washed with ethanol-ethyl acetate and dissolved in 6 mol L-1 guanidine hydrochloride66. The absorbance values were registered at 370 nm (Molecular Devices, Menlo Park, CA, USA) and the carbonyl content was calculated using a molar absorbance of 22,000 mol L−1 cm−1; results were expressed as the ratio of nmols of DNPH reacted. Free thiol content was determined in BSA and in glycated BSA in the presence or absence of fractions according to an established method using 5,5′-dithiobisnitrobenzoic acid (DTNB)67. Free thiol concentration was calculated using 14,150 mol L−1 cm−1 as the molar extinction coefficient of DTNB.
Crosslinked advanced glycation end-products (AGEs)
BSA or lysozyme (10 mg mL−1) were incubated in the dark with 0.5 mol L−1 fructose or 53.3 mmol L−1 methylglyoxal with or without the fractions in 200 mmol L−1 sodium phosphate buffer containing 0.02% sodium azide, pH 7.4, at 37 °C for 30 days. Control samples without fructose and/or fractions were incubated under the same conditions. Crosslinked AGEs were assessed by crosslinking of the protein following sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) using 10% gels as described previously37. These gels were stained with Coomassie blue, destained and photographed (Amersham Imager 600 system, GE Healthcare Bio-Sciences AB, Uppsala, Sweden).
Measurement of glycated catalase activity and fluorescence
Measurement of glycated catalase activity was performed according to Mofidi Najjar, et al.49, with modifications. Bovine liver catalase (0.175 mg mL−1) was incubated with 16 mmol L−1 fructose and with or without the fractions in 10 mmol L−1 potassium phosphate buffer, pH 7.0, at 37 °C in the dark for 96 h. Control samples without fructose and/or fractions was incubated under the same conditions. Catalase activity evaluation was based upon hydrogen peroxide (0.2%) decomposition68 at 240 nm for 5 min (Perkin-Elmer LS 55, Massachusetts, USA). In addition, the fluorescence intensity of catalase and glycated catalase in the presence and absence of the fractions was measured at 350 nmex/420 nmem 96 h after incubation (Perkin-Elmer LS 55, Massachusetts, USA).
In Silico pharmacokinetic properties prediction
The pharmacokinetic properties of the procyanidins identified in A. crassiflora fruit peel were predicted by pKCSM54. These signatures were used to evaluate intestinal absorption, steady state volume of distribution (Vdss), cytochrome p inhibition, total clearance, mutagenicity and hepatotoxicity.
Statistical analysis
The statistical analyzes and graphics were done using GraphPad Prism 6.0 software. All analyzes were performed in triplicate and the data were expressed as mean ± standard deviation. The significance of difference was calculated using one-way and two-way ANOVA, and Dunnett’s and Tukey’s post-tests for multiple comparisons. Pearson’s correlation was used to examine linear relationships between the results. Values of p < 0.05 were considered significant.
Data availability
All data generated or analyzed during the current study are included.
References
Gkogkolou, P. & Böhm, M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinol 4, 259–270, https://doi.org/10.4161/derm.22028 (2012).
Ahmed, N. Advanced glycation endproducts–role in pathology of diabetic complications. Diabetes research and clinical practice 67, 3–21, https://doi.org/10.1016/j.diabres.2004.09.004 (2005).
Jakus, V. & Rietbrock, N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiological research / Academia Scientiarum Bohemoslovaca 53, 131–142 (2004).
Nowotny, K., Jung, T., Höhn, A., Weber, D. & Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 5, 194–222, https://doi.org/10.3390/biom5010194 (2015).
Miyata, T. et al. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney international 51, 1170–1181 (1997).
Burton, G. J. & Jauniaux, E. Oxidative stress. Best Pract Res Clin Obstet Gynaecol 25, 287–299, https://doi.org/10.1016/j.bpobgyn.2010.10.016 (2011).
Baynes, J. W. & Thorpe, S. R. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48, 1–9 (1999).
Brownlee, M., Cerami, A. & Vlassara, H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. The New England journal of medicine 318, 1315–1321, https://doi.org/10.1056/nejm198805193182007 (1988).
Elosta, A., Ghous, T. & Ahmed, N. Natural products as anti-glycation agents: possible therapeutic potential for diabetic complications. Current diabetes reviews 8, 92–108 (2012).
Plumb, G. W., De Pascual-Teresa, S., Santos-Buelga, C., Cheynier, V. & Williamson, G. Antioxidant properties of catechins and proanthocyanidins: effect of polymerisation, galloylation and glycosylation. Free radical research 29, 351–358 (1998).
Bagchi, D. et al. Free radicals and grape seed proanthocyanidin extract: importance in human health and disease prevention. Toxicology 148, 187–197 (2000).
Yang, L. et al. Proanthocyanidins against Oxidative Stress: From Molecular Mechanisms to Clinical Applications. BioMed research international 2018, 8584136–8584136, https://doi.org/10.1155/2018/8584136 (2018).
Chen, Y. et al. Structure-activity relationship of procyanidins on advanced glycation end products formation and corresponding mechanisms. Food chemistry 272, 679–687, https://doi.org/10.1016/j.foodchem.2018.08.090 (2019).
Muthenna, P. et al. Inhibition of protein glycation by procyanidin-B2 enriched fraction of cinnamon: delay of diabetic cataract in rats. IUBMB life 65, 941–950, https://doi.org/10.1002/iub.1214 (2013).
Justino, A. B. et al. Peel of araticum fruit (Annona crassiflora Mart.) as a source of antioxidant compounds with α-amylase, α-glucosidase and glycation inhibitory activities. Bioorganic Chemistry 69, 167–182, https://doi.org/10.1016/j.bioorg.2016.11.001 (2016).
Vilar, J. B., Ferreira, F. L., Ferri, P. H., Guillo, L. A. & Chen Chen, L. Assessment of the mutagenic, antimutagenic and cytotoxic activities of ethanolic extract of araticum (Annona crassiflora Mart. 1841) by micronucleus test in mice. Brazilian journal of biology = Revista brasleira de biologia 68, 141–147 (2008).
Almeida, S. P., Proença, C. E. B. & Ribeiro, J. F. Cerrado: espécies vegetais úteis. (1994).
Lin, L. Z., Sun, J., Chen, P., Monagas, M. J. & Harnly, J. M. UHPLC-PDA-ESI/HRMSn profiling method to identify and quantify oligomeric proanthocyanidins in plant products. Journal of agricultural and food chemistry 62, 9387–9400, https://doi.org/10.1021/jf501011y (2014).
Huang, D., Ou, B. & Prior, R. L. The chemistry behind antioxidant capacity assays. Journal of agricultural and food chemistry 53, 1841–1856, https://doi.org/10.1021/jf030723c (2005).
Liu, Z.-Q. Chemical Methods To Evaluate Antioxidant Ability. Chemical reviews 110, 5675–5691, https://doi.org/10.1021/cr900302x (2010).
Liang, N. & Kitts, D. D. Antioxidant property of coffee components: assessment of methods that define mechanisms of action. Molecules 19, 19180–19208, https://doi.org/10.3390/molecules191119180 (2014).
Kazeem, M. I., Akanji, M. A., Hafizur, R. M. & Choudhary, M. I. Antiglycation, antioxidant and toxicological potential of polyphenol extracts of alligator pepper, ginger and nutmeg from Nigeria. Asian Pacific journal of tropical biomedicine 2, 727–732, https://doi.org/10.1016/S2221-1691(12)60218-4 (2012).
Zhou, Q. et al. Free radical-scavenging activities of oligomeric proanthocyanidin from Rhodiola rosea L. and its antioxidant effects in vivo. Natural product research 28, 2301–2303, https://doi.org/10.1080/14786419.2014.921786 (2014).
Gonçalves, C., Dinis, T. & Batista, M. T. Antioxidant properties of proanthocyanidins of Uncaria tomentosa bark decoction: a mechanism for anti-inflammatory activity. Phytochemistry 66, 89–98, https://doi.org/10.1016/j.phytochem.2004.10.025 (2005).
Niki, E. Lipid peroxidation products as oxidative stress biomarkers. BioFactors 34, 171–180, https://doi.org/10.1002/biof.5520340208 (2008).
Yoshida, Y., Umeno, A. & Shichiri, M. Lipid peroxidation biomarkers for evaluating oxidative stress and assessing antioxidant capacity in vivo. Journal of clinical biochemistry and nutrition 52, 9–16, https://doi.org/10.3164/jcbn.12-112 (2013).
Mansouri, E., Panahi, M., Ghaffari, M. A. & Ghorbani, A. Effects of grape seed proanthocyanidin extract on oxidative stress induced by diabetes in rat kidney. Iran Biomed J 15, 100–106 (2011).
Lee, Y. A., Kim, Y. J., Cho, E. J. & Yokozawa, T. Ameliorative effects of proanthocyanidin on oxidative stress and inflammation in streptozotocin-induced diabetic rats. Journal of agricultural and food chemistry 55, 9395–9400, https://doi.org/10.1021/jf071523u (2007).
Lunceford, N. & Gugliucci, A. Ilex paraguariensis extracts inhibit AGE formation more efficiently than green tea. Fitoterapia 76, 419–427, https://doi.org/10.1016/j.fitote.2005.03.021 (2005).
Wang, W., Yagiz, Y., Buran, T. J., do Nascimento Nunes, C. & Gu, L. Phytochemicals from berries and grapes inhibited the formation of advanced glycation end-products by scavenging reactive carbonyls. Food research international 44, 2666–2673 (2011).
Kim, J. et al. Accumulation of argpyrimidine, a methylglyoxal-derived advanced glycation end product, increases apoptosis of lens epithelial cells both in vitro and in vivo. Experimental & molecular medicine 44, 167–175, https://doi.org/10.3858/emm.2012.44.2.012 (2012).
McPherson, J. D., Shilton, B. H. & Walton, D. J. Role of fructose in glycation and cross-linking of proteins. Biochemistry 27, 1901–1907, https://doi.org/10.1021/bi00406a016 (1988).
Allaman, I., Bélanger, M. & Magistretti, P. J. Methylglyoxal, the dark side of glycolysis. Frontiers in neuroscience 9, 23–23 (2015).
Schemmel, K. E., Padiyara, R. S. & D’Souza, J. J. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: a review. Journal of Diabetes and its Complications 24, 354–360 (2010).
Hammes, H.-P. et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nature medicine 9, 294–294 (2003).
Stadtman, E. R. Protein oxidation and aging. Free radical research 40, 1250–1258, https://doi.org/10.1080/10715760600918142 (2006).
Elosta, A., Slevin, M., Rahman, K. & Ahmed, N. Aged garlic has more potent antiglycation and antioxidant properties compared to fresh garlic extract in vitro. Scientific Reports 7, 39613, https://doi.org/10.1038/srep39613 (2017).
Barbosa, J. H., Oliveira, S. L. & Seara, L. T. The role of advanced glycation end-products (AGEs) in the development of vascular diabetic complications. Arquivos brasileiros de endocrinologia e metabologia 52, 940–950 (2008).
Wu, C. H., Huang, S. M., Lin, J. A. & Yen, G. C. Inhibition of advanced glycation endproduct formation by foodstuffs. Food & function 2, 224–234, https://doi.org/10.1039/c1fo10026b (2011).
Ramkissoon, J. S., Mahomoodally, M. F., Ahmed, N. & Subratty, A. H. Antioxidant and anti-glycation activities correlates with phenolic composition of tropical medicinal herbs. Asian Pacific journal of tropical medicine 6, 561–569, https://doi.org/10.1016/s1995-7645(13)60097-8 (2013).
Sadowska-Bartosz, I. & Bartosz, G. Prevention of protein glycation by natural compounds. Molecules 20, 3309–3334, https://doi.org/10.3390/molecules20023309 (2015).
Xie, Y. & Chen, X. Structures required of polyphenols for inhibiting advanced glycation end products formation. Current drug metabolism 14, 414–431 (2013).
Spranger, I., Sun, B., Mateus, A. M., Freitas, V. & Ricardo-da-Silva, J. M. Chemical characterization and antioxidant activities of oligomeric and polymeric procyanidin fractions from grape seeds. Food chemistry 108, 519–532, https://doi.org/10.1016/j.foodchem.2007.11.004 (2008).
Yeh, W.-J., Hsia, S.-M., Lee, W.-H. & Wu, C.-H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. Journal of food and drug analysis 25, 84–92 (2017).
Mesias, M., Navarro, M., Gokmen, V. & Morales, F. J. Antiglycative effect of fruit and vegetable seed extracts: inhibition of AGE formation and carbonyl-trapping abilities. Journal of the science of food and agriculture 93, 2037–2044, https://doi.org/10.1002/jsfa.6012 (2013).
Kusirisin, W. et al. Antioxidative activity, polyphenolic content and anti-glycation effect of some Thai medicinal plants traditionally used in diabetic patients. Medicinal chemistry 5, 139–147 (2009).
Dey, A. & Lakshmanan, J. The role of antioxidants and other agents in alleviating hyperglycemia mediated oxidative stress and injury in liver. Food & function 4, 1148–1184, https://doi.org/10.1039/c3fo30317a (2013).
Bakala, H., Hamelin, M., Mary, J., Borot-Laloi, C. & Friguet, B. Catalase, a target of glycation damage in rat liver mitochondria with aging. Biochimica et biophysica acta 1822, 1527–1534, https://doi.org/10.1016/j.bbadis.2012.05.016 (2012).
Mofidi Najjar, F., Taghavi, F., Ghadari, R., Sheibani, N. & Moosavi-Movahedi, A. A. Destructive effect of non-enzymatic glycation on catalase and remediation via curcumin. Archives of biochemistry and biophysics 630, 81–90, https://doi.org/10.1016/j.abb.2017.06.018 (2017).
Yan, H. & Harding, J. J. Glycation-induced inactivation and loss of antigenicity of catalase and superoxide dismutase. The Biochemical journal 328(Pt 2), 599–605, https://doi.org/10.1042/bj3280599 (1997).
Anwar, S., Khan, M. A., Sadaf, A. & Younus, H. A structural study on the protection of glycation of superoxide dismutase by thymoquinone. International journal of biological macromolecules 69, 476–481, https://doi.org/10.1016/j.ijbiomac.2014.06.003 (2014).
Zhang, L. et al. The absorption, distribution, metabolism and excretion of procyanidins. Food & function 7, 1273–1281, https://doi.org/10.1039/c5fo01244a (2016).
Spencer, J. P. et al. Epicatechin is the primary bioavailable form of the procyanidin dimers B2 and B5 after transfer across the small intestine. Biochemical and biophysical research communications 285, 588–593, https://doi.org/10.1006/bbrc.2001.5211 (2001).
Pires, D. E., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. Journal of medicinal chemistry 58, 4066–4072, https://doi.org/10.1021/acs.jmedchem.5b00104 (2015).
Yamakoshi, J., Saito, M., Kataoka, S. & Kikuchi, M. Safety evaluation of proanthocyanidin-rich extract from grape seeds. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 40, 599–607 (2002).
Bak, M. J. et al. Antioxidant and Hepatoprotective Effects of Procyanidins from Wild Grape (Vitis amurensis) Seeds in Ethanol-Induced Cells and Rats. International journal of molecular sciences 17, 758, https://doi.org/10.3390/ijms17050758 (2016).
Yu, C. L. & Swaminathan, B. Mutagenicity of proanthocyanidins. Food and Chemical Toxicology 25, 135–139, https://doi.org/10.1016/0278-6915(87)90146-3 (1987).
Li, Q. et al. Proanthocyanidins, Isolated from Choerospondias axillaris Fruit Peels, Exhibit Potent Antioxidant Activities in Vitro and a Novel Anti-angiogenic Property in Vitro and in Vivo. Journal of agricultural and food chemistry 64, 3546–3556, https://doi.org/10.1021/acs.jafc.6b00236 (2016).
Zou, Y., Chang, S. K., Gu, Y. & Qian, S. Y. Antioxidant activity and phenolic compositions of lentil (Lens culinaris var. Morton) extract and its fractions. Journal of agricultural and food chemistry 59, 2268–2276, https://doi.org/10.1021/jf104640k (2011).
Justino, A. B. et al. Annona muricata Linn. leaf as a source of antioxidant compounds with in vitro antidiabetic and inhibitory potential against alpha-amylase, alpha-glucosidase, lipase, non-enzymatic glycation and lipid peroxidation. Biomed Pharmacother 100, 83–92, https://doi.org/10.1016/j.biopha.2018.01.172 (2018).
Benzie, I. F. & Strain, J. J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Analytical biochemistry 239, 70–76, https://doi.org/10.1006/abio.1996.0292 (1996).
Larmonier, N. et al. Freshly isolated bone marrow cells induce death of various carcinoma cell lines. International journal of cancer 107, 747–756 (2003).
Lee, J. Y. & Park, W. Anti-inflammatory effects of oroxylin A on RAW 264.7 mouse macrophages induced with polyinosinic-polycytidylic acid. Experimental and therapeutic medicine 12, 151–156 (2016).
Shih, D. et al. P-189 atg16l1 Deficiency Leads to Mitochondria Defect and Increased Oxidative State in Mice and Human Macrophages. Inflammatory Bowel Diseases 22, S66–S67 (2016).
Franco, R. R. et al. Antioxidant and anti-glycation capacities of some medicinal plants and their potential inhibitory against digestive enzymes related to type 2 diabetes mellitus. Journal of ethnopharmacology 215, 140–146, https://doi.org/10.1016/j.jep.2017.12.032 (2018).
Justino, A. B. et al. Hepatoprotective properties of a polyphenols-enriched fraction from Annona crassiflora Mart. fruit peel against diabetes-induced oxidative and nitrosative stress. Journal of agricultural and food chemistry. https://doi.org/10.1021/acs.jafc.7b01355 (2017).
Ellman, G. L. Tissue sulfhydryl groups. Archives of biochemistry and biophysics 82, 70–77 (1959).
Aebi, H. Catalase in vitro. Methods in enzymology 105, 121–126 (1984).
Acknowledgements
The authors gratefully acknowledge the Institute of Biotechnology and Institute of Chemistry of the Federal University of Uberlândia for infrastructural support and Mário M. Martins for technical assistance in LC-MS analysis. A.B.J., R.R.F. and H.C.G.S. received graduate fellowships from Coordination for the Improvement of Higher Education Personnel (CAPES), A.L.S., F.S.E. received post-doctoral fellowships from CAPES and F.S.E is a grant recipient of National Council for Scientific and Technological Development (CNPq). This research was funded by the National Institute of Science and Technology in Theranostics and Nanobiotechnology—INCT-TeraNano (CNPq/CAPES/FAPEMIG, CNPq-465669/2014-0 e FAPEMIG-CBB-APQ-03613-17).
Author information
Authors and Affiliations
Contributions
A.B.J., R.M.F.S. and F.S.E. conceived the experiment(s), A.B.J., R.R.F., H.C.G.S. and A.L.S. conducted the experiment(s), A.B.J., R.R.F., H.C.G.S., A.L.S., R.M.F.S. and F.S.E. analyzed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Justino, A.B., Franco, R.R., Silva, H.C.G. et al. B procyanidins of Annona crassiflora fruit peel inhibited glycation, lipid peroxidation and protein-bound carbonyls, with protective effects on glycated catalase. Sci Rep 9, 19183 (2019). https://doi.org/10.1038/s41598-019-55779-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55779-3
- Springer Nature Limited