
Abstract
Schizophrenia (Scz), autism spectrum disorder (ASD) and intellectual disability are common complex neurodevelopmental disorders. Kainate receptors (KARs) are ionotropic glutamate ion channels involved in synaptic plasticity which are modulated by auxiliary NETO proteins. Using UK10K exome sequencing data, we interrogated the coding regions of KAR and NETO genes in individuals with Scz, ASD or intellectual disability and population controls; performed follow-up genetic replication studies; and, conducted in silico and in vitro functional studies. We found an excess of Loss-of-Function and missense variants in individuals with Scz compared with control individuals (p = 1.8 × 10−10), and identified a significant burden of functional variants for Scz (p < 1.6 × 10−11) and ASD (p = 6.9 × 10−18). Single allele associations for 6 damaging missense variants were significantly replicated (p < 5.0 × 10−15) and confirmed GRIK3 S310A as a protective genetic factor. Functional studies demonstrated that three missense variants located within GluK2 and GluK4, GluK2 (K525E) and GluK4 (Y555N, L825W), affect agonist sensitivity and current decay rates. These findings establish that genetic variation in KAR receptor ion channels confers risk for schizophrenia, autism and intellectual disability and provide new genetic and pharmacogenetic biomarkers for neurodevelopmental disease.
Similar content being viewed by others


Introduction
Schizophrenia and autism are common, highly heritable debilitating neurodevelopmental disorders which are often comorbid with intellectual disability. Advances in genomic technology suggest a role for both common and rare variants contributing to genetic risk1,2,3,4. For instance, whole genome and exome sequencing studies have indicated that ultra-rare Loss-of-Function (LoF) point mutations and indel and CNV variants that truncate proteins, are enriched in individuals with neuropsychiatric disease and are both inherited and de novo5,6,7,8,9. Such studies commonly find that the disrupted genes are involved in synaptic function, which has led to the increasingly used term ‘synaptopathy’ for brain disorders that are thought to arise from synaptic dysfunction10,11,12.
Kainate receptors (KARs) are ionotropic glutamatergic receptors which form functional ion channels by tetrameric combinations of five different subunits. GluK1-3 subunits form functional homomeric or heteromeric receptors while GluK4 and GluK5 participate as functional receptors when combined with GluK1-3 subunits. Each subunit consists of an extracellular amino terminal domain, an extracellular ligand binding domain, three transmembrane domains and a loop M2, and a C-terminal domain. Mutating amino acids in these conserved regions gives rise to changes in channel properties13 and can alter the affinity for external cations such as sodium and lithium14,15. KAR channel properties are also regulated by the presence of the KAR auxiliary subunits Neuropilin And Tolloid Like 1, NETO1, and Neuropilin And Tolloid Like 2, NETO216,17,18.
KARs contribute to generating post-synaptic excitatory responses and to shorter term synaptic plasticity mechanisms by influencing presynaptic transmitter release19. In recent years, their action in non-canonical G protein coupled signalling, new forms of Long Term Potentiation, and synaptic targeting mechanisms have also been recognised20,21,22,23. NETO1 and NETO2 are single pass transmembrane CUB domain-containing proteins which influence the trafficking of KAR subunits and kinetics of KAR function24. During development KARs and Netos are highly expressed in the brain and are important for synaptogenesis, neurite outgrowth and glutamatergic pathway connectivity25,26,27. Rodent knock out and transgenic models have provided some evidence that loss of these receptors influences brain function and behaviour in a manner analogous to human disease28,29,30,31,32,33. In humans, common, non-coding, intronic variants and private de novo mutations located within specific GluK subunit genes (e.g. GRIK1, GRIK2, GRIK3, GRIK4, GRIK5) have been associated with a broad spectrum of neurological diseases34,35,36,37,38,39 and GluK genotype-dependent changes in cognition, brain activation and response to antidepressant and antipsychotic treatments have been reported40,41,42,43. However, as yet no comprehensive screen of coding variants across all GluK genes or KAR auxiliary proteins have been performed in cohorts with intellectual disability and neurodevelopmental conditions.
We hypothesize that damaging coding alleles within KAR subunit and NETO genes contribute to risk for developing neurodevelopmental disorders. Here we characterised LoF variants, performed single allele association and burden enrichment analysis of damaging coding variants within KAR subunit and NETO genes, in individuals with schizophrenia, psychosis, autism and intellectual disabilities available from the UK10K project and ExAC study44. We subsequently examined the functional effect of predicted damaging missense variants within GluK2 and GluK2/GluK4 receptors using in silico modelling tools and in vitro electrophysiological assays.
Results
LoF and rare missense variants are increased within individuals with neurodevelopmental disorders
The pipeline followed for the genetic analysis is presented in Fig. 1. In a first discovery phase, we identified 154 non-synonymous variants which included 4 LoF and 150 missense variants and 143 regulatory variants within GRIK1-5 and NETO1-2 genes (Table 1). 265 variants had a MAF < 1% and were classified as rare or ultra-rare. We postulated that genes, which in the general population, are characterised as having few LoF and missense variants would carry high numbers of LoF and damaging variants in individuals with neurodevelopmental disorders as these variants putatively contribute to disease risk. In the ExAC project database, GRIK2, GRIK3, GRIK5 and NETO1 are all classified as LoF intolerant genes (LoF pLI > 0.90) indicating that these genes are extremely intolerant of Loss-of-Function variation and hence few LoF mutations are present in the general population. As detailed in Table 1, we identified 4 LoF variants within affected individuals who had either ID or ASD or comorbid with Scz (GRIK1 L411X, Scz comorbid with ID; GRIK4 S98X, ID; GRIK5 Q848X, ASD; and GRIK5 19:42546908 splice acceptor, variant, ASD), and two of which were within the LoF intolerant gene GRIK5. No LoF variants were identified in the control cohort.

Genetic analysis workflow. Data sets investigated are shown in blue, methods and analysis in green and findings are colored orange. Discovery phase one analyzed WES and WGS from cohorts with neurodevelopmental disorders (including severe neuropsychiatric conditions and ASD, ID and dual Scz-ID) and general population control cohorts. The second discovery phase investigated two additional schizophrenia cohorts. Details of the cohorts are presented in Supplementary Table S1. Single allele associations and burden analysis was performed. Associated alleles were followed up by performing case-control studies using the ExAC cohort non psychiatric control and psychiatric case populations. Abbreviations: ASD, autism spectrum disorders; ExAC, Exome Aggregation Consortium; FINSCZ, Finnish schizophrenia samples; ID, Intellectual Disability; MAF, Minor Allele Frequency; Scz, schizophrenia; SNV, Single-Nucleotide Variant; WES, Whole-Exome Sequencing; WGS, Whole-Genome Sequencing.
ExAC also categorizes GRIK2, GRIK3, GRIK4, GRIK5 and NETO1 as missense intolerant genes (missense z > 2.80) again suggesting that fewer missense mutations are present in the general population within these genes than expected. Of the 150 missense variants identified in the current study, 75 were considered as protein damaging, 40 as possibly damaging variants and 35 as benign. The number of rare and ultra-rare nonsynonymous variants within affected individuals was found to be higher than the number of rare nonsynonymous variants found within control individuals or shared despite the fact that the control population has twice the number of individuals (affected case frequency 0.58; control frequency 0.26; variants found in both cases and controls frequency 0.16) (Supplementary Tables S4–S7). As indicated in Fig. 2, the majority of LoF and predicted damaging missense variants were identified in individuals with Scz and ASD whilst most of the predicted benign missense variants were found either in controls or were shared between cases and controls subjects (Supplementary Table S8). In contrast to rare LoF and missense mutations, an equal frequency of common non-synonymous, synonymous and regulatory variants was found in both case and control individuals. The most common variants identified (MAF > 0.1) were synonymous variants (87.5%) and present in both case and control individuals.

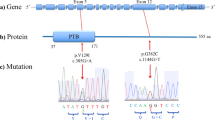
Frequency and effect size of functional variants and the location of non-synonymous variants within GRIK2 and GRIK4. (A) Individuals with severe neuropsychiatric disease (i.e. conditions with psychosis) and ASD have a higher percentage of rare functional variants compared to control individuals in the first discovery study. (B) Individuals with schizophrenia have a have a higher percentage of rare functional variants compared to control individuals in the second discovery study. (C) LoF and missense variants have a larger risk effect size than regulatory variants in genes which are classified as LoF and missense intolerant. (D) Risk effect sizes are smaller for regulatory variants and benign missense variants compared to LoF and damaging missense variants. (E) The location of LoF and damaging missense variants identified within GRIK2 and GRIK4 proteins in affected and control individuals. Protein domains are colour coded with the ATD as blue; LBDs (S1 and S2) as black; transmembrane domains M1-M3-M4 red; M2 loop as red and the C-terminal domain (CTD), green. Damaging missense variants carried in cases or controls or are colour coded as blue and black respectively. (F) The location of three damaging missense variants, GluK2 K525E, GluK4 Y555N, and GluK4 L825W. The term ‘shared’ denotes variants found within both case and control groups. Abbreviations: ATD, amino terminal domain; LoF pLI, propability for LoF intolerance score.
We also found that the effect size of rare LoF and missense variants within LoF intolerant genes (LoF pLI > 0.90), was larger than the effect size (0.3 compared to −0.2) of rare LoF and missense variants within genes with a low LoF tolerance metric (Fig. 2d). In addition, as shown in Fig. 2 and Supplementary Fig. S1, rare LoF and missense variants were indicated to have a larger effect size compared to variants classified as regulatory and the effect size of rare regulatory variants did not differ between LoF intolerant and LoF tolerant genes.
Single coding alleles associated with risk or protection against developing disease
Two single coding variants were found to be significantly associated with disease (Table 1 and Supplementary Figs. S2, S3). GRIK3 S310A, located in the amino terminal domain (ATD), was found to be protective against a broader neurodevelopmental phenotype, e.g. conditions with psychosis and ASD/ID phenotypes (p = 1.01 × 10−18; OR = 0.59, CI 0.52–0.66,). GRIK3 F586V, was associated with risk of developing ASD (p = 2.84 × 10−7; OR = Inf). In addition, A895G located within the cytoplasmic protein domain of GRIK5, was found protective at a nominal level of significance against developing schizophrenia (p = 4.06 × 10−5; OR = 44.83, CI 2.70–765).
Enrichment of coding variants within GRIK and NETO genes
We analysed the burden and accumulation rates of functional variants and found a significantly increased burden of LoF, missense and regulatory variants which included all allelic frequencies in the case population (p = 3.38 × 10−20). Similarly, and as detailed in Table 2, we also found an increased burden of ultra-rare and rare LoF, missense and regulatory variants (p = 2.07 × 10−15). When comparing a broader neurodevelopmental phenotype, e.g. ASD, psychosis and ID, or a narrower psychosis phenotype with control individuals, we observed a significantly increased burden of LoF and missense variants within all GRIK and NETO genes (broad neurodevelopmental, all allele frequencies, p = 2.97 × 10−10; broad neurodevelopmental, ultra-rare and rare p = 6.02 × 10−7; psychosis, all frequencies; p = 6.17 × 10−7; psychosis, ultra-rare and rare p = 1.83 × 10−7). We also found a significantly increased burden of LoF, missense and regulatory variants at all allele frequencies (p = 6.86 × 10−18) and for ultra-rare and rare variants alone (p = 1.30 × 10−9) for the combined intellectual disability/ASD cohorts. However, although we found a significant burden of common and rare variants (p = 3.15 × 10−11) for ASD/ID, ultra-rare and rare variants alone did not reach genome-wide level of significance (p = 0.026).
Burden analysis was also performed for each individual gene (Supplementary Tables S9–S11). GRIK3, GRIK5 and NETO1, three genes classified as LoF and missense intolerant, were indicated as having an increased burden of functional variants (common and rare variants combined for GRIK3 p = 1.26 × 10−5; NETO1 p = 4.66 × 10−16; and rare variants only for GRIK5 p = 9.99 × 10−6). We also assessed variant load per gene level grouped by either severe neuropsychiatric phenotypes, i.e. conditions with psychosis, or ASD/ID phenotypes. For the psychosis grouping, we observed a genome-wide significant burden of rare LoF and missense variants within GRIK5 (p = 7.83 × 10−10) and an increased burden of common and rare functional variants within NETO1 (p = 6.76 × 10−6). For ASD and ID samples, we identified a genome-wide or nominal significant burden of common and rare LoF, missense and regulatory variants within GRIK3 (p = 3.31 × 10−13), GRIK1 (p = 1.20 × 10−5) and NETO1 (p = 2.79 × 10−12).
GRIK and NETO genetic associations in two additional schizophrenia cohorts
In a second discovery phase, we investigated the exomes of two additional schizophrenia datasets, the UKSCZ (N = 553) and FSZNK (N = 285) cohorts. Unlike in the first discovery phase where LoF variants were identified in individuals with ID or ASD, we did not identify any LoF variants in this second phase. This may relate to the fact that all affected individuals had a diagnosis of Scz and not ID or ASD. However, we detected 197 coding variants of which 97 were missense variants. 58 missense variants were predicted damaging and 34 (59%) of these damaging missense variants were identified within affected individuals only (Supplementary Tables S12–S16). As before, we investigated GRIK and NETO single coding alleles for association with risk or protection against Scz (Figs. S1–S3). A splice variant within GRIK2 6:102337505, c.1525–10 C > T (p = 4.43 × 10−8; OR = 42.26, CI 2.53–704) and a missense variant within GRIK3, R865G, (p = 6.8 × 10−6; OR = 73, CI 4–1226) showed a significant nominal association with risk for schizophrenia (Table 1).
Consistent with our previous findings for a psychosis phenotype, we observed that GRIK and NETO genes had high accumulation rates of functional coding variants (Table 2, Supplementary Table S17). For instance, we found a significantly increased burden of missense and regulatory variants at all allele frequencies (p = 1.26 × 10−25), and a significantly increased burden of just missense variants (p = 4.39 × 10−15). In addition, a higher burden of common and rare missense variants were found within GRIK3 (p = 5.24 × 10−10) and both NETO1 and NETO2 genes had higher accumulation rates of common and rare regulatory variants (p = 1.60 × 10−28 and p = 8.03 × 10−10). We also observed a burden of rare regulatory variants within GRIK5 (p = 3.37 × 10−5).
Evidence for the robustness of allele associations
To assess the robustness of significantly associated single alleles identified in discovery phases, we compared allelic frequencies of rare and common coding or splicing variants in the UK10K discovery phase affected cases with non-affected exomes from ExAC cohorts (N = 45,376). We identified 8 genome-wide significant associations (3 missense, 4 synonymous and 1 splice site) listed in Table 1 and Supplementary Table S18 validating our previous findings. Of the 3 missense variants, two had significant associations for protection against neuropsychiatric disease, GRIK3 S310A (p = 6.49 × 10−50; OR = 0.51, CI 0.47–0.56) and GRIK1 L902S (p = 4.83 × 10−15; OR = 0.40, CI 0.32–0.48), whilst one showed a significant association for risk for neuropsychiatric disease, GRIK5 A895G (p = 8.55 × 10−24; OR = Inf). We also identified 6 variants associated at the nominal level of significance (3 missense, 1 synonymous and 2 splice site variants), Supplementary Table S18.
Finally, using data from ExAC for well individuals (N = 45,376) and from the psychiatric disease arm of the ExAC study (N = 15,328) which includes individuals with additional neurological and psychiatric conditions, e.g. Tourette’s syndrome, and hence relates to a yet broader neurodevelopmental phenotype, we compared allele frequencies for all damaging coding variants which we had previously identified in the earlier phases of the study. We observed nine significant associations (p < 2 × 10−34); 5 missense variants, 3 synonymous and one splice variant, details of which are provided in Table 1 and Supplementary Table S19. The levels of significance (1 × 10−6 to 1 × 10−108) reflects the power to detect associations with very large sample numbers, e.g. N = 45,000, and is consistent with values reported for individual variants studied in large cohorts44,45,46.
Consistent with our previous findings, we again observed a significant difference in allele frequencies for the GRIK3 S310A variant (non-affected individuals MAF 0.29, disease MAF 0.25; p = 4.07 × 10−35; OR 0.81, CI = 0.80–0.84) and nominal significance for GRIK3 R865G variant (non-affected individuals MAF 0.004, disease MAF 0.006; p = 2.07 × 10−5; OR 1.49, CI = 1.22–1.73).
In silico and in vitro assays of rare variants within GluK2 and GluK4 TMD and LBD domains support a functional effect
Three predicted damaging missense mutations identified in individuals with schizophrenia and located within ‘key’ ligand binding (GluK2 K525E) and transmembrane (GluK4 L825W; GluK4 Y555N) domains of GluK2 and GluK4 subunits, were examined using in silico modelling tools. We found that GluK4 Y555N disrupted a hydrogen bond and resulted in a significant destabilizing thermodynamic effect (∆∆G = 1.65). GluK4 L825W did not affect the formation of hydrogen bonds but did however, have a slightly destabilizing effect on the total energy (∆∆G = 0.755). The GluK2 LBD K525E mutation led to creation of a hydrogen bond but no predicted observable thermodynamic effect was predicted (∆∆G = 0.06), shown in Supplementary Figure S4. However, a decrease in predicted positive electrostatic potential was observed over the ligand binding domain area of the GluK2 K525E variant (Supplementary Figure S5) and which could influence cell surface expression47. Taken together, in silico protein modeling analysis suggests that these three predicted damaging mutations could affect protein conformation, structural relationships or electrostatic potential.
To further study functional changes, we expressed wild-type and mutated GluK2 homomers and GluK2/GluK4 heteromers in Xenopus oocytes and measured their current responses to application of glutamate using a voltage-clamp. Currents rose to a peak then decayed to a steady state (Fig. 3b). Because of the temporal limitations of this system, currents represent a mixture of activation, desensitization and deactivation processes but presumably with activation dominating the rising phase and desensitization and deactivation dominating the decaying phase. We measured the agonist sensitivity of the peak current (EC50) and the current decay rates (τdecay) and although these current kinetics are not physiological, we compared changes in responses between wild-type and mutant receptors (Fig. 3; Supplementary Table 20).

Altered channel properties of GluK2 and GluK4 mutants. (A) Top: Glutamate concentration-response curves for wild type GluK2 and GluK2/GluK4 receptors and the mutated GluK2/GluK2 K525E, GluK2/GluK4 Y555N and GluK2/GluK4 L825W subunit combinations. Points are mean % of maximum peak response ± SEM and curves are fits of the Hill equation. Bottom: Comparisons of pEC50 for wild type and mutated GluK2 K525E, GluK4 Y555N, GluK4 L825W KARs. (B) Top: Two microelectrode voltage clamp (TEVC) traces for wild type and mutant GluK2 K525E, GluK4 Y555N, GluK4 L825W KARs in response to a 10-s application (bar) of 1 mM glutamate (for wild type and mutated GluK2 receptors) or 0.1 mM glutamate (for wild type and mutated GluK2/GluK4 receptors) at −80 mV. The responses have been scaled such that the peak responses are equal to aid comparison of the current decay kinetics, hence there are no vertical scale bars. Bottom: Comparisons of current decay τ1 values for wild type and mutated GluK2 K525E, GluK4 Y555N, GluK4 L825W KARs. Statistically significant differences are indicated by *(p < 0.05), **(p < 0.01) or ****(p < 0.0001). Abbreviations: Glu, glutamate; hGluK2, human GluK2; KA, kainic acid, pEC50: negative logarithm of the EC50 value.
GluK4 L825W reduced glutamate potency of peak current generation for GluK2/GluK4 receptors by 42.6 -fold (p = 0.0001, n = 10–18). Similarly, the GluK4 Y555N mutant decreased glutamate potency for GluK2/GluK4 receptor peak currents by 15 -fold (p = 0.0001, n = 9–13). The GluK2 K525E LBD variant was not functional by itself but when co-expressed with GluK2, glutamate potency of GluK2 homomers was decreased by 4.5-fold (p < 0.05, n = 15–17) (Fig. 3a, b). These results imply decreased KAR channel activity.
The rate of current decay of GluK2/GluK4 L825W and GluK2/GluK4 Y555N heteromers was found to be 2.3-fold slower (p = 0.047 and p = 0.002 respectively) than for GluK2/GluK4 wild type receptors upon application of 0.1 mM glutamate (Fig. 3, Supplementary Table S20). This would suggest that both GluK4 mutations may result in mildly increased function through the channels potentially remaining open for a prolonged time. GluK2 K525E did not significantly affect the rate of current decay. Taken together, our observations support that there is likely an overall decreased function in all mutants.
Discussion
Our findings from an integrated analysis of ~4,580 genomes investigated in two discovery phases, supports the hypothesis that LoF and damaging variants within KAR subunit and NETO genes are enriched in individuals with schizophrenia, autism and ID. Our observations of a specific candidate gene set are congruent with recent large scale whole genome and exome studies of individuals with schizophrenia and schizophrenia with ID which report an increased burden of ultra-rare coding and common variants in genes characterised as missense and LoF variant depleted genes9,48. Likewise, our findings provide further support that, in addition to rare de novo variation as a strong causative factor for autism5,49, inherited LoF and damaging mutations can confer risk for autism and ID. We also confirm evidence that Scz, ASD and ID phenotypes share genetic predisposing factors and neuropathology50,51,52, and that variants with a spectrum of allele frequencies and effect size within GRIK and NETO genes contribute to these phenotypes.
We identified LoF and damaging missense variants across key protein domains of KAR subunits involved in specific functions. The majority of the significantly associated replicated alleles were found to be protective, e.g. GRIK3 S310A and are novel targets for future genetic studies. Our electrophysiological findings supported the idea that both LBD and TMD mutants altered channel gating behavior. However, variants may also impact upon KAR function by a number of alternative means. For instance, disruption of KAR and NETO interaction may affect the synaptic localisation of KARs53,54. Similarly, KAR CTD alterations could inhibit N-cadherin interaction and thereby influence synaptic compartmentalization and recruitment of KARs to the membrane55 and mutations disrupting C terminal PDZ ligand binding might influence secretory pathway processes, feedback systems and neuronal activity20. These synaptic-population and subcellular specific processes highlight the importance of KAR subunit availability and how variants, whether coding or acting through epigenetic mechanisms, could affect the KAR spatio-temporal patterns which may cause downstream alterations in the glutamate neurotransmission.
As with many NGS studies, a limitation of this study was that genomic coverage was dependent on read depth and quality of sequencing. Several regulatory UTR variants had to be excluded as they were not consistently called across cohorts and hence our results may have missed important regulatory KAR and NETOs alleles contributing to disease risk. For instance, we excluded an indel within the 3′UTR of GRIK4 which we previously reported confers protection against developing bipolar disorder through altering GluK4 RNA and protein abundance36,37. Furthermore, intronic variants which were not assessed within GRIK4 and GRIK3 have also shown significantly association with response to antidepressant and antipsychotic medication42,56,57,58.
Case-control GWAS studies of individuals with schizophrenia have recently indicated common alleles within transcripts of the C4 genes, which encodes a complement component 4 protein, and SNAP25, a vesicle fusion protein, contribute to risk of disease59,60. Both SNAP25 and members of a second complement cascade protein family (C1ql2 and C1ql3) are located at postsynaptic sites and bind to KAR subunits and thereby regulate KAR ion channel behaviour61,62. Further exploration of this emerging genetic risk pathway may aid in the development of new drugs to target neurodevelopmental conditions. Based on our findings, and with the need for translation from genetic risk factors to clear biomarkers of treatment response and disease prognosis, future studies using large population cohorts with collated phenotypic, genomic, medical and medication data (e.g. the UK biobank and USA ‘All of Us research program’) should involve the detailed characterisation of KAR and NETO risk alleles.
Methods and Materials
Individuals with a clinical diagnosis of neurodevelopmental disorders were exome sequenced as part of the neurodevelopmental collections in the UK10K sequencing project (further details are provided in Supplementary Table S1). Ethical approval for genomic research and informed consent from all participants and/or their legal guardians was obtained previously by the UK10K consortium committee. The UK10K project was conducted in compliance with the Declaration of Helsinki statement of ethical principles. Access to the sequencing datasets was granted to Dr Knight under the UK10K Project access agreement ID5574. All individuals sequenced were of European ancestry.
In a first discovery phase, we examined approximately 846 individuals with schizophrenia or psychosis, 550 individuals with ASD, 124 individuals with intellectual disability, and individuals with a dual diagnosis of either ASD comorbid with ID (77) or psychosis comorbid with ID (175) (Fig. 1). The numbers assessed vary owing to exclusion of poor sequencing data within different KAR/NETO genes. In a second discovery phase, two additional schizophrenia cohorts, (the NEURO UKSCZ N = 553; and NEURO FSZNK N = 285) were investigated. Population controls came from the population control arms of the UK10K project (TwinsUK10K, Obesity UK10K; N = 2,095). In a follow-up phase of the study we attempted to replicate allelic associations by first comparing MAFs of variants identified in the case discovery cohorts with MAFs reported for the ExAC general population (N = 45,376). Subsequently, we compared alleles of interest in the psychiatric (N = 15,328) and non-psychiatric (N = 45,376) arms of the ExAC cohort.
Variant call files (VCF) were obtained from the European Genome-phenome Archive. VCF files for the non-psychiatric arm of ExAC was available from the ExAC website. The Genotype-Tissue Expression Project was used to identify primary transcripts expressed in brain and both brain-expressed and canonical transcripts were examined (Supplementary Table S2). A minimum of 12x read depth was accepted as a quality control. Functional annotation of coding variants was performed using snpEff, snpSift and dbNSFP. Variants were classified as: LoF variants (stop-gained, frameshift and splice-disrupting variants); missense; and regulatory (synonymous, non-damaging splicing site variants within 10 bp surrounding the exon and 3′UTR or 5′UTR variants). LoF annotation was conducted using the LoF Transcript Effect Estimator (LOFTEE, version 0.2). Mutation Taster, Panther DB, Align GVGD and PolyPhen2 were used to predict whether missense mutations were damaging. Splicing effects were assessed using the Human Splicing Finder (HSF 3.0). Minor allele frequencies (MAFs) were calculated for variants identified in the discovery phases and compared with MAFs derived from general population databases, e.g. GnoMAD. Variants were classified as common (MAF > 0.05), low frequency (MAF = 0.05–0.01), rare (MAF = 0.01–0.001) and ultra-rare (MAF = 0.001–0.0001).
Single allele association analysis was performed using either the Fisher’s exact test or the chi square tests and were two tailed. P-values were adjusted for correction using the Holm-Bonferroni method and significance was set at two levels; a genome wide level (p < 5 × 10−8) and a less conservative nominal level (p < 1 × 10−6). Odds Ratios (ORs) and confidence interval values were calculated using R software (v.3.4.1). Kernel methods SKAT and SKAT-O were implemented to identify genes carrying a significant burden of common, rare, and rare damaging variants63. Imputation of missing wild-type genotypes was conducted by using IMPUTE2 software64. Structural templates used for in silico protein modelling were acquired either from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank or generated from Uniprot amino acid sequences using RaptorX software65- further details are provided in Supplementary Table S3. In silico mutagenesis was performed using the mutagenesis function of Pymol (PyMOL Version 2.0, Schrödinger, LLC) and predicted hydrogen bonds within 8 A were examined in wild type and mutated structures. Free energy calculations (ΔG) were performed using FoldX v3.066. The Adaptive Poisson-Boltzmann Solver (APBS) software package was used to study surface potential changes in the electrostatic surface potential in mutant proteins67.
Wild-type and mutant KARs (GluK2 K525E, GluK4 Y555N, and GluK4 L825W) were expressed in Xenopus oocytes using methods described previously68. Xenopus oocytes were supplied as ovarian lobes by the European Xenopus Resource Centre, University of Portsmouth, UK. Animal care and treatment were conducted in compliance with national and international laws and policies. The electrophysiology research protocol was performed in accordance with the University of Nottingham institutional guidelines and regulations. Human cDNA clones for GluK2 and GluK4 were obtained from GenScript (USA). Mutations were introduced into constructs using the QuikChange II Mutagenesis kit (Agilent Technologies) and cRNA generated using a mMessage mMachine kit (Invitrogen). Oocytes were injected with 50.6 nL cRNA (250–300 ng/μL) and incubated in GTP solution. Oocytes were perfused with frog Ringer solution at 10 mL/min and two-electrode voltage-clamped at −80 mV (Geneclamp 500, Axon Instruments). Glutamate was perfused at a flow rate of 10 mL/min at concentrations ranging between 10−9 M and 10−3 M for 10 s. Peak current amplitudes were normalised and plotted against glutamate concentration to determine EC50. Current decay time constants (τ) were estimated over the 10-s glutamate application by fitting exponential equations. Experiments were performed in multiple cells (n ≥ 5).
Data availability
The datasets generated during and/or analysed during the current study are not publicly available due access being granted to Dr Knight under the UK10K Project access agreement ID5574 but further information concerning identified variants is available from the corresponding author on reasonable request.
References
Cross-Disorder Group of the Psychiatric Genomics, C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379, https://doi.org/10.1016/S0140-6736(12)62129-1 (2013).
Cross-Disorder Group of the Psychiatric Genomics, C. et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nature genetics 45, 984–994, https://doi.org/10.1038/ng.2711 (2013).
Ganna, A. et al. Quantifying the Impact of Rare and Ultra-rare Coding Variation across the Phenotypic Spectrum. American journal of human genetics 102, 1204–1211, https://doi.org/10.1016/j.ajhg.2018.05.002 (2018).
International Schizophrenia, C. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752, https://doi.org/10.1038/nature08185 (2009).
Genovese, G. et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nature neuroscience 19, 1433–1441, https://doi.org/10.1038/nn.4402 (2016).
Hamdan, F. F. et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. American journal of human genetics 88, 306–316, https://doi.org/10.1016/j.ajhg.2011.02.001 (2011).
Kirov, G. et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Molecular psychiatry 17, 142–153, https://doi.org/10.1038/mp.2011.154 (2012).
Moreno-De-Luca, D. et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. American journal of human genetics 87, 618–630, https://doi.org/10.1016/j.ajhg.2010.10.004 (2010).
Singh, T. et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nature genetics 49, 1167–1173, https://doi.org/10.1038/ng.3903 (2017).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215, https://doi.org/10.1038/nature13772 (2014).
Gilman, S. R. et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907, https://doi.org/10.1016/j.neuron.2011.05.021 (2011).
The, N. & Pathway Analysis Subgroup of the Psychiatric Genomics, C. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nature neuroscience 18, 199, https://doi.org/10.1038/nn.3922 https://www.nature.com/articles/nn.3922#supplementary-information (2015).
Weston, M. C., Schuck, P., Ghosal, A., Rosenmund, C. & Mayer, M. L. Conformational restriction blocks glutamate receptor desensitization. Nature structural & molecular biology 13, 1120–1127, https://doi.org/10.1038/nsmb1178 (2006).
Dawe, G. B. et al. Defining the structural relationship between kainate-receptor deactivation and desensitization. Nature structural & molecular biology 20, 1054–1061, https://doi.org/10.1038/nsmb.2654 (2013).
Paramo, T., Brown, P., Musgaard, M., Bowie, D. & Biggin, P. C. Functional Validation of Heteromeric Kainate Receptor Models. Biophys J 113, 2173–2177, https://doi.org/10.1016/j.bpj.2017.08.047 (2017).
Straub, C. et al. Distinct functions of kainate receptors in the brain are determined by the auxiliary subunit Neto1. Nature neuroscience 14, 866–873, https://doi.org/10.1038/nn.2837 (2011).
Straub, C. & Tomita, S. The regulation of glutamate receptor trafficking and function by TARPs and other transmembrane auxiliary subunits. Curr Opin Neurobiol 22, 488–495, https://doi.org/10.1016/j.conb.2011.09.005 (2012).
Zhang, W. et al. A transmembrane accessory subunit that modulates kainate-type glutamate receptors. Neuron 61, 385–396, https://doi.org/10.1016/j.neuron.2008.12.014 (2009).
Lerma, J. & Marques, J. M. Kainate receptors in health and disease. Neuron 80, 292–311, https://doi.org/10.1016/j.neuron.2013.09.045 (2013).
Evans, A. J., Gurung, S., Wilkinson, K. A., Stephens, D. J. & Henley, J. M. Assembly, Secretory Pathway Trafficking, and Surface Delivery of Kainate Receptors Is Regulated by Neuronal Activity. Cell reports 19, 2613–2626, https://doi.org/10.1016/j.celrep.2017.06.001 (2017).
Negrete-Diaz, J. V., Sihra, T. S., Flores, G. & Rodriguez-Moreno, A. Non-canonical Mechanisms of Presynaptic Kainate Receptors Controlling Glutamate Release. Front Mol Neurosci 11, 128, https://doi.org/10.3389/fnmol.2018.00128 (2018).
Petrovic, M. M. et al. Metabotropic action of postsynaptic kainate receptors triggers hippocampal long-term potentiation. Nature neuroscience 20, 529–539, https://doi.org/10.1038/nn.4505 (2017).
Valbuena, S. & Lerma, J. Non-canonical Signaling, the Hidden Life of Ligand-Gated Ion Channels. Neuron 92, 316–329, https://doi.org/10.1016/j.neuron.2016.10.016 (2016).
Sheng, N., Shi, Y. S. & Nicoll, R. A. Amino-terminal domains of kainate receptors determine the differential dependence on Neto auxiliary subunits for trafficking. Proceedings of the National Academy of Sciences of the United States of America 114, 1159–1164, https://doi.org/10.1073/pnas.1619253114 (2017).
Jack, A. et al. Development of Cortical Pyramidal Cell and Interneuronal Dendrites: a Role for Kainate Receptor Subunits and NETO1. Molecular neurobiology, https://doi.org/10.1007/s12035-018-1414-0 (2018).
Orav, E. et al. NETO1 Guides Development of Glutamatergic Connectivity in the Hippocampus by Regulating Axonal Kainate Receptors. eNeuro 4, https://doi.org/10.1523/ENEURO.0048-17.2017 (2017).
Vernon, C. G. & Swanson, G. T. Neto2 Assembles with Kainate Receptors in DRG Neurons during Development and Modulates Neurite Outgrowth in Adult Sensory Neurons. J Neurosci 37, 3352–3363, https://doi.org/10.1523/JNEUROSCI.2978-16.2017 (2017).
Aller, M. I., Pecoraro, V., Paternain, A. V., Canals, S. & Lerma, J. Increased Dosage of High-Affinity Kainate Receptor Gene grik4 Alters Synaptic Transmission and Reproduces Autism Spectrum Disorders Features. J Neurosci 35, 13619–13628, https://doi.org/10.1523/JNEUROSCI.2217-15.2015 (2015).
Arora, V. et al. Increased Grik4 Gene Dosage Causes Imbalanced Circuit Output and Human Disease-Related Behaviors. Cell reports 23, 3827–3838, https://doi.org/10.1016/j.celrep.2018.05.086 (2018).
Catches, J. S., Xu, J. & Contractor, A. Genetic ablation of the GluK4 kainate receptor subunit causes anxiolytic and antidepressant-like behavior in mice. Behavioural brain research 228, 406–414, https://doi.org/10.1016/j.bbr.2011.12.026 (2012).
Micheau, J., Vimeney, A., Normand, E., Mulle, C. & Riedel, G. Impaired hippocampus-dependent spatial flexibility and sociability represent autism-like phenotypes in GluK2 mice. Hippocampus 24, 1059–1069, https://doi.org/10.1002/hipo.22290 (2014).
Shaltiel, G. et al. Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Molecular psychiatry 13, 858–872, https://doi.org/10.1038/mp.2008.20 (2008).
Xu, J. et al. Complete Disruption of the Kainate Receptor Gene Family Results in Corticostriatal Dysfunction in Mice. Cell reports 18, 1848–1857, https://doi.org/10.1016/j.celrep.2017.01.073 (2017).
Griswold, A. J. et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Human molecular genetics 21, 3513–3523, https://doi.org/10.1093/hmg/dds164 (2012).
Guzman, Y. F. et al. A gain-of-function mutation in the GRIK2 gene causes neurodevelopmental deficits. Neurol Genet 3, e129, https://doi.org/10.1212/NXG.0000000000000129 (2017).
Knight, H. M. et al. GRIK4/KA1 protein expression in human brain and correlation with bipolar disorder risk variant status. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 159B, 21–29, https://doi.org/10.1002/ajmg.b.31248 (2012).
Pickard, B. S. et al. A common variant in the 3′UTR of the GRIK4 glutamate receptor gene affects transcript abundance and protects against bipolar disorder. Proceedings of the National Academy of Sciences of the United States of America 105, 14940–14945, https://doi.org/10.1073/pnas.0800643105 (2008).
Pickard, B. S. et al. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Molecular psychiatry 11, 847–857, https://doi.org/10.1038/sj.mp.4001867 (2006).
Schiffer, H. H. & Heinemann, S. F. Association of the human kainate receptor GluR7 gene (GRIK3) with recurrent major depressive disorder. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 144B, 20–26, https://doi.org/10.1002/ajmg.b.30374 (2007).
Drago, A. et al. AKAP13, CACNA1, GRIK4 and GRIA1 genetic variations may be associated with haloperidol efficacy during acute treatment. Eur Neuropsychopharmacol 23, 887–894, https://doi.org/10.1016/j.euroneuro.2012.08.013 (2013).
Koromina, M., Flitton, M., Mellor, I. R. & Knight, H. M. A kainate receptor GluK4 deletion, protective against bipolar disorder, is associated with enhanced cognitive performance across diagnoses in the TwinsUK cohort. World J Biol Psychiatry, 1-9, https://doi.org/10.1080/15622975.2017.1417637 (2018).
Paddock, S. et al. Association of GRIK4 with outcome of antidepressant treatment in the STAR*D cohort. The American journal of psychiatry 164, 1181–1188, https://doi.org/10.1176/appi.ajp.2007.06111790 (2007).
Whalley, H. C. et al. A GRIK4 variant conferring protection against bipolar disorder modulates hippocampal function. Mol Psychiatry 14, 467–468, https://doi.org/10.1038/mp.2009.7 (2009).
Consortium, U. K. et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90, https://doi.org/10.1038/nature14962 (2015).
Ligthart, S. et al. Genome Analyses of>200,000 Individuals Identify 58 Loci for Chronic Inflammation and Highlight Pathways that Link Inflammation and Complex Disorders. American journal of human genetics 103, 691–706, https://doi.org/10.1016/j.ajhg.2018.09.009 (2018).
Xue, A. et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nature communications 9, 2941, https://doi.org/10.1038/s41467-018-04951-w (2018).
Scholefield, C. L., Atlason, P. T., Jane, D. E. & Molnar, E. Assembly and Trafficking of Homomeric and Heteromeric Kainate Receptors with Impaired Ligand Binding Sites. Neurochem Res 44, 585–599, https://doi.org/10.1007/s11064-018-2654-0 (2019).
Pardinas, A. F. et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nature genetics 50, 381–389, https://doi.org/10.1038/s41588-018-0059-2 (2018).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221, https://doi.org/10.1038/nature13908 (2014).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184, https://doi.org/10.1038/nature12929 (2014).
Gandal, M. J. et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697, https://doi.org/10.1126/science.aad6469 (2018).
McCarthy, S. E. et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Molecular psychiatry 19, 652–658, https://doi.org/10.1038/mp.2014.29 (2014).
Sheng, N., Shi, Y. S., Lomash, R. M., Roche, K. W. & Nicoll, R. A. Neto auxiliary proteins control both the trafficking and biophysical properties of the kainate receptor GluK1. eLife 4, https://doi.org/10.7554/eLife.11682 (2015).
Wyeth, M. S. et al. Neto auxiliary protein interactions regulate kainate and NMDA receptor subunit localization at mossy fiber-CA3 pyramidal cell synapses. J Neurosci 34, 622–628, https://doi.org/10.1523/JNEUROSCI.3098-13.2014 (2014).
Fievre, S. et al. Molecular determinants for the strictly compartmentalized expression of kainate receptors in CA3 pyramidal cells. Nature communications 7, 12738, https://doi.org/10.1038/ncomms12738 (2016).
Horstmann, S. et al. Polymorphisms in GRIK4, HTR2A, and FKBP5 show interactive effects in predicting remission to antidepressant treatment. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 35, 727–740, https://doi.org/10.1038/npp.2009.180 (2010).
Mas, S. et al. Pharmacogenetic study of antipsychotic induced acute extrapyramidal symptoms in a first episode psychosis cohort: role of dopamine, serotonin and glutamate candidate genes. The pharmacogenomics journal 16, 439–445, https://doi.org/10.1038/tpj.2016.44 (2016).
Pu, M. et al. Influence of genetic polymorphisms in the glutamatergic and GABAergic systems and their interactions with environmental stressors on antidepressant response. Pharmacogenomics 14, 277–288, https://doi.org/10.2217/pgs.13.1 (2013).
Li, Z. et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nature genetics 49, 1576–1583, https://doi.org/10.1038/ng.3973 (2017).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183, https://doi.org/10.1038/nature16549 (2016).
Matsuda, K. et al. Transsynaptic Modulation of Kainate Receptor Functions by C1q-like Proteins. Neuron 90, 752–767, https://doi.org/10.1016/j.neuron.2016.04.001 (2016).
Selak, S. et al. A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron 63, 357–371, https://doi.org/10.1016/j.neuron.2009.07.017 (2009).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. American journal of human genetics 91, 224–237, https://doi.org/10.1016/j.ajhg.2012.06.007 (2012).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics 5, e1000529, https://doi.org/10.1371/journal.pgen.1000529 (2009).
Peng, J. & Xu, J. Raptor X: exploiting structure information for protein alignment by statistical inference. Proteins 79(Suppl 10), 161–171, https://doi.org/10.1002/prot.23175 (2011).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic acids research 33, W382–388, https://doi.org/10.1093/nar/gki387 (2005).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America 98, 10037–10041, https://doi.org/10.1073/pnas.181342398 (2001).
Kachel, H. S., Patel, R. N., Franzyk, H. & Mellor, I. R. Block of nicotinic acetylcholine receptors by philanthotoxins is strongly dependent on their subunit composition. Scientific reports 6, 38116, https://doi.org/10.1038/srep38116 (2016).
Acknowledgements
The UK10K project is a major collaboration among several leading academic and research institutions including Bristol University, King’s College London, the Medical Research Council, UK Department of Health and the Wellcome Trust Sanger Insititute. This study makes use of data generated by the UK10K Consortium derived from samples from: UK10K RARE FIND, UK10K NEURO ASD GALLAGHER, UK10K NEURO ASD SKUSE, UK10K NEURO IOP COLLIER, UK1OK NEURO MUIR, UK10K NEURO EDINBURGH, UK10K NEURO ABERDEEN, UK10K NEURO GURLING, UK10K OBESITY GS, UK10K COHORT TWINSUK, UK10K COHORT IMGSAC, UK10K COHORT MGAS, UK10K NEURO UKSCZ, UK10K NEURO FSZNK. A full list of the investigators who contributed to the generation of the data is available from www.UK10K.org44. Funding for UK10K was provided by the Wellcome Trust under award WT091310. TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. This study was funded by the University of Nottingham and MK was funded by a University of Nottingham Vice-Chancellor’s Scholarship for Research Excellence (PhD European Union) scholarship.
Author information
Authors and Affiliations
Contributions
Conceptualization, H.M.K., I.R.M., M.K. Methodology, M.K., M.F., A.B., I.R.M., H.M.K. Software, M.K., M.F. Validation, M.K., M.F. Formal Analysis, M.K., M.F; Investigation, M.K., M.F., A.B., I.R.M., H.M.K; Resources, H.M.K., I.R.M. Data Curation, M.K., M.F. H.M.K. Writing – Original Draft, H.M.K., M.K., I.R.M. Writing – Review & Editing, H.M.K., M.K., I.R.M. Visualization, M.K. Supervision, H.M.K., I.R.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koromina, M., Flitton, M., Blockley, A. et al. Damaging coding variants within kainate receptor channel genes are enriched in individuals with schizophrenia, autism and intellectual disabilities. Sci Rep 9, 19215 (2019). https://doi.org/10.1038/s41598-019-55635-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55635-4
- Springer Nature Limited
This article is cited by
-
GluK1 kainate receptors are necessary for functional maturation of parvalbumin interneurons regulating amygdala circuit function
Molecular Psychiatry (2024)
-
Changes in ADAR RNA editing patterns in CMV and ZIKV congenital infections
BMC Genomics (2023)
-
Kainate receptor subunit 1 (GRIK1) risk variants and GRIK1 deficiency were detected in the Indian ADHD probands
Scientific Reports (2022)