
Abstract
Epigenetic modifications are known to play critical roles in the expression of genes related to differentiation and dedifferentiation. Histone lysine demethylase KDM5B (PLU-1) catalyzes the demethylation of histone H3 on Lys 4 (H3K4), which results in the repression of gene expression. KDM5B is involved in regulation of luminal and basal cell specific gene expression in breast cancers. However, the mechanisms by which KDM5B is regulated in breast cancer, in particular in response to post-translational signals is not well-defined. Here, we demonstrate that KDM5B is phosphorylated at Ser1456 by the cyclin-dependent kinase 1 (CDK1). Phosphorylation of KDM5B at Ser1456 attenuated the occupancy of KDM5B on the promoters of pluripotency genes. Moreover, KDM5B inhibited the expression of pluripotency genes, SOX2 and NANOG, and decreased the stem cell population in triple-negative breast cancer cell lines (TNBC). We previously reported that the tumor suppressor HEXIM1 is a mediator of KDM5B recruitment to its target genes, and HEXIM1 is required for the inhibition of nuclear hormone receptor activity by KDM5B. Similarly, HEXIM1 is required for regulation of pluripotency genes by KDM5B.
Similar content being viewed by others


Introduction
Triple Negative Breast Cancer (TNBC) is a breast cancer subtype that does not express the estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor-2 (HER2)1. Clinically, 70% of TNBC tumors belong to the basal subtype, which are more aggressive, less differentiated, and have enhanced metastatic capacity2. Although TNBC tumors have been reported to be more responsive to neoadjuvant chemotherapy than receptor-positive tumors, development of resistance to cytotoxic chemotherapy and early relapse are more common3. Due to the lack of effective therapies resulting in poor prognosis for patients with TNBC, understanding the underlying mechanisms of TNBC aggressiveness is of key importance and thus a major focus of research in the hopes that new therapeutic targets will be elucidated.
Cancer stem cells (CSCs) are a population of cells within tumors that exhibit multipotency, self-renewal, clonogenicity, and proliferative properties4. CSCs can be sorted by a unique phenotype of surface proteins such as CD44+CD24- and characterized by increased aldehyde dehydrogenase (ALDH) activity5,6. Published reports indicate that CSCs contribute to therapeutic-resistance and metastasis of malignant breast cancer cells7,8. Furthermore, biopsy samples from cancer patients that have higher percentages of CSCs are more likely to metastasize9. The CD44+/CD24−/ALDH+ population is enriched in certain TNBC subtypes6,10,11, and may contribute to the aforementioned phenotype of TNBC tumors.
Epigenetic mechanisms are key factors in the emergence of CSCs12. At the initial stage of cell fate determination, maintaining the balance of H3K4 methylation is essential13,14, and the choice between differentiation versus dedifferentiation partly depends on this dynamic balance of methylation markers. Epigenetic enzymes, such as KDM5B, play essential roles in this decision. Along this line, KDM5B, a demethylase of histone 3 methylated on lysine 4, directly regulates genes that control cell cycle, cell differentiation, and cell lineage in murine embryonic stem cells (mECs) during the early epiblast, thus influencing cell fate decisions15.
High expression of KDM5B in prostate and breast cancer cells supports a proliferative role of KDM5B16,17,18. shRNA knockdown of KDM5B inhibits proliferation of several cancer cell lines and xenograft models19,20,21. However, a tumor-suppressive role of KDM5B has also been reported in TNBC, partly due to its ability to bind and stabilize hypophosphorylated pRb22,23,24. We previously reported that a functional interaction between KDM5B and the tumor suppressor, HEXIM1, is required for HEXIM1 inhibition of nuclear hormone receptor activity25. These divergent roles of KDM5B suggest that the role of KDM5B is cell-type dependent and dictated by KDM5B interacting partners.
Cyclin-dependent kinase 1 (CDK1) is a critical regulator of the cell cycle and its activity is highest during G2/M transition. CDK1 inhibition resulted in attenuated growth of TNBC tumor xenografts that have elevated Myc expression by inducing the apoptotic response26. Paradoxically, high expression of CDK1 in clinical invasive breast cancer is associated with better DRFSC (Distant relapse-free survival)27, suggesting the CDK1 may also have divergent roles in cancer.
CDK1 has been reported to be a critical factor in maintaining pluripotency of human stem cells28. In this report, we show that CDK1 phosphorylated KDM5B and in doing so downregulated KDM5B recruitment to genes encoding SOX2 and NANOG. Downregulation of KDM5B expression resulted in increased Sox2 and Nanog expression, and increase in CSC population. However, the role of CDK1 is more complex, and reflect CDK1 regulation of the recruitment of another histone demethylase, KDM4B.
Results
Phosphorylation of KDM5B by CDK1
KDM5B, a H3K4me2/3 histone demethylase, has been reported to be overexpressed in human breast tumors and proposed to have an oncogenic role21,29,30. As a histone demethylase, KDM5B is involved in the transcriptional regulation of several genes, in particular those involved in differentiation and dedifferentiation30,31,32. Although phosphorylation has been reported to play roles in the regulation of other histone demethylases, regulation of KDM5B activity via phosphorylation has not been reported. Our immunoprecipitation experiments supported serine phosphorylation of KDM5B in a triple negative breast cancer cell line, MDA-MB-231 (Fig. 1a). Potential KDM5B phosphorylation sites were determined using KinasePhos. The CDK1 phosphorylation site was of specific interest due to its high score, and CDK1 was therefore explored for its involvement in the regulation of KDM5B phosphorylation. Serine phosphorylation of KDM5B was decreased in cells transfected with CDK1 shRNA when compared to control shRNA (Fig. 1a). Moreover, the phosphorylation of KDM5B was attenuated in RO3306-treated cells, a selective CDK1 inhibitor (Fig. 1b). Further support for phosphorylation of KDM5B by CDK1 is the interaction between CDK1 and KDM5B based on co-immunoprecipitation of endogenous CDK1 and KDM5B (Fig. 1c). In vitro CDK1 kinase assays were utilized to demonstrate that CDK1 catalyzed the phosphorylation of KDM5B, but not glutathione S-transferase (GST) alone (Fig. 1d).

KDM5B is serine phosphorylated by CDK1. MDA-MB-231 cells were infected with control shRNA lentiviruses or CDK1 shRNA lentiviruses. (a) Lysates from MDA-MB-231 cells were immunoprecipitated using antibodies against phosphorylated serine (pS) and analyzed for KDM5B by Western blotting. (b) MDA-MB-231 cells were treated with kinase inhibitors (Erk inhibitor, Vx: Vx-11e, 100 μM or CDK1 inhibitor, RO: RO3306, 10 μM) and lysates were immunoprecipitated using antibodies against KDM5B and analyzed for phosphorylated serine by Western blotting. (c) Lysates from MDA-MB-231 cells were immunoprecipitated using antibodies against KDM5B and CDK1 and analyzed for co-immunoprecipitation of CDK1 and KDM5B, respectively, by Western blotting. Normal rabbit IgG was used as a negative control. Input lanes represent 25% of the total protein. (d) Upper panel: In vitro kinase assay wherein recombinant cyclin B1 were incubated with purified GST-KDM5B in the absence or presence of CDK1 or ATP. Phosphoserine signal was detected by Western blotting. Lower panel: Western blot analyses of purified GST-KDM5B used in kinase assays. Figures are representative of at least 3 independent experiments.
Identification of CDK1 phosphorylation sites
To identify residues phosphorylated by CDK1 we used both mass spectrometry as well as in silico/predictive approaches. We used both approaches due to limitations of mass spectrometry33 and reports of functionally relevant phosphorylation sites not detected by mass spectrometry34,35,36,37,38.
In preparation for mass spectrometry analyses, recombinant cyclin B and CDK1 were incubated with purified GST-KDM5B in the presence of ATP. Reaction products were electrophoresed on a SDS-PAGE gel. The resulting gel was visualized with SYPRO Ruby, and gel bands were in-gel digested using trypsin prior to LC-MS analysis. Mass spectrometry analyses revealed S1328 as a putative phosphorylation site of CDK1 (Fig. 2a).

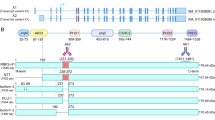
KDM5B is phosphorylated at S1456 and S1328. (a) Cyclin B, CDK1 and GST–KDM5B (1156–1544) were subjected to an in vitro kinase assay and analyzed by mass spectrometry. Shown is tandem mass spectra of phosphorylated peptides from KDM5B. Observed b- and y-series ions are shown in each spectrum. MS/MS spectrum of a peptide containing phospho-Ser1328 (precursor ion: m/z 716.8, +2 charge). (b) PRABI sequence alignment of orthologous KDM5B C-terminal region. MDA-MB-231 cells were transfected with expression vectors for FLAG-KDM5BWT, FLAG-KDM5BS1384A, FLAG-KDM5BS1456A, or FLAG-KDM5BS1328A. (c) Left panels: Lysates were immunoprecipitated using antibodies against phosphorylated serine. FLAG-KDM5B WT and mutants were detected by Western blotting using FLAG antibody. Center and right panels: Lysates were immunoprecipitated using FLAG antibody and the phosphoserine signal was detected by Western blotting. Normal rabbit IgG was used as a negative control. Input lanes represent 25% of the total protein. Figures are representative of at least 3 independent experiments.
As mentioned above, in silico prediction of KDM5B residues phosphorylated by CDK1 was carried out using KinasePhos, and the highest scoring sites identified using KinasePhos were also selected for further study. Common properties of CDK1 recognition motifs include localization in loops or highly disordered regions39. Among the predicted phospho-acceptor sites, S1384 and S1456, are conserved across different vertebrate species and are located in disordered region (Fig. 2b). Putative phosphorylation sites identified via the two approaches, serines at 1328, 1384, and 1456 were substituted with alanines. While phosphorylation of KDM5B was detected in cells transfected with expression vectors for wild type and KDM5BS1384A, phosphorylation of KDM5B was attenuated upon mutation of S1328 or S1456 (Fig. 2c).
Phosphorylation of KDM5B did not alter nuclear localization but attenuated target KDM5B occupancy and its ability to inhibit expression of pluripotency genes
It has been previously reported that AKT phosphorylated KDM5A, resulting in cytoplasmic retention of KDM5A. KDM5B was reported to be localized in cytoplasm during phases of the cell cycle phases wherein CDK1 is most active19. To investigate whether KDM5B phosphorylation by CDK1 alters KDM5B nuclear localization, subcellular fractionation was performed. Cytoplasmic localization of KDM5BS1456A (which cannot be phosphorylated by CDK1) was slightly increased compared to the wild type (Fig. 3a). Increased cytoplasmic localization of endogenous KDM5B was observed in shCDK1-transfected cells (Fig. 3b). Pharmacological inhibition of CDK1 using RO3306 also resulted in increased cytoplasmic localization of KDM5B. However, in both cases the levels of nuclear KDM5B were not significantly altered. These data suggest that CDK1 plays a minor role in the regulation of nuclear localization of KDM5B.

Phosphorylation of KDM5B by CDK1 does not significantly alter the nuclear localization of KDM5B. (a) Cell fractionation assay was conducted using lysates from MDA-MB-231 cells transfected with expression vectors for KDM5BWT, KDM5BS1456A, or KDM5BS1328A. The samples were analyzed by western blotting using the indicated antibodies. (b) Cell fractionation assay was conducted using lysates from MDA-MB-231 cells treated with RO3306 or infected with shCDK1 lentiviruses. The samples were analyzed by western blotting using the indicated antibodies. Tubulin served as cytoplasmic marker and HDAC1 served as nuclear marker. Figures are representative of at least 3 independent experiments.
It has been reported that KDM5B binds to the promoters and regulate expression of the core pluripotency regulators, Sox2 and Nanog, in mouse embryonic stem cells31. High expression of Sox2 has been associated with the early stages of breast tumor initiation and the development of cancer stem cell properties40. To determine whether phosphorylation impacts KDM5B recruitment and histone modifications on SOX2 and NANOG promoters, Chromatin IPs were performed. Downregulation of CDK1 resulted in enhanced recruitment of KDM5B and decreased H3K4me3 levels on SOX2 and NANOG promoters (Fig. 4a). To determine whether the altered recruitment can be attributed to KDM5B phosphorylation, relative recruitment of FLAG-tagged KDM5BWT and KDM5BS1456A were determined using the FLAG antibody. Increased occupancy of KDM5BS1456A was observed on both SOX2 and NANOG promoters when compared to the KDM5BWT cells (Fig. 4b). Mutation at S1328A did not alter KDM5B occupancy on the SOX2 and NANOG promoters. Consistent with phosphorylation at S1456 resulting in perturbation of KDM5B recruitment to the promoter regions of pluripotency genes, the phosphomimetic mutant KDM5BS1456D exhibited attenuated recruitment to the NANOG promoter (Fig. 4c). Altered recruitment of KDM5BS1456A and KDM5BS1456D when compared to KDM5BWT cannot be attributed to differences in expression levels of mutant and WT KDM5B (Fig. 4d).

Phosphorylation of KDM5B perturbs the occupancy of KDM5B on SOX2 and NANOG promoters. MDA-MB-231 cells were transfected as indicated with expression vectors for shCDK1, FLAG-KDM5BWT, FLAG-KDM5BS1456A, FLAG-KDM5BS1456D, or KDM5BS1328A. (a) ChIP analyses of lysates from MDA-MB-231 cells immunoprecipitated with antibodies against KDM5B, H3K4me3, or control non-specific rabbit immunoglobin, followed by PCR amplification of the promoter region of SOX2 and NANOG. (b,c) ChIP analyses of lysates from MDA-MB-231 cells immunoprecipitated with antibodies against FLAG or control non-specific mouse immunoglobin, followed by PCR amplification of the promoter region of SOX2 and/or NANOG. (d) Western blot analyses of cells transfected with expression vectors for FLAG-KDM5BWT, FLAG-KDM5BS1456A, FLAG-KDM5BS1456D, or KDM5BS1328A. Figures are representative of at least 3 independent experiments.
To verify that CDK1 mediated phosphorylation of KDM5B regulates expression of pluripotency regulators, quantitative real time PCR and western blot analyses were conducted. Expression of both Sox2 and Nanog were significantly decreased in cells with increased KDM5B expression. Conversely, expression of Sox2 and Nanog were increased in shKDM5B-transfected MDA-MB-231 cells (Fig. 5a,b). Different clones of shKDM5B were used to verify the effect of KDM5B and to delimit the role off-target effects (Fig. 5b). Downregulation of KDM5B also increased Nanog expression in other TNBC lines, MDA-MB-453, HCC70 and HCC1937 (Fig. 5c). However, downregulation of KDM5B had the opposite effect on luminal epithelial breast cancer cell lines, MCF-7 and T47D, and resulted in downregulation of Nanog expression in these cells (Supplemental Fig. S1). We were unable to detect Sox2 in these cells. Our observation of divergent actions of KDM5B on Nanog expression in luminal epithelial cells and TNBC are consistent with a report by Yamamoto et. al. that supports differences in KDM5B function in mammary luminal and basal cells21. Their study also provides support for a role of KDM5B as a luminal lineage driving oncogene. It is possible that KDM5B contributes to the maintenance of the luminal cell lineage, while repressing the basal cell lineage.

KDM5B mediates expression of pluripotency genes and role of CDK1 in KDM5B action. TNBC cells were transfected as indicated above with expression vectors for WT or mutant FLAG-KDM5B, shKDM5B, and/or shCDK1. Two clones each of shKDM5B (14758 and 14761) and shCDK1 (585 and 603) were used. (a) RNA was isolated and subjected to qRT-PCR to assess SOX2 and NANOG mRNA levels using GAPDH as a control. (b–d) Western blotting analyses of endogenous and/or transfected KDM5B, CDK1, Nanog, and/or Sox2 relative to loading control (GAPDH) in TNBC cell lines. Figures are representative of at least 3 independent experiments.
Unexpectedly, when CDK1 expression was downregulated, Sox2 and Nanog levels were significantly increased when compared to control shRNA-transfected cells. Downregulation of CDK1 also attenuated KDM5B-induced decreases in Sox2 and Nanog expression (Fig. 5a,c). Our data support repressive roles for KDM5B and CDK1 in the expression of Sox2 and Nanog in MDA-MB-231 cells.
Consistent with enhanced ability of the phosphodefective mutant, KDM5BS1456A, to bind to the NANOG promoter, this mutant was the most effective at attenuating Nanog expression (Fig. 5d). Mutation at S1328 did not alter the ability of KDM5B to downregulate Sox2 and Nanog expression.
CDK1-mediated phosphorylation of KDM5B is cell-cycle dependent
Because CDK1 is a key regulator of the cell cycle, we examined whether phosphorylation of KDM5B is altered during the phases of the cell cycle. For these studies our focus is on phosphorylation of S1456, because the phosphorylation of this residue was more relevant in the ability of KDM5B to regulate Nanog and Sox2 expression when compared to other residues. We first used in vitro and ELISA analyses to validate the phosphorylation of KDM5B at S1456 by CDK1, and to justify the generation of a S1456 phosphospecific antibody. The phosphorylation of KDM5B by CDK1 in an in vitro kinase was attenuated upon mutation at S1456 (Fig. 6a). These results were also evident from an ELISA assay (Supplemental Fig. S2) wherein GST antibody, which has been immobilized onto the solid surface of a 96-well plate, were complexed with in vitro kinase products, followed by the addition of the phosphoserine antibody. Phosphorylation of WT KDM5B, but not KDM5BS1456A, was evident from this colorimetric assay due to the ensuing interaction with HRP-conjugated secondary antibody, and the reaction between the HRP and the substrate tetramethylbenzidine, TMB.

Phosphorylation of KDM5B is cell-cycle dependent. (a) Upper panel: In vitro kinase assay wherein recombinant CDK1 and cyclin B1 were incubated with purified GST-KDM5BWT or GST-KDM5BS1456A. Phosphoserine signal was detected by Western blotting. Lower panel: Western blot analyses of purified GST-KDM5BWT or GST-KDM5BS1456A used in kinase assays. (b) Lysates from MDA-MB-231 and HCC1937 cells were analyzed by Western blotting using antibody against pKDM5B-S1456 in the absence or presence of phosphopeptide ERER[pS]YELVRSAETC. (c) Lysates from MDA-MB-231 cells treated with nocodazole (0.5 μg/ml, 18 hr) or hydroxyurea (2 mM, 12–16 hr) were analyzed by western blotting using indicated antibodies. Figures are representative of at least 3 independent experiments.
Examination of cell cycle dependent phosphorylation of KDM5B was aided by the development of an antibody to detect KDM5B phosphorylated at S1456, pKDM5B-S1456. pKDM5B-S1456 was detected in MDA-MB-231 cells and another TNBC line HCC1937 (Fig. 6b). The signal detected by the phosphospecific antibody was attenuated upon incubation with phosphopeptide encompassing pKDM5B-S1456 and surrounding residues. Activation of CDK1 by treatment of cells with nocodazole (which arrests cells in G2/M phase) resulted in the increased levels of pKDM5B-S1456 (Fig. 6c). Treatment of cells with hydroxyurea, which arrested cells near the G1/S boundary, did not result in increased levels of pKDM5B-S1456. Together our studies provided further support for phosphorylation of KDM5B by CDK1.
HEXIM1 attenuates KDM5B phosphorylation and is required for inhibition of the expression of pluripotency genes by KDM5B
We previously reported that the tumor suppressor HEXIM1 is a mediator of KDM5B recruitment to its target genes, and HEXIM1 is required for the inhibition of nuclear hormone receptor activity by KDM5B25. We also reported that induction of HEXIM1 expression by a small molecule inducer resulted in downregulation of Nanog expression and CSC fraction in MDA-MB-231 cells41. We determined if HEXIM1 is also a mediator of the regulation of the expression of pluripotency genes by KDM5B. Downregulation of HEXIM1 resulted in increased levels of pKDM5B-S1456 (Fig. 7a). Consistent with KDM5B phosphorylation as attenuating KDM5B occupancy and ability to inhibit expression of pluripotency genes, downregulation of HEXIM1 resulted in increased expression of Sox2 and Nanog (Fig. 7b). Moreover, ectopic expression of HEXIM1 rescued the effects of KDM5B downregulation, resulting in downregulation in expression of Sox2 and Nanog (Fig. 7c). These data further support the role of HEXIM1 as a critical mediator of the effects of KDM5B.

HEXIM1 counteracts is required for the inhibitory effects of KDM5B. MDA-MB-231 cells were transfected with expression vector for shHEXIM1, shKDM5B, and/or FLAG-HEXIM1. (a) Lysates were analyzed by Western blotting using indicated antibodies. (b) RNA was isolated and subjected to qRT-PCR or lysates were analyzed by Western blotting for expression of indicated genes or proteins, respectively. (c) RNA was isolated and subjected to qRT-PCR analyses of SOX2 and NANOG mRNA levels using GAPDH as a control. Figures are representative of at least 3 independent experiments.
KDM5B reduces the cancer stem cell frequency in TNBC
The regulation of the expression of the pluripotency regulators by CDK1 and KDM5B prompted us to investigate whether the cancer stem cell population is regulated by CDK1 and KDM5B. The CD44+/ALDH+ population was reduced in cells with increased KDM5B expression, and this reduction was reversed upon downregulation of CDK1 expression (Fig. 8a). Although the CD24- population was not altered by KDM5B (data not shown), mammosphere formation was regulated by KDM5B, and this action was dependent on the presence of CDK1 (Fig. 8b). Mammosphere formation was regulated by KDM5B in other TNBC lines, MDA-MB-453 and HCC1937 (Supplemental Fig. S3). Phosphodefective mutant KDM5BS1456A was more effective at inhibiting mammosphere formation than the phosphomimetic mutant KDM5BS1456D (Fig. 8c).

KDM5B induces a reduction in the CSC population in TNBC and role of CDK1 in KDM5B action. MDA-MB-231 cells were transfected as indicated with expression vectors for FLAG-KDM5BWT, shKDM5B, and/or shCDK1. Shown are (a) flow cytometry analyses of ALDH+ (aldehyde dehydrogenase) and CD44-APC (allophycocyanin) positive populations including mean fluorescence values (MFI) and (b) mammosphere formation assay. (c) MDA-MB-231 cells were transfected with expression vectors for FLAG-KDM5BWT, -KDM5BS1456A or -KDM5BS1456D. Transfected cells were cultured in mammosphere media and colonies were quantified. Figures are representative of at least 3 independent experiments.
We also assessed the functional role of KDM5B on TNBC cell invasion. We observed reduced invasion of MDA-MB-231 cells with increased KDM5B expression (Supplemental Fig. S4). Conversely, downregulation of KDM5B resulted in increased cell invasion. This effect was attenuated upon downregulation of CDK1 expression.
CDK1 also induces recruitment of KDM4B, which has inhibitory effects on Nanog expression and mammosphere formation
We then determined the basis for the unexpected finding that CDK1 inhibited expression of pluripotency genes and cancer stem cell frequency, despite inhibiting KDM5B recruitment to NANOG. A possibility is that KDM5B recruitment and attenuation of H3K4me3 levels on SOX2 and NANOG may not be the critical determinants of KDM5B regulation of the expression of pluripotency genes. However, this is unlikely because of the enhanced recruitment and repressive effects of KDM5BS1456A, indicating that CDK1-induced phosphorylation of KDM5B attenuated the inhibitory effects of KDM5B on the CSC population.
We examined the possibility that CDK1 induced recruitment of a transcription factor that inhibits Sox2 and Nanog expression, counterbalancing the negative effects of CDK1 on KDM5B recruitment. Another histone demethylase, KDM4B, has been shown to inhibit transcription by attenuating H3K36 methylation. CDK1 interacted with KDM4B and influenced recruitment of KDM4B to Nanog (Supplemental Fig. S5). Downregulation of CDK1 resulted in decreased KDM4B occupancy of Nanog, which can be correlated with increased H3K36me3 levels. An inhibitory effect of KDM4B on expression of Nanog is supported by our data that downregulation of KDM4B resulted in increased H3K36me3 levels on Nanog and increased Nanog expression. We did not find evidence for regulation of Sox2 expression by KDM4B. It is possible that other transcription factors, in addition to KDM5B, with similar functions as KDM4B are involved in the regulation of Sox2 expression by CDK1.
Discussion
Our findings support the repressive effects of KDM5B on the expression of pluripotency genes and the CSC population. CDK1 influenced this function by phosphorylating KDM5B, resulting in attenuated recruitment of KDM5B to pluripotency genes.
Patients with TNBC have poor clinical outcomes and limited therapeutic options3,42. The CD44+/CD24−/ALDH+ population is enriched in certain TNBC subtypes6,10,11, and CSCs are proposed to contribute to worse progression-free survival in TNBC patients6,43. CSCs are considered as major contributors to therapeutic resistance and metastasis of malignant breast cancer cells44,45. Chemotherapies have been demonstrated to stimulate CSC proliferation46, supported by the inhibition of mitotic kinase leading to the recurrence and enrichment of the CSC47,48. The high degree of plasticity in CSCs is maintained partly by reversible epigenetic modifications, including DNA methylation49,50.
Epigenetic modifications are known to play critical roles in the activation and repression of genes important for cell differentiation51,52. Perturbation of the balance among epigenetic modifications results in dysregulation of cancer related genes53. Published reports support a role for KDM5B in the maintenance or differentiation of normal and cancer stem cells31,54,55. In oral cancers, KDM5B enables the transition of cells into a stem-cell like phenotype56. Melanoma cells expressing high levels of KDM5B cycled slowly and possessed a high self‐renewal potential, while knockdown of KDM5B resulted in a reduction in tumorigenic activity57. On the other hand, KDM5B suppressed leukemogenesis and induced Acute Myeloid Leukemia (AML) cells to differentiate out of the leukemia stem cell compartment58. Other support for the pro-differentiation role of KDM5B include reports that knockdown of KDM5B in mESCs results in failure of neural differentiation and inability to efficiently silence stem- and germ-cell-related genes59. Depletion of KDM5B also impairs ESC differentiation to embryoid bodies, causing partial retention in a self-renewing stage. While conflicting, these findings support a role for KDM5B in cell fate decision. Our findings are consistent with a role for KDM5B in the reduction of the stem cell population in TNBC.
KDM5B is overexpressed in certain cancers, and has led to interest in the development of KDM5B inhibitors for clinical use60,61. In breast cancer, increased KDM5B expression drives a luminal cell-specific expression program21,62. Our data are consistent with the inhibition of basal-like cell enrichment by KDM5B24, given that a majority of TNBC belong to the basal subtype2. Along this line, KDM5B inhibited Sox2 expression that has been proposed to play a role in the development of a less differentiated phenotype of the basal subtype, with Sox2 expression being observed in 43% of basal-like breast carcinomas63.
While an oncogenic role for KDM5B in the luminal subtype of breast cancer has been reported, there are also reports of tumor suppressor action of KDM5B. Overexpression of KDM5B inhibited MDA-MB-231 cell migration and invasion by interacting with LSD1/NuRD complexes23. On the other hand, KDM5B has been reported to promote MDA-MB-231 cell migration and invasion by regulating MALAT1 and has-miR-448 expression64. However, the KDM5B species reported in this study does not appear to be full-length, and may complicate the interpretation of their findings on the effect of KDM5B on cell invasion. The conflicting reports on KDM5B action in cancer cells may be attributed to KDM5B target genes and interacting partners, which play diverse roles in development, differentiation, and the cell cycle15,30. Our data provide further support for HEXIM1 as critical in determining KDM5B function.
Post-translational modifications of histone methylases and demethylases have been receiving more attention. SKP2 inactivates KDM5B through ubiquitination in prostate cancer cells65. SUMOylation of KDM5B results in altered KDM5B occupancy at certain genes in HEK293T cells66. In luminal breast cancer cells, activated PI3K/Akt pathway phosphorylates another member of the KDM5B family, KDM5A, which results in increased localization of KDM5A to the cytoplasm and enhancement of its oncogenic role67. On the other hand, nuclear KDM5A functions as a tumor suppressor by exerting its demethylase activity68. Based on our observations, phosphorylation of KDM5B attenuates it ability to regulate expression of pluripotency genes and downregulate the CSC population. Whether the phosphorylation status of KDM5B results in different interaction partners and/or results in a conformational change in KDM5B that attenuate recruitment to target genes are subjects of our current investigation.
We observed that CDK1 phosphorylated KDM5B, which can be correlated with decreased recruitment to the promoter of SOX2 and NANOG. Thus, increased Sox2 and Nanog expression and the attenuation of the repressive effects of KDM5B on the expression of these pluripotency genes upon downregulation of CDK1 appears paradoxical. The paradoxical finding on CDK1 function may also be attributed to other targets of CDK169, which may counteract repressive actions of CDK1 on KDM5B. Our data suggest that this paradox may be attributed partly to CDK1-induced recruitment of another histone modifier, KDM4B, that has inhibitory effects on Nanog expression. Thus, the relative levels of KDM5B and KDM4B may determine how CDK1 influences the stem cell fraction.
While CDK1 has a well-known role in promoting cell proliferation, reports indicate divergent actions. CDK1 has been shown to inhibit the phosphorylation of PDK1, subsequently activating the PI3K/Akt pathway, and resulting in the development of pluripotency70. On the other hand, CDK1 phosphorylation inhibited the oncogenic activity of TAZ, a transcription factor that confers stem cell-like properties in breast cells71. CDK1 promoted mesenchymal stem cell differentiation in osteoblasts through phosphorylation of EZH2 and inhibition of its methyltransferase activity72. Divergent actions of CDK1 may contribute to the modest activity of CDK1 inhibitors exhibited in clinical trials73. This modest activity may be attributed to the development of drug resistance, which can be attributed to the CSC population. Our findings suggest a contributing factor to the unsatisfactory outcomes in clinical trials of CDK1 inhibitors.
Conclusions
In this study, we discovered that protein kinase CDK1 phosphorylates KDM5B at S1456, resulting in decrease occupancy of KDM5B on the promoter regions of pluripotency genes, leading to the suppression of H3K4 trimethylation. KDM5B inhibits the expression of these genes, which can be correlated with a reduction in the CD44+/CD24−/ALDH+ population in TNBC. Preclinical evidence suggests that CD44+/CD24−/ALDH+ population in TNBC is correlated with worse progression-free survival. Our findings provide insight into the pros and cons of targeting KDM5B or CDK1, in particular with regards to the development of drug resistance.
Methods
Cell culture, transfections and lentiviral infections
TNBC lines were obtained from the American Tissue Culture Collection on April 2017, and were maintained based on the instructions from ATCC. Cells were transfected with control or expression vector for FLAG-KDM5B (WT or mutants) using FuGENE HD (Promega) according to the manufacturer’s instructions. For treatment with Vx-11e and RO3306, concentrations of 100 μM and 10 μM were used, respectively. Plasmids used to generate KDM5B shRNA (shKDM5B), CDK1 shRNA (shCDK1) and KDM4B shRNA (shKDM4B) lentiviruses were obtained from Sigma-Aldrich. To generate lentiviral particles, 293FT cells were transfected with envelope expressing plasmid (pMD2.G), packaging plasmid (psPAX2) and shRNA expression plasmid (pLKO) using Lipofectamine 2000. Media containing lentiviral particles were collected 48–72 h post-transfection and used to transduce breast cancer cells for 12–16 hours. Puromycin was then used to select for cells expressing shRNAs.
Co-immunoprecipitations
Endogenous proteins were co-immunoprecipitated and analyzed as previously described25 and described in more detail in the Supplementary Methods.
Site-directed mutagenesis
pCMX-FLAG plasmid was derived from the pCMV mammalian expression plasmid74. Wild-type KDM5B cDNA was cloned into the pCMX plasmid. KDM5B mutants were generated using the QuikChange II XL Site-Directed Mutagenesis Kit according to the manufacturer’s protocol (Clontech; Palo Alto, CA). Primers sequences are provided in Table S1.
GST constructs and purification
To generate constructs for bacterial expression of wild-type and S1456A KDM5B in frame with glutathione-S-transferase (GST), cDNAs encoding C-terminal of KDM5B (1156–1544) were subcloned into the pGEX-4T vector. GST-KDM5B (wild-type and mutant) proteins were inducibly expressed in Escherichia coli strain BL21 and purified using Glutathione Sepharose beads (Amersham Biosciences). Expression of GST proteins were detected using GST monoclonal antibody (catalog number 2624, Cell Signaling Technology).
In vitro kinase assay
Recombinant cyclin B and CDK1 (0.1 μg, catalog number 14-450-D, Millipore) were incubated with 1 μg of purified GST-KDM5B (wild-type or mutant) in the presence of 100 μM ATP in kinase buffer (50 mM β-glycerophosphate, pH 7.4; 10 mM MgCl2; 10 mM NaF; 1 mM DTT) for 30 min at 30 °C. Reactions were stopped by adding equal volume of 10 mM EDTA and were kept in −20 °C until further analysis. Reaction products were analyzed using ELISA (as described below) or Western blotting and detected using phosphoserine antibody (catalog number AB1603, Millipore).
Determination of KDM5B phosphorylation sites via mass spectrometry
The in vitro phosphorylated KDM5B was separated on a SDS-PAGE gel and the protein band was visualized by Sypro Ruby. A band from the SDS-PAGE gel corresponding to KDM5B was excised, the protein was in-gel digested using trypsin75, and the digests were analyzed by data-dependent LC-MS/MS with collision-induced dissociation (CID) using a Thermo Fisher Scientific Fusion Lumos mass spectrometry system76. The HPLC column was a Dionex 15 cm × 75 μm id Acclaim Pepmap C18, 2 μm, 100 Å reversed-phase capillary chromatography column. The peptides were eluted from the column by an acetonitrile/0.1% formic acid gradient at a flow rate of 0.3 μL/min. The acquired mass spectrometry data was analyzed using MassMatrix database search software (Version 3.10, MassMatrix, Columbus, OH) to identify phosphorylated peptides and all the possibly phosphorylated peptides were manually verified by inspecting their MS/MS spectra.
ELISA assay
Ninety-six-well plates were coated (overnight, 4 °C) with 100 ng/well of GST monoclonal antibody (catalog number 2624, Cell Signaling Technology) and incubated with blocking buffer (2% BSA, 0.1% Tween 20) for 1 h. Serially diluted kinase products (generated as described above) were loaded onto the coated wells and incubated for 2 hours at room temperature, followed by the addition of 100 μl of phosphoserine antibody (1:500, catalog number 06-427, Millipore) and incubation for an additional 2 hours at room temperature. HRP-conjugated goat anti-rabbit IgG (1:10,000) in blocking buffer was added and incubated for 1 h at room temperature. Reaction products were detected by the addition of 100 μl chromogenic substrate (3,3’,5,5’- tetramethylbenzidine, TMB) for 30 min.
Plates were washed five times with washing buffer (PBS, pH 7.4, containing 0.02% (v/v) Tween 20). The reaction was stopped with 50 μl of 1 M HCl and absorbance at 450 nm was measured with reduction at 630 nm using an ELISA plate reader.
Cell fractionation assay
Cell were harvested and washed with 1X PBS, pelleted, and resuspended in cytosolic extraction buffer (CEB, 10 mM HEPES, 3 mM MgCl2, 20 mM KCl, 5% Glycerol, 0.5 mM DTT, 0.5% NP40, protease inhibitors and phosphatase inhibitors). Cytosolic extracts were obtained by passing the suspension through a 25-gauge needle ten times and by centrifugation at 6000 rpm for 10 min at 4 °C. The pellets were washed with CEB three times and resuspended in nuclear extraction buffer (NEB, 20 mM HEPES, 3 mM MgCl2, 225 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5 mM DTT, 0.5% NP40, protease inhibitors and phosphatase inhibitors). Nuclear extracts were obtained by passing the suspension through a 26-gauge syringe needle ten times and by centrifugation at 14,000 rpm for 10 min at 4 °C.
Chromatin immunoprecipitation and ChIP qRT-PCR
Cells were grown in 150 mm diameter dishes and processed for ChIP analyses as described previously77, and described in more detail in the Supplementary Methods. Primers sequences are provided in Table S178,79.
Real-time quantitative RT-PCR
Total mRNAs were extracted using TRIzol® reagent (Invitrogen) according to the manufacturer’s protocol. mRNAs were reverse transcribed using the M-MLV Reverse Transcriptase kit (Invitrogen) following the recommended protocol. Quantitative RT-PCR (qRT-PCR) reactions were performed in duplicate with SYBR Green Master Mix (catalog number 170-8882, Bio-Rad) and 10 μM forward and reverse primers. Products were then subjected to melting curve analysis. Gene expression was quantified relative to the expression of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primer sequences are provided in Supplementary Table 1, some of which are based on previous reports80,81,82.
Western blotting
Cell lysates were analyzed by Western blotting as described previously83, and described in more detail in the Supplementary Methods.
Generation of phosphorylated KDM5B, pKDM5B-S1456, antibody
Phosphopeptide ERER[pS]YELVRSAETC was coupled with keyhole limpet hemocyanin by m-maleimidobenzoic acid N-hydroxysuccinimide ester method. The conjugated phosphopeptide was used to immunize rabbits. The antibody is double purified from antiserum to ensure specificity. The crude antiserum is passed through a first column coupled with the phosphopeptide. The sequence specific and phosphate sequence specific antibodies recovered from the first purification were applied onto a second column coupled with non-phosphorylated peptide. Phosphospecific antibodies recovered in the flow through were used in ELISA tests with 2 peptides, the free phosphopeptide and non-phosphopeptide (to test for cross reaction). The antibody was validated by analyzing lysates from TNBC cells using Western blotting with pKDM5B-S1456 antibody in the absence or presence of 10 μM of phosphopeptide.
Flow cytometry and aldehyde dehydrogenase assay
Cells were dissociated using Accutase (Innovative Cell Technologies, catalog number AT106-550) and filtered through 40 micron filters. Cells (500,000) were then washed with FACS buffer (2% FBS, 0.2% sodium azide solution in 1x PBS) and incubated with anti-CD44 conjugated to allophycocyanin (APC, catalog number 560–890, BD PharmingenTM) and anti-CD24 conjugated to Fluorescein isothiocyanate (FITC, catalog number 560–992, BD PharmingenTM) for 30 minutes on ice. Isotype-matched conjugated non-immune antibodies were used as negative controls (APC-ISO, catalog number 17-4321-81; FITC-ISO, catalog number 11-4321-81, Thermo Fisher Scientific). Cells were then washed with FACS buffer, resuspended in fixative solution (3.7% PFA in 1X PBS), and incubated overnight at 4 °C before analysis by flow cytometry (BD LSR Fortessa). Data analysis was completed using FlowJo Software v10.
Aldehyde Dehydrogenase assays were performed using the ALDEFLUOR Assay Kit (StemCell Technologies, catalog number 01700) as described in the manufacturer’s protocol. Briefly, 106 cells were centrifuged and resuspended in 1 mL ALDH assay buffer, followed by addition of substrate BODIPY-aminoacetaldehyde. A portion of each reaction was transferred into a tube containing ALDH inhibitor, Diethylaminobenzaldehyde, to control for background fluorescence. Cells were then incubated for 40 min at 37 °C. Levels of ALDH expression were analyzed by flow cytometry and FlowJo Software v10.
Mammosphere assay
Cells were sorted using BD FACS Aria II and cultured in duplicate rows of serial dilutions into a 96-well plate and incubated for 10–14 days in DMEM media supplemented with 2% B27 (Invitrogen) in the presence of 20 ng/mL EGF and 10 ng/mL bFGF. Mammosphere-forming efficiency was calculated as the number of spheres divided by the number of single cells seeded.
Cell invasion assay
Transwell migration assay were performed as previously described25 and described in more detail in the Supplementary Methods.
Data analyses
Statistical significance was determined using Student’s t test comparison for unpaired data.
Data availability
The authors declare that all the materials, data and associated protocols related to this manuscript are available upon reasonable request.
References
Foulkes, W. D., Smith, I. E. & Reis-Filho, J. S. Triple-negative breast cancer. N Engl J Med 363, 1938–1948, https://doi.org/10.1056/NEJMra1001389 (2010).
Badve, S. et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol 24, 157–167, https://doi.org/10.1038/modpathol.2010.200 (2011).
Wahba, H. A. & El-Hadaad, H. A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med 12, 106–116, https://doi.org/10.7497/j.issn.2095-3941.2015.0030 (2015).
Frank, N. Y., Schatton, T. & Frank, M. H. The therapeutic promise of the cancer stem cell concept. J Clin Invest 120, 41–50 (2010).
Li, W. et al. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep 7, 13856 (2017).
Horimoto, Y. et al. Combination of Cancer Stem Cell Markers CD44 and CD24 Is Superior to ALDH1 as a Prognostic Indicator in Breast Cancer Patients with Distant Metastases. PloS one 11, e0165253 (2016).
Velasco-Velazquez, M. A., Popov, V. M., Lisanti, M. P. & Pestell, R. G. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol 179, 2–11 (2011).
Shiozawa, Y., Nie, B., Pienta, K. J., Morgan, T. M. & Taichman, R. S. Cancer stem cells and their role in metastasis. Pharmacol Ther 138, 285–293 (2013).
Balic, M. et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res 12, 5615–5621 (2006).
Kaur, P. et al. A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC cancer 12, 120 (2012).
Dai, M. et al. CDK4 regulates cancer stemness and is a novel therapeutic target for triple-negative breast cancer. Sci Rep 6, 35383 (2016).
Toh, T. B., Lim, J. J. & Chow, E. K. Epigenetics in cancer stem cells. Mol Cancer 16, 29 (2017).
Kosan, C. & Godmann, M. Genetic and Epigenetic Mechanisms That Maintain Hematopoietic Stem Cell Function. Stem Cells Int 2016, 5178965 (2016).
Lussi, Y. C. et al. Impaired removal of H3K4 methylation affects cell fate determination and gene transcription. Development (Cambridge, England) 143, 3751–3762 (2016).
Dey, B. K. et al. The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Mol Cell Biol 28, 5312–5327 (2008).
Xiang, Y. et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci USA 104, 19226–19231, https://doi.org/10.1073/pnas.0700735104 (2007).
Barrett, A. et al. PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: a new cancer/testis antigen? Int J Cancer 101, 581–588 (2002).
Li, J. et al. MiR-29a suppresses prostate cell proliferation and induces apoptosis via KDM5B protein regulation. Int J Clin Exp Med 8, 5329–5339 (2015).
Hayami, S. et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer 9, 59, https://doi.org/10.1186/1476-4598-9-59 (2010).
Yamane, K. et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell 25, 801–812, https://doi.org/10.1016/j.molcel.2007.03.001 (2007).
Yamamoto, S. et al. JARID1B Is a Luminal Lineage-Driving Oncogene in Breast Cancer. Cancer Cell 25, 762–777, https://doi.org/10.1016/j.ccr.2014.04.024 (2014).
Roesch, A. et al. Re-expression of the retinoblastoma-binding protein 2-homolog 1 reveals tumor-suppressive functions in highly metastatic melanoma cells. J Invest Dermatol 126, 1850–1859, https://doi.org/10.1038/sj.jid.5700324 (2006).
Cariou, S., Catzavelos, C. & Slingerland, J. M. Prognostic implications of expression of the cell cycle inhibitor protein p27Kip1. Breast Cancer Res. Treat. 52, 41 (1998).
Klein, B. J. et al. The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell reports 6, 325–335, https://doi.org/10.1016/j.celrep.2013.12.021 (2014).
Yeh, I. et al. HEXIM1 Plays a Critical Role in the Inhibition of the Androgen Receptor by Antiandrogens. Biochemical Journal 462, 315–327 (2014).
Horiuchi, D. et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med 209, 679–696 (2012).
Pietila, M. et al. FOXC2 regulates the G2/M transition of stem cell-rich breast cancer cells and sensitizes them to PLK1 inhibition. Sci Rep 6, 23070 (2016).
Neganova, I. et al. CDK1 plays an important role in the maintenance of pluripotency and genomic stability in human pluripotent stem cells. Cell Death Dis 5, e1508 (2014).
Catchpole, S. et al. PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int J Oncol 38, 1267–1277, https://doi.org/10.3892/ijo.2011.956 (2011).
Han, M., Xu, W., Cheng, P., Jin, H. & Wang, X. Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget 8, 8980–8991, https://doi.org/10.18632/oncotarget.13858 (2017).
Kidder, B. L., Hu, G. & Zhao, K. KDM5B focuses H3K4 methylation near promoters and enhancers during embryonic stem cell self-renewal and differentiation. Genome Biol 15, R32, https://doi.org/10.1186/gb-2014-15-2-r32 (2014).
Brier, A. S. B. et al. The KDM5 family is required for activation of pro-proliferative cell cycle genes during adipocyte differentiation. Nucleic Acids Research 45, 1743–1759, https://doi.org/10.1093/nar/gkw1156 (2017).
Dephoure, N., Gould, K. L., Gygi, S. P. & Kellogg, D. R. Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Molecular biology of the cell 24, 535–542, https://doi.org/10.1091/mbc.E12-09-0677 (2013).
Chan, P. C., Sudhakar, J. N., Lai, C. C. & Chen, H. C. Differential phosphorylation of the docking protein Gab1 by c-Src and the hepatocyte growth factor receptor regulates different aspects of cell functions. Oncogene 29, 698–710, https://doi.org/10.1038/onc.2009.363 (2010).
Lemaire, M. et al. CDC25B phosphorylation by p38 and MK-2. Cell Cycle 5, 1649–1653, https://doi.org/10.4161/cc.5.15.3006 (2006).
Papinski, D. et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell 53, 471–483, https://doi.org/10.1016/j.molcel.2013.12.011 (2014).
Shembade, N., Pujari, R., Harhaj, N. S., Abbott, D. W. & Harhaj, E. W. The kinase IKKalpha inhibits activation of the transcription factor NF-kappaB by phosphorylating the regulatory molecule TAX1BP1. Nature immunology 12, 834–843, https://doi.org/10.1038/ni.2066 (2011).
Sun, H. L. et al. ERK Activation Globally Downregulates miRNAs through Phosphorylating Exportin-5. Cancer Cell 30, 723–736, https://doi.org/10.1016/j.ccell.2016.10.001 (2016).
Holt, L. J. et al. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686, https://doi.org/10.1126/science.1172867 (2009).
Leis, O. et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 31, 1354–1365, https://doi.org/10.1038/onc.2011.338 (2012).
Ketchart, W. et al. Induction of HEXIM1 activities by HMBA derivative 4a1: functional consequences and mechanism. Cancer Lett. 379, 60–69 (2016).
Brunello, A., Borgato, L., Basso, U., Lumachi, F. & Zagonel, V. Targeted approaches to triple-negative breast cancer: current practice and future directions. Curr Med Chem 20, 605–612 (2013).
Yang, F. et al. Evaluation of Breast Cancer Stem Cells and Intratumor Stemness Heterogeneity in Triple-negative Breast Cancer as Prognostic Factors. Int J Biol Sci 12, 1568–1577, https://doi.org/10.7150/ijbs.16874 (2016).
Luo, M., Brooks, M. & Wicha, M. S. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des 21, 1301–1310 (2015).
Doherty, M. R., Smigiel, J. M., Junk, D. J. & Jackson, M. W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers (Basel) 8 (2016).
Kurtova, A. V. et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517, 209–213 (2015).
Lee, C. et al. Polo-like kinase 1 inhibition kills glioblastoma multiforme brain tumor cells in part through loss of SOX2 and delays tumor progression in mice. Stem Cells 30, 1064–1075 (2012).
Maire, V. et al. Polo-like kinase 1: a potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res 73, 813–823 (2013).
Wainwright, E. N. & Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 3, 372–386 (2017).
Easwaran, H., Tsai, H. C. & Baylin, S. B. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 54, 716–727 (2014).
Wu, H. & Sun, Y. E. Epigenetic regulation of stem cell differentiation. Pediatr Res 59, 21R–25R (2006).
Wutz, A. Epigenetic regulation of stem cells: the role of chromatin in cell differentiation. Adv Exp Med Biol 786, 307–328 (2013).
Shukla, S. & Meeran, S. M. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim Biophys Acta 1840, 3494–3502 (2014).
Schmitz, S. U. et al. Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J 30, 4586–4600, https://doi.org/10.1038/emboj.2011.383 (2011).
Kuo, Y. T. et al. JARID1B Expression Plays a Critical Role in Chemoresistance and Stem Cell-Like Phenotype of Neuroblastoma Cells. PloS one 10, e0125343 (2015).
Facompre, N. D. et al. JARID1B Enables Transit between Distinct States of the Stem-like Cell Population in Oral Cancers. Cancer Res 76, 5538–5549 (2016).
Roesch, A. et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594, https://doi.org/10.1016/j.cell.2010.04.020 (2010).
Wong, S. H. et al. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 28, 198–209, https://doi.org/10.1016/j.ccell.2015.06.003 (2015).
Vallianatos, C. N. & Iwase, S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 7, 503–519 (2015).
Johansson, C. et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nature chemical biology 12, 539–545, https://doi.org/10.1038/nchembio.2087 (2016).
Vinogradova, M. et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nature chemical biology 12, 531–538, https://doi.org/10.1038/nchembio.2085 (2016).
Zou, M. R. et al. Histone demethylase jumonji AT-rich interactive domain 1B (JARID1B) controls mammary gland development by regulating key developmental and lineage specification genes. J Biol Chem 289, 17620–17633 (2014).
Rodriguez-Pinilla, S. M. et al. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Mod Pathol 20, 474–481, https://doi.org/10.1038/modpathol.3800760 (2007).
Bamodu, O. A. et al. Aberrant KDM5B expression promotes aggressive breast cancer through MALAT1 overexpression and downregulation of hsa-miR-448. BMC Cancer 16, 160, https://doi.org/10.1186/s12885-016-2108-5 (2016).
Lu, W. et al. SKP2 inactivation suppresses prostate tumorigenesis by mediating JARID1B ubiquitination. Oncotarget 6, 771–788 (2015).
Bueno, M. T. & Richard, S. SUMOylation negatively modulates target gene occupancy of the KDM5B, a histone lysine demethylase. Epigenetics 8, 1162–1175 (2013).
Spangle, J. M. et al. PI3K/AKT Signaling Regulates H3K4 Methylation in Breast Cancer. Cell Rep 15, 2692–2704, https://doi.org/10.1016/j.celrep.2016.05.046 (2016).
Liefke, R. et al. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev 24, 590–601 (2010).
Lim, S. & Kaldis, P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development (Cambridge, England) 140, 3079–3093, https://doi.org/10.1242/dev.091744 (2013).
Wang, X. Q. et al. CDK1-PDK1-PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ 24, 38–48 (2017).
Zhao, Y. & Yang, X. Regulation of sensitivity of tumor cells to antitubulin drugs by Cdk1-TAZ signalling. Oncotarget 6, 21906–21917, https://doi.org/10.18632/oncotarget.4259 (2015).
Wei, Y. et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol 13, 87–94, https://doi.org/10.1038/ncb2139 (2011).
Asghar, U., Witkiewicz, A. K., Turner, N. C. & Knudsen, E. S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature reviews. Drug discovery 14, 130–146, https://doi.org/10.1038/nrd4504 (2015).
Chen, Y. S., Racca, J. D., Phillips, N. B. & Weiss, M. A. Inherited human sex reversal due to impaired nucleocytoplasmic trafficking of SRY defines a male transcriptional threshold. Proc Natl Acad Sci USA 110, E3567–3576 (2013).
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V. & Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1, 2856–2860, https://doi.org/10.1038/nprot.2006.468 (2006).
Vukoti, K. et al. Monitoring newly synthesized proteins over the adult life span of Caenorhabditis elegans. Journal of proteome research 14, 1483–1494, https://doi.org/10.1021/acs.jproteome.5b00021 (2015).
Wittmann, B. M., Wang, N. & Montano, M. M. Identification of a novel inhibitor of cell growth that is down-regulated by estrogens and decreased in breast tumors. Cancer Res. 63, 5151–5158 (2003).
Liang, S. et al. Isolation and characterization of human breast cancer cells with SOX2 promoter activity. Biochem Biophys Res Commun 437, 205–211 (2013).
Lai, D. et al. Optimization of culture conditions to support undifferentiated growth of human embryonic stem cells. Cell Reprogram 12, 305–314 (2010).
Fumoto, K., Takigawa-Imamura, H., Sumiyama, K., Kaneiwa, T. & Kikuchi, A. Modulation of apical constriction by Wnt signaling is required for lung epithelial shape transition. Development (Cambridge, England) 144, 151–162, https://doi.org/10.1242/dev.141325 (2017).
Hussenet, T. et al. SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PloS one 5, e8960, https://doi.org/10.1371/journal.pone.0008960 (2010).
Yang, X. et al. Rab5a promotes the migration and invasion of hepatocellular carcinoma by up-regulating Cdc42. Int J Clin Exp Pathol 11, 224–231 (2018).
Ogba, N., Chaplin, L., Doughman, Y. Q., Fujinaga, K. & Montano, M. M. HEXIM1 regulates E2/ERα-mediated expression of Cyclin D1 in mammary cells via modulation of P-TEFb. Cancer Research 68, 7015–7024 (2008).
Acknowledgements
This work was supported by grants from NIH (CA195558) to M.M.M. The mass spectrometer used was purchased via an NIH shared instrument grant, 1S10RR031537-01. We thank Belinda Willard and Ling Li for the acquisition of mass spectrometry data. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.M.M., O.R. and J.L. supervised the project. I.Y., M.M.M., M.M. and E.E. designed and performed experiments. I.Y. and M.M.M. wrote the manuscript text and prepared the figures. E.E. also helped with the preparation of Figure 7. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
Dr. Montano is co-founder of Oncostatyx. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yeh, IJ., Esakov, E., Lathia, J.D. et al. Phosphorylation of the histone demethylase KDM5B and regulation of the phenotype of triple negative breast cancer. Sci Rep 9, 17663 (2019). https://doi.org/10.1038/s41598-019-54184-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54184-0
- Springer Nature Limited
This article is cited by
-
The phosphorylation to acetylation/methylation cascade in transcriptional regulation: how kinases regulate transcriptional activities of DNA/histone-modifying enzymes
Cell & Bioscience (2022)
-
Drawing a line between histone demethylase KDM5A and KDM5B: their roles in development and tumorigenesis
Experimental & Molecular Medicine (2022)
-
Enhanced O-GlcNAc modification induced by the RAS/MAPK/CDK1 pathway is required for SOX2 protein expression and generation of cancer stem cells
Scientific Reports (2022)