
Abstract
The common management practices of dairy calves leads to increased starch concentration in feed, which subsequently may cause rumen acidosis while on milk and during weaning. Until recently, few attempts were undertaken to understand the health risks of prolonged ruminal acidosis in post weaning calves. Resultantly, the molecular changes in the digestive tracts in post-weaning calves with ruminal acidosis remain largely unexplored. In this study, we investigated the liver transcriptome changes along with its correlation with the rumen microbial rRNA expression changes in young calves using our model of feed induced ruminal acidosis. In this model, new born calves were fed a highly processed, starch-rich diet starting from one week of age through 16 weeks. A total of eight calves were involved in this study. Four of them were fed the acidosis-inducing diet (Treated) and the rest of the four were fed a standard starter diet (Control). Liver and rumen epithelial tissues were collected at necropsy at 17 weeks of age. Transcriptome analyses were carried out in the liver tissues and rRNA meta-transcriptome analysis were done using the rumen epithelial tissues. The correlation analysis was performed by comparing the liver mRNA expression with the rumen epithelial rRNA abundance at genus level. Calves with induced ruminal acidosis had significantly lower ruminal pH in comparison to the control group, in addition to significantly less weight-gain over the course of the experiment. In liver tissues, a total of 428 differentially expressed genes (DEGs) (fold-change, FC ≥ 1.5; adjusted P ≤ 0.1) were identified in treated group in comparison to control. Biological pathways enriched by these DEGs included cellular component organization, indicating the impact of ruminal acidosis on liver development in young calves. Specifically, the up-regulated genes were enriched in acute phase response (P < 0.01), pyruvate metabolic process (P < 0.01) and proton-acceptors (P ≪ 0.001), indicating the liver’s response to feed induced acidosis at the transcriptome level. Twelve transferase activity related genes had significant correlation with rumen microbial rRNA expression changes. Among these genes, two up-regulated genes were reported with involvement in lipid metabolism in the liver, implying the direct effect of feed-induced acidosis on both the rumen microbial community and liver metabolism. Our study provides insight into the physiological remodeling in the liver resultant from the prolonged acidosis in post weaning calves, which may facilitate future RNA-seq based diagnosis and precision management of rumen acidosis in dairy calves.
Similar content being viewed by others

Introduction
Weaning transition is a critical period for the functional rumen development for dairy calf, during which the dairy calves changes from liquid to solid feed consumption1. The rumen is not fully functional at birth. The rumen will go through significant development in size, morphology and function2 in order to provide sufficient protein and energy to the dairy calves at the time of weaning (at ~eight weeks of age). The solid feed fermentation lead to increased production of volatile fatty acid (VFA), which is reported as the primary stimulant for the functional rumen epithelial tissue development3,4,5. Thus, for the purpose of fostering rumen development and allowing the calves to be weaned at an earlier age, maximum intake of readily fermented calf starter is common during weaning transition period1 (NRC, 2001). However, calves fed starch-source displayed excessive VFA and lactic acid production, which leads to significant decrease in rumen pH6. An overall reduction in ruminal pH7,8 caused by the ingestion of diets rich in rapidly fermentable carbohydrates with insufficient amount of fiber required for efficient rumen buffering can lead to sub-acute Ruminal Acidosis (SARA), a common metabolic disorder in dairy cattle. SARA is a well-recognized, economically important disorder in dairy cattle. Studies in Wisconsin (US) reported an estimated 20–23% of cows with SARA9,10, while a large study in Australian found 10% of the cows less than 100 days in milk had acidosis11. Milk yield reduction, premature culling and increased mortality are among the direct consequences of SARA-induced digestive and metabolic disfunction.
Several other deleterious consequences have been associated with SARA. During SARA, free lipopolysaccharides (LPS) also increase in the rumen12,13. When the free LPS enters blood circulation, it activates immunosuppression and inflammation responses resultant from the depressed ruminal pH7,14. Once acidosis is developed, a sharp increase in the production of VFAs, especially lactic acid can further decrease the rumen pH15. Rapid fermentation caused pH reduction has been linked to the impairment of barrier function in the gut16, ruminal parakeratosis, erosion, and ulceration of the ruminal epithelium17.
There are no typical clinical signs of SARA in affected cows18,19, and the commonly defined clinical symptoms are generally delayed in onset from the time of low ruminal pH insult. Inflammations of different tissues and organs have been reported in cows with SARA. The associated pathophysiological cascade of events begin with decreased dry matter intake14, reduced in situ fiber degradation20, rumen epithelial damage21 and inflammation22. Once the ruminal epithelium is inflamed, the gut bacteria may enter into the portal circulation and into the liver. The subsequent bacterial leak into the lungs, kidneys, heart valves and joints can cause chronic inflammatory diseases that are hard to diagnose post-mortem (reviewed in Oetzel23). Most commonly, ruminal acidosis predisposes cattle to liver abscesses24,25, which are the primary liver abnormality of feedlot cattle seen at slaughter, averaging 67% of all liver abnormalities26. The mean prevalence of liver abscesses in conventionally managed feedlot cattle in the US ranges from 10% to 20%26,27, and as high as 90% to 95% in individual groups of grain fed cattle28,29. The widely accepted etiology of liver abscess was that acidosis induced damage to the rumen epithelium was the main causing factor, supported by previously reported high correlation between ruminal ulcers and the occurrence of liver abscess27,30,31. Consistent with this, early studies have reported several microorganisms as the causes for liver abscess, including the Fusobacterium necrophorum, Bacteroides spp., Peptostreptococcus spp., Staphylococcus spp. by Berg and Scanlan32; Clostridium spp., Pasteurella spp. and Streptococcus spp. by Simon and Stovell33, and Trueperella pyogenes by Calkins and Scrivner34. Among these, F. necrophorum (a common inhabitant of the rumen35) was the most commonly isolated pathogen in liver abscess (with its incident rate ranges from 85% to 100% of the studied cases)36. As part of the cell wall of gram-negative bacteria, lipopolysaccharide (LPS) is a form of endotoxin that can transport into the liver via the portal vein. Previous in vitro experiments indicated that the liver hepatocytes can excrete endotoxins present in the circulatory system in the bile, and detoxify LPS through the activity of liver macrophages (Kupffer cells)37. In this study, though we did not observe visible sighs of liver abscess at the time of tissue collection, we did observe significant physiological changes in the treated calves, including significantly lower ruminal pH and overall weight-gain, and rumen papillae degradation via histology analysis
The liver is a critical organ for nutrient metabolism. However, we currently have very limited knowledge about the impacts of feed-induced acidosis on the liver transcriptomics and associated molecular pathways. In this study, we specifically focused on the global transcriptome changes and impacted molecular pathways in liver tissues collected from four month old calves. This work is in conjunction with our recently published work38, where a highly-processed, starch-rich feed was used to induce ruminal acidosis in bull calves beginning at 1 week of age through 16 weeks. Liver tissues were collected after sacrifice at 17 weeks of age, followed by whole transcriptome sequencing analysis. We hypothesized that feed-induced acidosis in young calves was associated with significant changes in the liver transcriptome. And such changes in the liver were linked to the rumen microbial community alterations.
Material and Methods
Ethics statement and animal care
This study is part of one larger study where other portions of the study have been published39,40. All procedures for the animal study were reviewed and approved by the University of Wisconsin – Madison Institutional Animal Care and Use Committee (IACUC no. A005848). Throughout the experiment, all animals were maintained according to the standard herd practices approved at the USDA Dairy Forage Research Center farm.
All the Holstein bull calves were from the same study published by our group recently39,40. In brief, ten Holstein bull calves born at the Marshfield Agricultural Research Station (Marshfield, WI) between June 17 and July 5, 2017 were used for this experiment. Calves were housed in individual calf hutches (4.8 sq. m/calf) from birth to 8 weeks and then divided into larger hutches (5.0 sq. m/calf) through 16 weeks.
Study Design
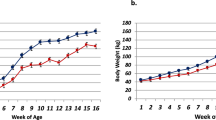
For the treated and the control diets, two grain starter diets were used in the study as reported in our recently published work39,40. Different from our previously published work where the rumen epithelial tissue38 was the focus, this research focuses on the liver transcriptome changes resultant from the dosing trial. In brief, the treated diet had a starch concentration of 42.7%, while the control diet had a starch concentration of 35.3% (Fig. 1). The detailed nutrient concentration is published in Gelsinger et al.39. For sequencing experiment, there were four calves in each treated and control groups. Treatments were randomly assigned and offered to calves beginning at 1 week of age (6.6 d ± 3.4). RNAseq power analysis using Scotty41 indicated that with an average of 60 M reads and four replicates per treatment condition, one can achieve the power of identifying 85% of the genes with at least 50% maximum power. And with this experimental design, one can identify 65% of the genes with at least 1.5X fold-change, 85% of the genes with at least 2X fold-change and 95% of the genes with at least 3X fold-change (Supplemental Fig. 1).

The concentration of starch and non-digestive fiber in the feed administered to the treated and control groups.
ad libitum access to their assigned starter was allowed for the calves throughout the duration of the trial up to 4500 g/d. On a daily basis, a measured amount of starter was offered at 0800 with refusals determined daily. Beginning at week 6 through week 16, rumen pH values were measured 7 times in a single day every other week. The seven time-points were: −8, −4, 0, 2, 4, 8 and 12 hours relative to grain feeding. For the measurement of rumen pH, the pH probes were placed in the ventral sac of the rumen via a rumen canula. Blood pH was measured on weeks 8, 10, 12 and 14. Starter intake and body weight were measured for all calves at a given time every week, beginning at week 1 through week 16. Calves were euthanized at 17 weeks of age for tissue collection. In conjunction with our recently published works38,39,40, we have observed lower (P < 0.01) rumen pH in the treated calves compared to the control group. Additionally, both the starter intake and weight gain were significantly lower in the treated animals across all the weeks during the experiment.
Calf liver tissue collection
Four calves from each treatment group were subjected to liver and rumen epithelial tissue collection. Liver tissues were collected from the right lobe. Before collection, the right lobe was cut into even halves with a sterile scalpel and the tissue sample was collected from the center part of the liver tissue. Rumen papillae tissues were taken from same location as described in our previous publication38. All tissues were collected right after animal euthanasia. Collected tissues were rinsed in 1X PBS and cut with sterilized scalpels into small fragments and put into Eppendorf safe-lock tubes (Eppendorf North America, US) followed by flash frozen in liquid nitrogen and stored at −80 °C for long-term storage.
RNA extraction, RNA-seq library preparation and sequencing
Liver and rumen epithelial tissues were homogenized into fine powders separately in liquid nitrogen using a mortar and pestle. RNAs were extracted using the miRNeasy protocol with a QIAcube instrument (Qiagen US). RNA samples with RIN value ≥ 8 were pursued for RNA quantification using Qbit Broad Range assay (Thermo Fisher, US). RNA-seq library prep followed the same procedure as described previously38. In brief, For each sample, an input amount of 1μg of total RNA was used for sequencing library preparation. RNA-sequencing library was prepared using a Illumina TruSeq ribo-zero gold kit (Illumina, San Diego, US). Concentration of each prepared library was quantified using a Kapa quantification kit (Kapa Systems, US) on an ABI7300 RT-qPCR instrument (Thermo Fisher, US). After Kapa quantification, libraries were further normalized and pooled using the Pooling Calculator (https://support.illumina.com/help/pooling-calculator/pooling-calculator.htm), an online tool offered by Illumina. An Illumina NextSeq 500, high-output kit was used to sequence the pooled libraries to generate 2 × 75 bp, paired-end reads.
Differential gene expression analysis
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to check the quality of raw reads. Additionally, sequencing raw reads shorter than 35 bp were excluded for further analysis. Bos taurus UMD3.1 was used as the genomic reference for sequence reads mapping. RNA-sequencing reads were aligned to the B. taurus reference genome using a two-step alignment approach as described previously38. The first step of read-mapping was done using Tophat242. The unmapped reads from the first step were further aligned by Bowtie243 by setting the “–very-sensitive-local” parameter. HTSeq (v0.6) HTseq44 was used to calculate the raw read-counts for each annotated gene in the B. taurus gene annotation file, using the combined (Tophat + bowtie2) sequence alignment file generated by the two-step alignment approach. The expression level of mRNAs in each sample were normalized to Fragments Per Kilobase of exon per Million fragments mapped (FPKM) using cufflinks45. Using a FPKM cutoff value of one, the total number of expressed genes were calculated.
Differential gene expression (DEG) analysis was performed using R/Bioconductor package DESeq246 with raw read-counts calculated by HTseq44 following the previously published procedure38. When using DEseq. 2, read-count normalization was performed using the regularized logarithm (rlog) method. An average of ten normalized read-counts was used as cutoff to exclude genes from further differential expression analysis. To be considered as DEGs, the following cutoff were imposed: adjusted p-value ≤ 0.05 and the fold change ≥1.5. DAVID47 and stringDB48,49 were employed for gene function annotation and pathway analysis. FPKM values were used to identify the top 1% most highly expressed genes in each sample. Using the top 1% most highly expressed genes, the shared, most abundantly expressed genes were identified for both treated and control groups and the list of the most highly expressed genes unique to treated group was also identified.
Expression correlation analysis of rumen epithelial microbial community and liver mRNA
RNA-sequencing reads used for rumen epimural microbial community expression were obtained using the method from previously published work by our group Li et al.38. In brief, raw-reads generated by total RNA sequencing using the rumen papillae tissues were used for the epithelial of rRNA reads for the rumen epithelial microbial community. Genus level expression quantification was done using Kraken50. Genus level raw-read counts generated by Kraken were further normalized by dividing the total number of raw-reads for each genus with the per million factor (PMF). To calculate the PMF, the total number of reads mapped to genus level for a given sample was divided by 1,000,000. Then, the mapped raw reads at each genus was divided by the PMF, yielding a normalized read count. The top 10% most highly expressed genus for the treated and control groups were identified by the following steps: (1) the average, normalized read-count for each group was calculated for each genus; (2) the genus with normalized read-counts at the 10% percentile were identified for the treated and control groups respectively.
To identify the correlation between liver mRNA and rumen epimural microbial rRNA abundance, we performed association analysis using pearsons’r from scipy.stats (SciPy v1.2.0) as described previously38. For rumen microbial rRNA abundance data, normalized read-counts at genus level were included in the correlation analysis. The list of significantly differentially expressed, microbial genus identified in our previously published work was included in the analysis38. p-values ≤ 0.001 and the absolute value of correlation coefficient more than 0.8 were used as the cutoffs.
Verification of target genes expression profile using RT-qPCR
The expression profile for four randomly selected DEGs identified by RNA-seq was analyzed in both treated and control groups using RT-qPCR. These genes were: ARHGDIA, SUSD2, IGF2R and RASSF4. ARHGDIA was reported with a key role in the regulation of cell motility through Rho GTPases51
(https://www.genecards.org/cgi-bin/carddisp.pl?gene = ARHGDIA); SUSD2 plays important roles in cell-to-cell and cell-matrix adhesion. This gene encodes a type I transmembrane protein of 820 amino acids consisting of a large extracellular region containing Somatomedin B52. IGF2R was reported with various functions, including the activation of transforming growth factor beta and the intracellular trafficking of lysosomal enzymes53,54. RASSF4 belongs to the Ras associated domain family. The RASSF proteins have reported roles in microtubule stability, regulating mitotic cell division, and modulating cell migration and adhesion55. The following Taqman probes were ordered from Thermo Fisher (Thermo Fisher, US): ARHGDIA, Bt03224507_g1; SUSD2, Bt04284484_m1; IGF2R, Bt03223452_m1; and RASSF4, Bt03241299_m1.
cDNA synthesis was performed using 2000 ng of total RNA with High Capacity cDNA master mix following manufacturer’s instruction (Thermo Fisher, US). All RT-qPCR reactions were performed using the QuantStudio 5, 396-well system (Thermo Fisher, USDA), using pre-designed Taqman assay probes along with the Taqman fast advanced master mix (Thermo Fisher, US). The thermocycler steps were set following the manufacturer’s instruction as the following: one step of uracil-N-glycosylase (UNG)56,57 treatment at 50 °C for 2 min, followed by an initial denaturation/activation step at 95 °C for 2 min, then 40 cycles at 95 °C for 1 s and 60 °C for 20 s. The experiments were carried out in triplicate for each targeted gene. Two reference genes, Beta-actin (ACTB) and hydroxymethylbilane synthase (HMBS) were used to normalize the expression quantification of targeted genes. Bovine specific, predesigned Taqman probes (Bt03279174_g1 for ACTB and Bt03234763_m1 for HMBS) for these two reference genes were used in the assay. The relative quantification of gene expression was determined using the 2−ΔΔCt method58.
Statistical analysis
Pearsons’r from scipy.stats (SciPy v1.2.0) was used to calculate the correlation between liver mRNA expression and rumen epimural microbial rRNA abundance. DEG analysis was done using DESeq246 using regularized log-transformation for read-count normalization. RT-qPCR data were done using t-test if the data were normally distributed. For non-normally distributed data, the statistical significance was calculated using mood’s median test. Starter intake, body weight gain, ruminal and blood pH parameters were analyzed as previously described38,40.
Results
Physiological effects of the animal model: blood and ruminal pH, body weight, starter intake and rumen papillae tissues
Mean ruminal pH differed by diet (P < 0.01), but not by age (P = 0.12); whereas, blood pH decreased linearly with age (P = 0.01), but was not different between diets (P = 0.20). There was no interaction between diet and age for either metric (P > 0.69). Mean ruminal pH reached a nadir for all calves during week 8. Mean ruminal pH ± S.E. (min, max) was 5.37 ± 0.24 (3.3, 7.2) and 5.63 ± 0.24 (3.5, 6.8) for treatment and control calves, respectively. Body weight and feed intake increased linearly (P < 0.01) with age. Calves fed the control diet consumed a greater amount of feed (P < 0.01) and attained greater body weight (P < 0.01) at weeks 4 and 5, respectively. These differences were maintained through week 16 (P < 0.001). For rumen papillae tissues: Papillae length and width were not different (P = 0.38), but a greater degree of tissue degradation was observed in acidotic calves (P < 0.01)40.
RNA quality and sequencing reads alignment for liver transcriptome
The extracted RNA samples of liver tissues were of high quality, with the average RNA integrity number (RIN) of 8.3 ± 0.11 (standard error (s.e.)). An average of 76.8M ± 1.29M (s.e.) reads were obtained for the sequenced samples, with a range of 62 M to 78 M. Using the FPKM cutoff value of one, the total number of expressed genes ranged from 11,684 to 12,606. All samples had similar distribution of gene expression, with majority of the genes expressed in the range of 0.2 to 15 FPKMs (Supplemental Dataset 1).
Liver transcriptome changes between treated and control groups
The expression profile of the selected genes was successfully confirmed by RT-qPCR method (Fig. 2). A list of 74 genes was identified as the top 1%, mostly abundantly expressed across all samples. Gene ontology (GO) analysis indicated that these genes were enriched in the cellular component in the extracellular region (GO:CC~GO:0005576; 41 genes, P ≪ 0.00001). For the top 1% most highly expressed genes in the control and treated group, 18 genes were uniquely expressed in the treated group (APOC2, A1BG, FMO1, P4HB, BRP44L, LDHB, HP, SAA3, APOA1, APOA5, MT1E-2, MT1A, ARG1, HSD17B10, ITIH3, HMGCS2, SERPINA3, ACADM). Pathway analysis indicated that these genes were significantly enriched in organic acid metabolic process (GO:BP~GO:0006082; 8 genes; P ≪ 0.00001), high-density lipoprotein (Uniprot, keywords_results; 4 genes; P ≪ 0.0001), and positive regulation of triglyceride metabolic process (GO:BP~GO:0090208; 3 genes; P ≪ 0.00001).

RT-qPCR confirmation of four differentially expressed genes identified by RNA-seq. Fold-change (Treated vs control) of target genes were calculated by both RNA-seq and RT-qPCR methods.
A total of 428 differentially expressed genes (DEGs) were identified between the treated and control groups (Supplemental Dataset 2). And the top 50 most significant DEGs clearly separated the two treatment groups (Fig. 3). Among the list of DEGs, 244 of them were up-regulated and 184 of them were down-regulated in the treated group. Pathways and GO analysis using the combined list of DEGs indicated the enrichment of pathways involving cell division and growth. They included cellular component organization or biogenesis (GO:0071840; 69 genes; P ≪ 0.0001) and membrane bounded organelle (GO:0043227; 120 genes; P ≪ 0.0001). For down-regulated genes, they were predominantly enriched in the GO terms related to cellular growth and signaling, including cellular component biogenesis (GO:BP, GO:0044085; 25 genes, P < 0.005); intracellular signal transduction (GO:BP, GO:0035556; 23 genes, P < 0.01); For up-regulated genes (Fig. 4), they showed enrichment in genes involved in acute phase response (GO:BP, GO:0006953, 4 genes; P < 0.002), pyruvate metabolic process (GO:BP, GO:0006090, 5 genes; P < 0.01), response to nutrient levels (GO:BP, GO:0031667, 10 genes; P < 0.01), and 15 genes have shared motif “proton acceptor” (Fig. 5) (Supplemental Dataset 3).

Clustering heat-map of top 50 most significant differentially expressed genes between the treated and control groups.

Gene ontology (GO) pathway analysis of up- and down-regulated genes. GO pathways enriched by up-regulated genes were indicated by the triangles, while these enriched by down-regulated genes were indicated by the circles. For each circle or triangle, the size is proportional to the number of genes in each GO pathway, as represented by log2(number of genes); and the gradient of color (from green to blue) is associated with the p-value.

Expression profile of 15 proton-acceptor genes. The log2FC (fold-change) is calculated by log2 transformation of expression fold-change between the treated and control animals.
rRNA transcriptome analysis of rumen epithelial microbial community and its association with liver mRNA expression changes
The expression changes of 95 genes were identified with significant correlation with the abundance variation with the rumen epithelial microbial community at the genus level. Among these, 77 of the genes (Supplemental Dataset 4) had positive correlation with the rumen microbial rRNA expression, while 18 of the genes (Supplemental Dataset 5) had negative correlative with the rumen microbial rRNA expression. For the genes with positive correlation with the microbial rRNA expression, they were enriched in the pathways of membrane-bounded organelle (GO:0043227; 43 genes; P < 0.001) and transferase activity (GO:0016740; 11 genes; P < 0.05) (SIK3, TAF6L, ACAT2, BHMT2, CAMK2N2, CSNK1G1, GPT, HIPK1, KCNH3, PIAS3 and SIRT6).
Discussion
Acidosis-inducing diet caused liver remodeling at the transcriptome level
In our study, a significant enrichment of genes involved in cellular morphogenesis were observed for the complete list of DEGs, indicating the potential effect of ruminal acidosis on liver cellular growth and development at the transcriptome level. Consistent with this finding, Guo et al.59 observed histopathologic changes in the liver tissue in dairy cows with SARA. These included the glycogenated nuclei, inflammatory cells infiltration and liver cells injury ballooning59. Reduced body condition was also associated with the occurrence of SARA despite the high energy intake60,61. Consistently, we observed significantly reduced weight gain in our study38. However, we did not observe visible signs of inflammation in the joints or any other visible signs of inflammation. Since the significant number of the DEGs identified in the liver tissues was involved in cellular component organization and biogenesis, our finding implied that before any fundamental signs related to inflammation, significant changes in the liver transcriptome have already occurred. The liver is a critical organ for nutrient metabolism and host health. Most importantly, it has been recognized as an important immune organ in vertebrates62,63,64. Besides receiving blood from the portal circulatory system, the liver receives blood coming back from the intestine. Thus, the liver is exposed to a wide array of antigens coming from the gut. Specifically during ruminal acidosis, once the ruminal epithelium is inflamed, the gut bacteria may enter into the portal circulation and into the liver, initiating a broad range of immune responses in immune-related cellular components. For example, different population of liver lymphocyte, including the macrophages, natural killer cells and T lymphocytes have been identified with response to different antigenic peptides64,65. For further follow up, comparative gene expression analysis between liver and circulation blood might help identify blood-based biomarkers indicative of host response to feed-induced acidosis. Such easily accessible biomarkers may improve feed management before the typical signs of ruminal acidosis to avoid serious complications.
Acidosis-inducing diet on most highly expressed genes in the liver
For the top 1%, most highly expressed genes between control and treated groups, 18 of them were uniquely expressed in the liver tissues of the treated group. These genes were enriched in high density lipoprotein (HDL) and positive regulation of triglyceride metabolism. Of interest, three genes encoding the class of high-density lipoproteins were among the most highly expressed genes in treated calves compared to the control group. HDLs are the most abundant lipoproteins in the cow’s serum66. With the induction of SARA, Stefanska et al.67 observed significantly higher concentration of HDL in the blood of the affected cows. And this increase of HDL in acidotic cows might be associated with the cow’s ability to fight against the pathological condition. Though we did not see any signs of ulcer in the liver of treated animals, the most highly expressed HDL-encoding genes (Apoa1, Apoa5 and SAA3) in treated animals are consistent with previously published work where high concentration of HDL was observed in acidotic animals. APOA1, APOA2 and APOA5 belong to the exchangeable apolipoprotein family.
APOA2 is the second most common proteins in high-density lipoproteins68. Its increased expression level has been linked to atherosclerosis69. Both APOA2 and APOA5 were linked to increased risk of obesity and metabolic syndrome70,71. Specifically, APOA5 is a liver-specific protein, which functions as an important modulator in lipoprotein metabolism72. The close relationship between plasma levels of APOA5 and obesity has been confirmed recently73. These uniquely, highly expressed genes identified in our study indicated that feed-induced ruminal acidosis impacted the lipoprotein metabolism in the liver as a potential buffering response before the development of bacteria-causing ulcers or sepsis in the liver. This response may function at the expense of the overall metabolic health of the liver. Thus, further investigation on liver lipid metabolism may shed light into the responsive mechanism associated with SARA in young calves.
Other biochemical molecules have also been reported with increased concentration in acidotic cows. One of them is rumen LPS, which is part of the cell wall of gram-negative bacteria. Gozho and coauthors7 reported the increased concentration of ruminal free LPS upon the induction of SARA via the feeding of high-concentrate diet74. Similarly, Shen et al.75 recorded a higher LPS (P < 0.05) in the rumen fluid from the cows fed a high-starch diet (39% starch, DM basis) versus a low-starch diet (24% starch, DM basis). And in dairy cows fed a high-concentrate diet, inflammatory injuries were observed in the liver due to the LPS traveled from the digestive tract back to the liver59. Once ruminal free LPS enters the blood stream through the portal vein, the LPS can initiate potent pro-inflammatory response, leading to laminitis and sudden death syndrome13. Previous studies increasingly indicated the role of HDL and other lipoproteins in controlling the host response to free LPS76, through the binding of HDL to LPS77. This binding inhibited the ability of LPS to interact with toll-like receptors and activate macrophages, and thus reduced the chances of septic shock and related death78. Furthermore, Read et al.79 indicated that the binding of LPS by HDL has a protective effect of increasing excretion of LPS via bile and prevention of immune response.
Genes encoding acute phase proteins and proton-acceptors in the liver
Traditionally measured by ruminal pH, the diagnosis of ruminal acidosis still needs further improvement to achieve higher precision and earlier prognosis. Acute phase response is a non-specific defense mechanism due to tissue injury, infection or exposure to pro-inflammatory molecules. This type of response constitutes a complex network of numerous cell types and organs that produce and react to a multitude of cytokines and other mediators80. In human clinical studies, the interplays between acidosis, altered inflammation/innate immunity and metabolic diseases (e.g., diabetes and obesity) has been established. By studying the effect of lowered pH on the response of murine macrophage-like cells, Kellum and co-authors indicated that acidosis was associated with the increased response to LPS stimulation81. This finding is consistent with other studies, where low pH was reported with a proinflammatory effect82,83. And low-grade inflammatory activity has also been reported with a link to increased mortality and predisposition to metabolic diseases. Thus, investigating the link between acidosis, inflammation and metabolism holds high potential to uncover the pathophysiology of severe metabolic disease and help devise new therapies (reviewed in Farwell and Taylor84 and Rizvi85).
As a class of primarily liver-produced proteins, acute phase proteins (APPs) are up-regulated during acute phase response. Several acute phase proteins (APP) have been reported in cattle, including serum amyloid A (SAA), haptoglobin, and fibrinogen86,87. Thus, the measurements of these APP provide a tool to detect infection, inflammation and to monitor inflammation status changes88. Consistent with this, in dairy heifers, ruminal acidosis has been linked to increased levels of serum APPs and leukocytes89. As a proof of concept, one of the APP-encoding genes identified in our study was SAA, which was considered by Gozho and coauthors7 as the most sensitive APP with faster response to inflammation stimuli with its early detection in blood.
Aside from the elevated expression of APPs in heifers with SARA or acute ruminal acidosis, investigation and development of other easily accessible molecular biomarkers holds great potential for precision diagnosis and real-time monitor of ruminal acidosis in cattle. Our comparative tissue transcriptomics work in both rumen and liver indicated that the proton related proteins might be a fruitful avenue. In our recent work38, the comparative analysis of whole transcriptome sequencing in the rumen epithelial tissue of the treated and control groups yielded an enrichment of highly expressed genes involved in proton-transport. Along with significantly lowered ruminal pH we observed in the treated calves, these most highly expressed genes in the rumen indicated the direct response from the rumen epithelium to the elevated accumulation of protons resultant from carbohydrate-concentrated feed. Consistently, we observed an enrichment of proton-acceptors in the up-regulated genes in treated calves. Along with the lungs and kidneys, the liver has been recognized as an important regulator of acid-base homeostasis90. The significantly increased expression of proton-acceptors suggested the direct response of liver to feed-induced acidosis. For future follow up studies, it might be valuable to investigate the timing at which these proton acceptors are up-regulated during the process of acidosis development. These genes carry the high potential to be developed into sensitive, early diagnosis biomarkers.
Liver transcriptome changes related to the alterations in rumen epimural microbial community
Out of the genes that showed significant expression correlation with the microbial genus abundance in the epimural microbial community, 11 of them were involved in the pathway of transferase activity. Acetate, butyrate and propionate are the primary VFAs produced by bacterial fermentation within the gastrointestinal tract91. VFAs produced by the microbiota in the gut can be found in hepatic, portal and peripheral blood92,93. To prevent high VFA concentration in blood, the liver clears major portion of the VFAs from the portal circulation94. In human studies, up to 70% of the acetate (one major form of VFA) is taken up by the liver as a substrate for the synthesis of cholesterol in addition to being used as a source of energy94. Three major liver transferase enzymes, gamma–glutamyltransferase (GGT), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) are important for cholesterol metabolism. Increased level of GGT is independently correlated with elevated serum cholesterol95. Additionally, maximal induction of GGT was reported in butyrate-treated cells, indicating the responsive reaction of GGT activity to butyrate96. Different ratios of AST:ALT have also been reported as a potential clinical risk marker of liver metabolic syndrome97.
Two genes involved in transferase activity were associated with the most number of rumen microbial genera. SIK3 (Salt-inducible kinase 3) was linked to 10 genera, and KCNH3 (Potassium Voltage-Gated Channel Subfamily H Member 3) was linked to seven genera. SIK3 is an AMP-activated protein kinase-related kinase. In a mouse SIK3-deficent (SIK3 −/−) model, SIK3 −/− mice showed low expression levels for the gene components involved in the fatty acids synthesis pathways. Additionally, when fed a high cholesterol diet, SIK3 −/− mice lacked the ability to adapt to the increased cholesterol and developed clear liver damage98. Consistent with these findings, another study reported SIK3 as a new regulator of lipid homeostasis in the mouse liver by regulating the clearance of cholesterol and bile acids99. A growing number of studies have reported the beneficial roles of VFAs (also referred as short chain fatty acids) in energy homeostasis and lipid metabolism via stimulating several hormonal and neural signals in multiple tissues100,101. Of note, short chain fatty acids have been reported with positive impact in prevention and treatment of the metabolic syndrome102,103, ulcerative colitis104,105, Crohn’s disease106, and antibiotic-associated diarrhea107. Due to these potentially beneficial effects, the VFA producing ability of the associated microbial genera identified in this study may yield fruitful insights into theirs roles in liver metabolism and associated metabolic disease in dairy cattle.
Other genes with positive correlation with the rumen microbial community included SIRT6 and ACAT2. SIRT6 was previously reported with an anti-inflammatory role in the liver108. ACAT2 is specifically expressed in the hepatocytes. As a major cholesterol esterification enzyme, it controls the amount of hepatic free cholesterol available to secrete into the portal blood109. The potential protective roles of these genes in liver damage resultant from ruminal acidosis or excessive accumulation of VFAs due to high-concentrated feed warrant future follow-up studies.
Future perspectives
Our study captured liver transcriptome changes in response to the feed-induced acidosis in young calves. Importantly, our study identified significant expression changes in several groups of genes with the high potential of being further developed into biomarkers. These genes included acute phase proteins, proton-acceptors in liver tissues, and these involved in HDL and transferase activity. For future follow-up studies, the identification of host liver genes associated with lipid metabolism and VFA producing microbial species are of particular interest. As these genes sit at the direct junction between liver metabolism and the VFA production of gut microbial species. Specifically, functional confirmation of the genes involved in liver lipid metabolism and their association with VFA producing microbes will help the development of VFA-producing microbes as a new method to promote energy metabolism and liver health when a highly concentrated diet is used. Knowledge gained through these studies can be used to formulate precision ruminant feed, with the goals of improving the productivity and performance of ruminants while maintaining optimal liver health are met. More importantly, the optimized feed will facilitate the improved balances between the host’s metabolism and gut microbial ecology.
Data availability
Gene raw read-counts of liver tissues were included in the supplemental data. rRNA raw reads rumen papilla tissues were submitted to NCBI with project accession number of PRJNA493225.
References
Council, N. R. Nutrient Requirements of Dairy Cattle: Seventh Revised Edition, https://doi.org/10.17226/9825 (2001).
NAHMS. NAHMS Beef Cow-calf Studies, https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/monitoring-and-surveillance/nahms/nahms_beef_cowcalf_studies (2017).
Sander, E. G., Warner, R. G., Harrison, H. N. & Loosli, J. K. The Stimulatory Effect of Sodium Butyrate and Sodium Propionate on the Development of Rumen Mucosa in the Young Calf. Journal of Dairy Science 42, 1600–1605, https://doi.org/10.3168/jds.S0022-0302(59)90772-6 (1959).
Stobo, I. J. F., Roy, J. H. B. & Gaston, H. J. Rumen Development in Calf .1. Effect of Diets Containing Different Proportions of Concentrates to Hay on Rumen Development. Brit J Nutr 20, 171–&, https://doi.org/10.1079/Bjn19660021 (1966).
Mentschel, J., Leiser, R., Mulling, C., Pfarrer, C. & Claus, R. Butyric acid stimulates rumen mucosa development in the calf mainly by a reduction of apoptosis. Arch Tierernahr 55, 85–102 (2001).
Laarman, A. H. & Oba, M. Short communication: Effect of calf starter on rumen pH of Holstein dairy calves at weaning. J Dairy Sci 94, 5661–5664, https://doi.org/10.3168/jds.2011-4273 (2011).
Gozho, G. N., Plaizier, J. C., Krause, D. O., Kennedy, A. D. & Wittenberg, K. M. Subacute ruminal acidosis induces ruminal lipopolysaccharide endotoxin release and triggers an inflammatory response. J Dairy Sci 88, 1399–1403, https://doi.org/10.3168/jds.S0022-0302(05)72807-1 (2005).
Zebeli, Q. et al. Modeling the adequacy of dietary fiber in dairy cows based on the responses of ruminal pH and milk fat production to composition of the diet. J Dairy Sci 91, 2046–2066, https://doi.org/10.3168/jds.2007-0572 (2008).
Garrett, E. F. et al. Diagnostic methods for the detection of subacute ruminal acidosis in dairy cows. J Dairy Sci 82, 1170–1178, https://doi.org/10.3168/jds.S0022-0302(99)75340-3 (1999).
Oetzel, G. R. Monitoring and testing dairy herds for metabolic disease. Vet Clin North Am Food Anim Pract 20, 651–674, https://doi.org/10.1016/j.cvfa.2004.06.006 (2004).
Bramley, E. et al. The definition of acidosis in dairy herds predominantly fed on pasture and concentrates. J Dairy Sci 91, 308–321, https://doi.org/10.3168/jds.2006-601 (2008).
Gozho, G. N., Krause, D. O. & Plaizier, J. C. Ruminal lipopolysaccharide concentration and inflammatory response during grain-induced subacute ruminal acidosis in dairy cows. J Dairy Sci 90, 856–866, https://doi.org/10.3168/jds.S0022-0302(07)71569-2 (2007).
Emmanuel, D. G., Dunn, S. M. & Ametaj, B. N. Feeding high proportions of barley grain stimulates an inflammatory response in dairy cows. J Dairy Sci 91, 606–614, https://doi.org/10.3168/jds.2007-0256 (2008).
Kleen, J. L., Hooijer, G. A., Rehage, J. & Noordhuizen, J. P. Subacute ruminal acidosis (SARA): a review. J Vet Med A Physiol Pathol Clin Med 50, 406–414 (2003).
Uhart, B. A. & Carroll, F. D. Acidosis in beef steers. J Anim Sci 26, 1195–1198 (1967).
Gaebel, G., Martens, H., Suendermann, M. & Galfi, P. The effect of diet, intraruminal pH and osmolarity on sodium, chloride and magnesium absorption from the temporarily isolated and washed reticulo-rumen of sheep. Q J Exp Physiol 72, 501–511 (1987).
Garry, F. B. Indigestion in ruminants. Large Animal Internal Medicine, Mosby-Year Book, Mosby, St. Louis, Missouri (pp. 722–747 (2002).
Krause, K. M. & Oetzel, G. R. Inducing subacute ruminal acidosis in lactating dairy cows. Journal of Dairy Science 88, 3633–3639, https://doi.org/10.3168/jds.S0022-0302(05)73048-4 (2005).
Mutsvangwa, T. et al. Effects of a monensin controlled-release capsule or premix on attenuation of subacute ruminal acidosis in dairy cows. Journal of Dairy Science 85, 3454–3461, https://doi.org/10.3168/jds.S0022-0302(02)74433-0 (2002).
Plaizier, J. C., Keunen, J. E., Walton, J. P., Duffield, T. F. & McBride, B. W. Effect of subacute ruminal acidosis on in situ digestion of mixed hay in lactating dairy cows. Can. J. Anim. Sci. 81, 421–423.
Steele, M. A. et al. Bovine rumen epithelium undergoes rapid structural adaptations during grain-induced subacute ruminal acidosis. Am J Physiol Regul Integr Comp Physiol 300, R1515–1523, https://doi.org/10.1152/ajpregu.00120.2010 (2011).
Khafipour, E., Krause, D. O. & Plaizier, J. C. A grain-based subacute ruminal acidosis challenge causes translocation of lipopolysaccharide and triggers inflammation. J Dairy Sci 92, 1060–1070, https://doi.org/10.3168/jds.2008-1389 (2009).
Oetzel, G. R. Diagnosis and Management of Subacute Ruminal Acidosis in Dairy Herds. Vet Clin North Am Food Anim Pract 33, 463–480, https://doi.org/10.1016/j.cvfa.2017.06.004 (2017).
Dirksen, G. U., Liebich, H. G. & Mayer, E. Adaptive changes of the ruminal mucosa and their functional and clinical significance. Bovine Practitioner 20, 116–120 (1985).
Nagaraja, T. G. & Titgemeyer, E. C. Ruminal acidosis in beef cattle: the current microbiological and nutritional outlook. J Dairy Sci 90(Suppl 1), E17–38, https://doi.org/10.3168/jds.2006-478 (2007).
Brown, T. R. & Lawrence, T. E. Association of liver abnormalities with carcass grading performance and value. J Anim Sci 88, 4037–4043, https://doi.org/10.2527/jas.2010-3219 (2010).
Rezac, D. J. et al. Prevalence, severity, and relationships of lung lesions, liver abnormalities, and rumen health scores measured at slaughter in beef cattle. J Anim Sci 92, 2595–2602, https://doi.org/10.2527/jas.2013-7222 (2014).
Brink, D. R., Lowry, S. R., Stock, R. A. & Parrott, J. C. Severity of liver abscesses and efficiency of feed utilization of feedlot cattle. J Anim Sci 68, 1201–1207 (1990).
Nagaraja, T. G. & Chengappa, M. M. Liver abscesses in feedlot cattle: a review. J Anim Sci 76, 287–298 (1998).
Jensen, R., Deane, H. M., Cooper, L. J., Miller, V. A. & Graham, W. R. The rumenitis-liver abscess complex in beef cattle. Am J Vet Res 15, 202–216 (1954).
Smith, H. A. Ulcerative lesions of the bovine rumen and their possible relation to hepatic abscesses. Am J Vet Res. 5, 234–242 (1944).
Berg, J. N. & Scanlan, C. M. Studies of Fusobacterium necrophorum from bovine hepatic abscesses: biotypes, quantitation, virulence, and antibiotic susceptibility. Am J Vet Res 43, 1580–1586 (1982).
Simon, P. C. & Stovell, P. L. Isolation of Sphaerophorus necrophorus from bovine hepatic abscesses in British Columbia. Can J Comp Med 35, 103–106 (1971).
Calkins, H. E. & Scrivner, L. H. Isolation of Sphaerophorus necrophorus from Bovine Liver Abscesses. Appl Microbiol 15, 1492–1493 (1967).
Langworth, B. F. Fusobacterium necrophorum: its characteristics and role as an animal pathogen. Bacteriol Rev 41, 373–390 (1977).
Scanlan, C. M. & Hathcock, T. L. Bovine rumenitis - liver abscess complex: a bacteriological review. Cornell Vet 73, 288–297 (1983).
Yoshioka, M., Ito, T., Miyazaki, S. & Nakajima, Y. The release of tumor necrosis factor-alpha, interleukin-1, interleukin-6 and prostaglandin E2 in bovine Kupffer cells stimulated with bacterial lipopolysaccharide. Vet Immunol Immunopathol 66, 301–307 (1998).
Li, W., Gelsinger, S., Edwards, A., Riehle, C. & Koch, D. Transcriptome analysis of rumen epithelium and meta-transcriptome analysis of rumen epimural microbial community in young calves with feed induced acidosis. Sci Rep 9, 4744, https://doi.org/10.1038/s41598-019-40375-2 (2019).
Gelsinger, S. L., Coblentz, W. K., Zanton, G. I., Ogden, R. K. & Akins, M. S. Ruminal in situ disappearance and whole-tract digestion of starter feeds in calves before, during, and after weaning. J Dairy Sci 102, 2196–2206, https://doi.org/10.3168/jds.2018-15551 (2019).
Gelsinger, S., Coblentz, W., Zanton, G., Ogden, R. & Akins, M. Physiological effects of starter-induced ruminal acidosis in calves before, during, and after weaning. Journal of Dairy Science In Press (2019).
Busby, M. A., Stewart, C., Miller, C. A., Grzeda, K. R. & Marth, G. T. Scotty: a web tool for designing RNA-Seq experiments to measure differential gene expression. Bioinformatics 29, 656–657, https://doi.org/10.1093/bioinformatics/btt015 (2013).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36, https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, https://doi.org/10.1093/bioinformatics/btu638 (2015).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Dennis, G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3 (2003).
Szklarczyk, D. et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39, D561–568, https://doi.org/10.1093/nar/gkq973 (2011).
Szklarczyk, D. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–D368, https://doi.org/10.1093/nar/gkw937 (2017).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15, R46, https://doi.org/10.1186/gb-2014-15-3-r46 (2014).
Boulter, E. et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol 12, 477–483, https://doi.org/10.1038/ncb2049 (2010).
Watson, A. P., Evans, R. L. & Egland, K. A. Multiple functions of sushi domain containing 2 (SUSD2) in breast tumorigenesis. Mol Cancer Res 11, 74–85, https://doi.org/10.1158/1541-7786.MCR-12-0501-T (2013).
Kornfeld, S. & Mellman, I. The biogenesis of lysosomes. Annu Rev Cell Biol 5, 483–525, https://doi.org/10.1146/annurev.cb.05.110189.002411 (1989).
Kornfeld, S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu Rev Biochem 61, 307–330, https://doi.org/10.1146/annurev.bi.61.070192.001515 (1992).
Scrace, S. F. & O’Neill, E. RASSF Signalling and DNA Damage: Monitoring the Integrity of the Genome? Mol Biol Int 2012, 141732, https://doi.org/10.1155/2012/141732 (2012).
Schormann, N., Ricciardi, R. & Chattopadhyay, D. Uracil-DNA glycosylases-Structural and functional perspectives on an essential family of DNA repair enzymes. Protein Science 23, 1667–1685, https://doi.org/10.1002/pro.2554 (2014).
Tetzner, R., Dietrich, D. & Distler, J. Control of carry-over contamination for PCR-based DNA methylation quantification using bisulfite treated DNA. Nucleic Acids Research 35, doi:ARTN e4 10.1093/nar/gkl955 (2007).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Guo, J. et al. Rumen-derived lipopolysaccharide provoked inflammatory injury in the liver of dairy cows fed a high-concentrate diet. Oncotarget 8, 46769–46780, https://doi.org/10.18632/oncotarget.18151 (2017).
Kleen, J. L., Upgang, L. & Rehage, J. Prevalence and consequences of subacute ruminal acidosis in German dairy herds. Acta Vet Scand 55, 48, https://doi.org/10.1186/1751-0147-55-48 (2013).
Nordlund, K. Herd-based diagnosis of subacute ruminal acidosis. Proceedings of the 36th Annual Conference of American Association of Bovine, 1–6 (2003).
Hofmann, J., Greter, M., Du Pasquier, L. & Becher, B. B-cells need a proper house, whereas T-cells are happy in a cave: the dependence of lymphocytes on secondary lymphoid tissues during evolution. Trends Immunol 31, 144–153, https://doi.org/10.1016/j.it.2010.01.003 (2010).
Plitas, G. et al. Dendritic cells are required for effective cross-presentation in the murine liver. Hepatology 47, 1343–1351, https://doi.org/10.1002/hep.22167 (2008).
Ebrahimkhani, M. R., Mohar, I. & Crispe, I. N. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 54, 1379–1387, https://doi.org/10.1002/hep.24508 (2011).
Warren, A. et al. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology 44, 1182–1190, https://doi.org/10.1002/hep.21378 (2006).
Crociati, M. et al. Influence of lipoproteins at dry-off on metabolism of dairy cows during transition period and on postpartum reproductive outcomes. Theriogenology 94, 31–36, https://doi.org/10.1016/j.theriogenology.2017.02.007 (2017).
Stefanska, B. et al. Subacute ruminal acidosis affects fermentation and endotoxin concentration in the rumen and relative expression of the CD14/TLR4/MD2 genes involved in lipopolysaccharide systemic immune response in dairy cows. J Dairy Sci 101, 1297–1310, https://doi.org/10.3168/jds.2017-12896 (2018).
Martin-Campos, J. M., Escola-Gil, J. C., Ribas, V. & Blanco-Vaca, F. Apolipoprotein A-II, genetic variation on chromosome 1q21-q24, and disease susceptibility. Curr Opin Lipidol 15, 247–253 (2004).
Warden, C. H., Hedrick, C. C., Qiao, J. H., Castellani, L. W. & Lusis, A. J. Atherosclerosis in transgenic mice overexpressing apolipoprotein A-II. Science 261, 469–472 (1993).
Su, X., Kong, Y. & Peng, D. Q. New insights into apolipoprotein A5 in controlling lipoprotein metabolism in obesity and the metabolic syndrome patients. Lipids Health Dis 17, 174, https://doi.org/10.1186/s12944-018-0833-2 (2018).
Zaki, M. E., Amr, K. S. & Abdel-Hamid, M. APOA2 Polymorphism in Relation to Obesity and Lipid Metabolism. Cholesterol 2013, 289481, https://doi.org/10.1155/2013/289481 (2013).
Shu, X., Nelbach, L., Ryan, R. O. & Forte, T. M. Apolipoprotein A-V associates with intrahepatic lipid droplets and influences triglyceride accumulation. Biochim Biophys Acta 1801, 605–608, https://doi.org/10.1016/j.bbalip.2010.02.004 (2010).
Huang, X. S. et al. Decreased apolipoprotein A5 is implicated in insulin resistance-related hypertriglyceridemia in obesity. Atherosclerosis 210, 563–568, https://doi.org/10.1016/j.atherosclerosis.2009.12.004 (2010).
Dougherty, R. W., Coburn, K. S., Cook, H. M. & Allison, M. J. Preliminary study of appearance of endotoxin in circulatiory system of sheep and cattle after induced grain engorgement. Am J Vet Res 36, 831–832 (1975).
Shen, Y., Yang, W., Chen, L., Xu, J. & Wang, H. Effects of grain source and starch concentration in dairy goat diet on ruminal fermentation, milk production and inflammation. ASAS-CSAS Annual Meeting & Trade Show, Baltimore, MD., 318 (2017).
Contreras-Duarte, S., Varas, P., Awad, F., Busso, D. & Rigotti, A. Protective role of high density lipoproteins in sepsis: basic issues and clinical implications. Rev Chilena Infectol 31, 34–43, https://doi.org/10.4067/S0716-10182014000100005 (2014).
Khovidhunkit, W. et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 45, 1169–1196, https://doi.org/10.1194/jlr.R300019-JLR200 (2004).
Beutler, B., Hoebe, K., Du, X. & Ulevitch, R. J. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol 74, 479–485, https://doi.org/10.1189/jlb.0203082 (2003).
Read, T. E. et al. The protective effect of serum lipoproteins against bacterial lipopolysaccharide. Eur Heart J 14(Suppl K), 125–129 (1993).
Baumann, H. & Gauldie, J. The Acute-Phase Response. Immunol Today 15, 74–80, https://doi.org/10.1016/0167-5699(94)90137-6 (1994).
Kellum, J. A., Song, M. & Li, J. Lactic and hydrochloric acids induce different patterns of inflammatory response in LPS-stimulated RAW 264.7 cells. Am J Physiol Regul Integr Comp Physiol 286, R686–692, https://doi.org/10.1152/ajpregu.00564.2003 (2004).
Heming, T. A. et al. Effects of extracellular pH on tumour necrosis factor-alpha production by resident alveolar macrophages. Clin Sci (Lond) 101, 267–274 (2001).
Bellocq, A. et al. Low environmental pH is responsible for the induction of nitric-oxide synthase in macrophages - Evidence for involvement of nuclear factor-kappa B activation. Journal of Biological Chemistry 273, 5086–5092, https://doi.org/10.1074/jbc.273.9.5086 (1998).
Farwell, W. R. & Taylor, E. N. Serum anion gap, bicarbonate and biomarkers of inflammation in healthy individuals in a national survey. Can Med Assoc J 182, 134–141, https://doi.org/10.1503/cmaj.090329 (2010).
Rizvi, A. A. Cytokine Biomarkers, Endothelial Inflammation, and Atherosclerosis in the Metabolic Syndrome: Emerging Concepts. Am J Med Sci 338, 310–318, https://doi.org/10.1097/MAJ.0b013e3181a4158c (2009).
Eckersall, P. D. Recent advances and future prospects for the use of acute phase proteins as markers of disease in animals. Rev Med Vet-Toulouse 151, 577–584 (2000).
Eckersall, P. D. & Conner, J. G. Bovine and Canine Acute Phase Proteins. Veterinary Research Communications 12, 169–178, https://doi.org/10.1007/Bf00362798 (1988).
Jacobsen, S., Andersen, P. H., Toelboell, T. & Heegaard, P. M. Dose dependency and individual variability of the lipopolysaccharide-induced bovine acute phase protein response. J Dairy Sci 87, 3330–3339, https://doi.org/10.3168/jds.S0022-0302(04)73469-4 (2004).
Danscher, A. M., Thoefner, M. B., Heegaard, P. M. H., Ekstrom, C. T. & Jacobsen, S. Acute phase protein response during acute ruminal acidosis in cattle. Livestock Science 135, 62–69, https://doi.org/10.1016/j.livsci.2010.06.009 (2011).
Scheiner, B. et al. Acid-base disorders in liver disease. J Hepatol 67, 1062–1073, https://doi.org/10.1016/j.jhep.2017.06.023 (2017).
den Besten, G. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. Journal of Lipid Research 54, 2325–2340, https://doi.org/10.1194/jlr.R036012 (2013).
Murase, M., Kimura, Y. & Nagata, Y. Determination of Portal Short-Chain Fatty-Acids in Rats Fed Various Dietary-Fibers by Capillary Gas-Chromatography. J Chromatogr B 664, 415–420, https://doi.org/10.1016/0378-4347(94)00491-M (1995).
Cummings, J. H., Pomare, E. W., Branch, W. J., Naylor, C. P. E. & Macfarlane, G. T. Short Chain Fatty-Acids in Human Large-Intestine, Portal, Hepatic and Venous-Blood. Gut 28, 1221–1227, https://doi.org/10.1136/gut.28.10.1221 (1987).
Bloemen, J. G. et al. Short chain fatty acids exchange across the gut and liver in humans measured at surgery. Clin Nutr 28, 657–661, https://doi.org/10.1016/j.clnu.2009.05.011 (2009).
Paolicchi, A. et al. Beta-lipoprotein- and LDL-associated serum gamma-glutamyltransferase in patients with coronary atherosclerosis. Atherosclerosis 186, 80–85, https://doi.org/10.1016/j.atherosclerosis.2005.07.012 (2006).
Oster, T. et al. Induction of recombinant human gamma-glutamyl transferase by sodium butyrate in transfected V79 and CHO Chinese hamster cells. Biochem Biophys Res Commun 193, 406–412, https://doi.org/10.1006/bbrc.1993.1638 (1993).
Yadav, D. et al. Incremental Predictive Value of Serum AST-to-ALT Ratio for Incident Metabolic Syndrome: The ARIRANG Study. Plos One 11, https://doi.org/10.1371/journal.pone.0161304 (2016).
Uebi, T. et al. Involvement of SIK3 in Glucose and Lipid Homeostasis in Mice. Plos One 7, https://doi.org/10.1371/journal.pone.0037803 (2012).
Ito, Y., Uebi, T., Kumagai, A., Sanosaka, M. & Takemori, H. SIK3 is a new regulator of lipid homeostasis in the mouse liver. Faseb Journal 26 (2012).
Gao, Z. G. et al. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 58, 1509–1517, https://doi.org/10.2337/db08-1637 (2009).
Kimura, I. et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nature Communications 4, https://doi.org/10.1038/ncomms2852 (2013).
Hu, G. X., Chen, G. R., Xu, H., Ge, R. S. & Lin, J. Activation of the AMP activated protein kinase by short-chain fatty acids is the main mechanism underlying the beneficial effect of a high fiber diet on the metabolic syndrome. Med Hypotheses 74, 123–126, https://doi.org/10.1016/j.mehy.2009.07.022 (2010).
Donohoe, D. R. et al. The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon. Cell Metabolism 13, 517–526, https://doi.org/10.1016/j.cmet.2011.02.018 (2011).
Harig, J. M., Soergel, K. H., Komorowski, R. A. & Wood, C. M. Treatment of Diversion Colitis with Short-Chain Fatty-Acid Irrigation. New Engl J Med 320, 23–28, https://doi.org/10.1056/Nejm198901053200105 (1989).
Breuer, R. I. et al. Rectal irrigation with short-chain fatty acids for distal ulcerative colitis. Preliminary report. Dig. Dis. Sci. 36, 185-187 (1991).
Di Sabatino, A. et al. Oral butyrate for mildly to moderately active Crohn’s disease. Aliment Pharmacol Ther 22, 789–794, https://doi.org/10.1111/j.1365-2036.2005.02639.x (2005).
Binder, H. J. Role of colonic short-chain fatty acid transport in diarrhea. Annu Rev Physiol 72, 297–313, https://doi.org/10.1146/annurev-physiol-021909-135817 (2010).
Xiao, C. Y. et al. Progression of Chronic Liver Inflammation and Fibrosis Driven by Activation of c-JUN Signaling in Sirt6 Mutant Mice. Journal of Biological Chemistry 287, 41903–41913, https://doi.org/10.1074/jbc.M112.415182 (2012).
Parini, P. et al. ACAT2 is localized to Hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation 110, 2017–2023, https://doi.org/10.1161/01.Cir.0000143163.76212.0b (2004).
Acknowledgements
W.L. and S.G. were supported by appropriated project 5090-31000-026-00-D from the USDA Agriculture Research Service (US Dairy Forage Research Center). Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation by the US Department of Agriculture. The USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Contributions
W.L. and S.G. conceived and designed the Experiment. S.G. carried out the feeding trial, measurements of the feed intake, weight gain and ruminal pH values. A.E., C.R., S.G. and W.L. performed animal tissue collection and processing. S.G. performed the analysis of non-sequencing data. A.E., C.R. and W.L. performed RNA extraction, RNA sequencing library preparation and sequencing and RT-qPCR. experiment. A.E., C.R. and W.L. performed the analysis of data generated from RT-qPCR. W.L. and D.K. performed bioinformatics data analysis of RNA-sequencing data. W.L. and S.G. wrote and prepared the figures/tables of the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, W., Gelsinger, S., Edwards, A. et al. Changes in meta-transcriptome of rumen epimural microbial community and liver transcriptome in young calves with feed induced acidosis. Sci Rep 9, 18967 (2019). https://doi.org/10.1038/s41598-019-54055-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54055-8
- Springer Nature Limited