
Abstract
The foundation of food web analysis is a solid understanding of predator-prey associations. Traditional dietary studies of fishes have been by stomach content analysis. However, these methods are not applicable to Critically Endangered species such as the smalltooth sawfish (Pristis pectinata). Previous research using the combination of stable isotope signatures from fin clips and 18S rRNA gene sequencing of fecal samples identified the smalltooth sawfish as piscivorous at low taxonomic resolution. Here, we present a high taxonomic resolution molecular technique for identification of prey using opportunistically acquired fecal samples. To assess potential biases, primer sets of two mitochondrial genes, 12S and 16S rRNA, were used alongside 18S rRNA, which targets a wider spectrum of taxa. In total, 19 fish taxa from 7 orders and 11 families native to the Gulf of Mexico were successfully identified. The sawfish prey comprised diverse taxa, indicating that this species is a generalist piscivore. These findings and the molecular approach used will aid recovery planning for the smalltooth sawfish and have the potential to reveal previously unknown predator-prey associations from a wide range of taxa, especially rare and hard to sample species.
Similar content being viewed by others

Introduction
The smalltooth sawfish (Pristis pectinata) currently inhabits southwest Florida and the Florida Keys but was once widely distributed on both coasts of the Atlantic Ocean and in the Gulf of Mexico1. Decades of human activity, including bycatch mortality in commercial and recreational fishing and loss of red mangrove (Rhizophora mangle) shorelines associated with residential and commercial development, have greatly reduced the size of the smalltooth sawfish population1,2. Thus, in 2003 the species was listed as endangered under the U.S. Endangered Species Act.
Despite recent expansion of knowledge about the smalltooth sawfish3, its feeding ecology is poorly understood. To maximize the effectiveness of ongoing recovery planning, more detailed knowledge of the trophic ecology of smalltooth sawfish is needed to better understand what specific prey they rely on. Traditional dietary studies on piscivores typically involve lethal sampling for stomach content analysis, gastric lavage, and morphological hard part analysis of indigestible prey remains4,5,6,7. Morphological characterization of prey remains found during gastric lavage or lethal sampling permits conclusions to be drawn about the number and size of fish prey consumed8,9; however, lethal sampling is not feasible for endangered species and gastric lavage, though considered non-invasive, may not be a permitted activity for endangered species10. Therefore, alternative approaches to identify predator-prey associations are warranted. Recent research using the combination of stable isotope signatures from fin clips and 18S rRNA gene sequencing of fecal samples identified the smalltooth sawfish as piscivorous at low taxonomic resolution11. However, a higher taxonomic resolution understanding of this species’ diet is needed to implement effective recovery planning through species-specific management of its prey base, which may include popular commercial and sport fishes.
Here we present a high-resolution molecular method for identification of smalltooth sawfish prey using fecal samples collected over a six-year period. To assess potential biases that might be caused by the selection of primer sets, two mitochondrial genes, 12S and 16S rRNA, were used together with 18S rRNA, a more evolutionarily conserved gene that targets a wider spectrum of taxa11. Based on Poulakis and colleagues11 previous findings, we expected to find that smalltooth sawfish fed upon rays and various teleost fish species, some of which may be of anthropogenic interest, putting them in direct competition with humans. These data will improve recovery planning for this Critically Endangered species and the analysis technique has the potential to reveal previously unknown predator-prey associations of a wide range of taxa, especially rare and hard to sample species.
Results
Smalltooth sawfish fecal sample description
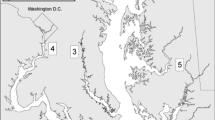
During field sampling from 2010 through 2015, 16 fecal samples were opportunistically obtained from primarily juvenile (<2500 mm) smalltooth sawfish (780–4355 mm stretch total length; mean = 1398 mm) in a roughly equal proportion of males (n = 8) and females (n = 7) in southwest Florida from the Charlotte Harbor estuarine system and the Ten Thousand Islands National Wildlife Refuge (Fig. 1; Table 1). DNA was unable to be extracted from one sample due to an insufficient amount of feces.

Map of smalltooth sawfish fecal sample collection locations in southwest Florida with stacked bars showing relative composition of fish prey taxa (minimum of 5%), after removal of host sequences. Each 100% stacked bar shows data from mitochondrial 12S (black border) and 16S (grey border) rRNA genes. No fish prey taxa were detected in 16S rRNA gene analysis of samples SF3, SF4, and SF12. DNA sequencing of 12S rRNA gene failed in SF4 and SF5. SF2 did not yield enough DNA for analysis from its extraction. Map generated with QGIS Desktop 2.18 (https://www.qgis.org/en/site/) using data provided by the Florida Fish and Wildlife Conservation Commission-Fish and Wildlife Research Institute.
18S rRNA gene analysis
The mean number of analyzed reads for each sample was 110,843 ± 39,647 (±SE; Supplementary Table S1). After normalization (scaled to 10,000 reads) and subsequent removal of host sequences (i.e., smalltooth sawfish), four Kingdoms were identified: Animalia (91.3%), Chromalveolata (6.2%), Plantae (2.4%), and Fungi (0.1%) (Fig. 2). All non-animal taxa were microscopic and considered to be of sediment or ambient water origin. Within Animalia, fishes comprised the majority of sequences (Actinopterygii: 83.1%; Elasmobranchii: 3.3%). In the majority of samples, Arthropoda sequences made up <1% of Animalia sequences; however, considerable numbers were identified in one sample, SF4, with 99.2% of Animalia sequences and 100% of Arthropoda sequences identified as penaeid shrimp (Supplementary Table S2), with 91.5% of these sequences exhibiting 99% similarity to reference sequences (Litopenaeus setiferus: JX403844.1; Farfantepenaeus duorarum: JX403828.1). SF4 was the smallest individual in our analysis (780 mm) and had no fish prey detected in our mitochondrial 16S rRNA gene analysis (Fig. 3).

Abundance of normalized 18S rRNA gene sequence reads by Kingdom (left) and composition of Animalia (right) from smalltooth sawfish fecal samples after removal of host sequences. Animalia composed 91.3% of sequence reads (left), with fishes (Actinopterygii and Elasmobranchii) composing 86.4% of Animalia sequence reads (right). Others includes Mollusca and Platyhelminthes.

Comparison of fish prey taxa detection in smalltooth sawfish fecal samples using mitochondrial 12S and 16S rRNA gene sequences. Sample locations indicated next to sample code (see footnote). SF2 did not yield enough DNA for analysis from its extraction.
Mitochondrial 12S and 16S rRNA gene analysis
Preliminary results revealed large amounts of unidentified sequences present in our samples and a lack of local fish species reference sequences in GenBank, indicating the need for additional sequencing effort. Thus, we sequenced 24 fish species, including nine orders and 13 families, known to inhabit our sampling area12, yielding 16 and 23 additional sequences for the mitochondrial 12S and 16S rRNA genes, respectively (Supplementary Table S3). The high specificity of these sequences allowed closely related species to be distinguished in most cases, which was beneficial in determining the highest resolution (i.e., species level) identifications for Operational Taxonomic Units (OTUs) with Basic Local Alignment Search Tool (BLAST) results that did not meet our identification criterion (98% similarity) or exhibited ties. To provide even more robust analysis, species signature sequences consisting of ~40 bp within 12S and 16S rRNA were identified for 150 and 128 species (Supplementary Table S4), respectively, to conduct manual alignments of unidentified sequences during the final step of our identification protocol. This tag-sequencing strategy attributed to 17.4% and 2.8% of identified 12S and 16S rRNA sequences, respectively (Supplementary Table S5).
The mean (±SE) number of analyzed reads for all samples was 8,963 ± 1,074. Two samples (SF4, SF5) failed to sequence for 12S rRNA analysis due to unsuccessful PCR amplicon generation. After removal of host sequences, our analysis identified 91.3 ± 1.8% of 12S rRNA and 88.9 ± 4.9% of 16S rRNA gene sequence reads at a minimum of genus (Table 2). Of 19 total fish taxa detected, including 7 orders and 11 families, 17 were identified to species, with a mean of 15.1 ± 3.2 OTUs and 2.3 ± 0.3 fishes identified in 12S rRNA fecal samples and 6.8 ± 2.3 OTUs and 1.7 ± 0.4 fishes identified in 16S rRNA fecal samples. Silver perch (Bairdiella chrysoura), bay anchovy (Anchoa mitchilli), tidewater mojarra (Eucinostomus harengulus), spotted seatrout (Cynoscion nebulosus), ladyfish (Elops saurus), and spot (Leiostomus xanthurus) were most prevalent in our analysis (Figs. 3 and 4; Supplementary Table S6). For each gene, Bray Curtis similarity indices were calculated after removing individuals in which no prey taxa were detected. A hierarchical clustering based on these results showed that similar river originated samples (i.e., Peace River and Caloosahatchee River) exhibited similarities and clustered together (Supplementary Fig. S1).

Normalized abundance of detected fish prey taxa in mitochondrial 12S (A) and 16S (B) rRNA gene sequence reads from smalltooth sawfish fecal samples. In the 12S rRNA gene analysis, 18 taxa were identified, and in the 16S rRNA gene analysis, 16 taxa were identified. The same taxa were included in both graphs for comparative purposes.
The 16S rRNA primer set appeared to be less sensitive than the 12S rRNA primer set when comparing samples sequenced for both genes. In 16S rRNA analysis, only three (all tidewater mojarra) of 26 identification instances were not corroborated by the other gene, whereas for 12S rRNA, 16 of 34 identification instances were not corroborated. Additionally, the 16S rRNA primer set was unable to identify any fish taxa for two samples (SF3, SF12), and striped mojarra (Eugerres plumieri) and pinfish (Lagodon rhomboides) were only identified using the 12S rRNA primer set (Fig. 3). While two seatrout species, sand seatrout (C. arenarius) and spotted seatrout (C. nebulosus), were identified in both analyses, species were not always distinguishable in our 12S rRNA analysis with some identifications relegated to Cynoscion sp., indicating that multiple primer sets may be desirable to overcome potential biases.
Discussion
Traditional dietary studies of fishes have mainly focused on stomach content analysis, using invasive methods that cannot be used to study Critically Endangered species such as the smalltooth sawfish. DNA-based prey identification can overcome many of these limitations, and a variety of molecular methods have been developed over the past decade to enable the investigation of feeding ecology at unprecedented resolution13,14,15,16. Moreover, even heavily digested prey remains can be identified by these methods to track trophic links17. Molecular techniques have been used to assess the diet of piscivores with a focus on marine predators such as pinnipeds18, squids19, and seabirds20; however, this is the first study achieving species level resolution for elasmobranchs. In addition, there has been limited application of these techniques for studying the feeding ecology of fishes more generally21.
In the present study, we combined multiple molecular markers to identify fish prey to species. Mitochondrial 12S and 16S rRNA genes were used to target fishes, which were previously identified as the largest fraction of smalltooth sawfish prey taxa via the 18S rRNA gene11. Using a set of high-resolution mitochondrial genes expanded our results by accounting for differences between the two primer sets, with samples exhibiting variation in taxa detection and proportions between them. An example of the need for multiple genes occurred with silver perch and tidewater mojarra which were often detected together but exhibited drastic differences in sequence proportions between genes. In some samples, the silver perch was only detected by 12S rRNA while tidewater mojarra was only detected using 16S rRNA. At first, we thought this indicated an error in our analysis or sample preparation for Sanger sequencing, but after comparing identified query sequences of both species for both genes, our sequences of both species, and reference sequences of congeners, all sequences identified as one or the other species via multiple sequence alignment. This process indicated that both sequences belong to these two species, were well differentiated from each other, were similar to other species within their respective genera, and met our threshold for valid identifications. Without the use of two overlapping high taxonomic resolution primer sets, there would have been undetected or undervalued taxa due to biases of individual primer sets and their respective genes.
We also used species-signature sequences consisting of around 40 bp to refine our data, increasing the number of successfully identified sequences and OTUs. However, in two samples, we were unable to differentiate species in the genus Cynoscion for 12S rRNA OTUs. The only additional Cynoscion species not identified in the study was the silver seatrout (C. nothus), which is not known to use the estuary where these samples were collected12,22. In one sample, we were unable to differentiate species in the genus Menticirrhus for 16S rRNA OTUs. Although our identification protocol identified these as M. littoralis, this species is not known to use the estuary where this sample was collected22, and relevant reference sequences of the two possible other Menticirrhus species (i.e., M. americanus and M. saxatilus) were absent from DNA databases. Further, Pompanon and colleagues14 discussed various potential biases of this new technology that could hamper quantitative questions, such as differences in amplification efficiency or DNA survival during digestion. Because the methodology is progressing rapidly, these problems might be minimized in the near future; however, some issues, such as variation in prey size and the time the prey was consumed, are not easily addressed. For example, we were unable to rule out instances of secondary predation (i.e., consumption of a predator which has consumed the target prey), which has not been addressed in any vertebrate predator13,23. These challenges are generally present in feeding ecology analyses using traditional techniques and are not limited to molecular methods24.
A high-throughput sequencing approach, able to detect trace amounts of prey, was first used on sawfish as a complementary method to assist studies using stable isotopes, a powerful and widely accepted technique in which trophic levels can be identified, but species level prey identification is impossible11,25,26,27,28,29. Poulakis and colleagues11 studied the trophic ecology of the smalltooth sawfish in South Florida using a combination of stable isotopes and 18S rRNA gene sequencing of fecal samples, providing evidence that the species feeds primarily on teleost and elasmobranch fishes. In the present study, we replicated and built on this technique with a larger sample size, resulting in successful identification of more prey and reaffirming the previous conclusion that this species is piscivorous. Furthermore, the prey identified comprised diverse taxa (19 fish taxa from 7 orders and 11 families), indicating that this species is a generalist piscivore. Fish prey items were detected in fecal samples from all individuals with the exception of one, which fed primarily on penaeid shrimp, which we speculate was scavenged discarded bait.
The fish prey we identified mirrored the habitats juvenile smalltooth sawfish are known to use within the Charlotte Harbor estuary30. Sawfish preyed on species such as the tidewater mojarra that tends to be found along mangrove shorelines, silver perch and spotted seatrout that are typically found on offshore seagrass flats, and bay anchovies and pinfish that are found in both habitats31. Acoustic tracking and monitoring in the study area has shown that smalltooth sawfish use all habitats available to them32,33, and the present study suggests that they feed on fishes in all of these habitats. Additionally, we did not observe an ontogenetic dietary shift in our data, which is a trait common amongst elasmobranchs34,35. No clear differences in prey were observed between juvenile age classes (less than 1 year old: <150 cm STL; greater than 1 year old: >150 cm STL36), with fishes such as bay anchovy, ladyfish, and silver perch found in each. A dietary shift may become evident if more samples from larger juveniles and adults could be collected as these age classes are known to use more diverse habitats3,11.
Although we know little about sawfish feeding behavior and diet beyond anecdotal reports and previous stable isotope analysis11, this study presents multiple new findings, such as the presence of anchovies, one of the smallest fish prey items identified, and the southern stingray, which was anticipated by the 18S rRNA analysis that identified Myliobatiformes in the diet11. Recent behavioral experiments have shown captive sawfish feeding with rapid lateral swipes of their rostra in the water column and close to the bottom37. Our data suggest that smalltooth sawfish are generalist piscivores that feed on pelagic and benthic fishes.
These findings will aid recovery planning for the smalltooth sawfish and the molecular approach used has the potential to reveal previously unknown predator-prey associations from a wide range of taxa, especially protected species. This study shows that smalltooth sawfish consume a variety of fish prey species found across a diverse range of estuarine habitats. Thus, maintaining healthy estuaries, including healthy fish populations, will be important for promoting recovery of the smalltooth sawfish population and should be more effective than species-specific management approaches. Additionally, climate change and associated environmental impacts may destabilize current habitats for the species and alter habitat use, further emphasizing the need for ecosystem level conservation38.
Materials and Methods
Smalltooth sawfish fecal sample collection and DNA extraction
From 2010 to 2015, 16 fecal samples were opportunistically obtained from primarily juvenile smalltooth sawfish in southwest Florida during ongoing field sampling or from necropsies (Table 1). All samples were stored at −20 °C until further analysis. DNA extractions were performed using the Quick-DNA Fecal/Soil Microbe Kits (Zymo Research, Irvine, CA, USA) according to manufacturer instructions. DNA was unable to be extracted from one sample (SF2) due to an insufficient amount of feces and 15 fecal samples were used in further sequencing analysis. All methods were carried out in accordance with relevant guidelines and regulations.
High-throughput sequencing
The extracted DNA samples were used for Illumina sequencing to determine the 18S rRNA, mitochondrial 12S and 16S rRNA genes. Amplification of the 18S rRNA gene was conducted using a universal primer pair (TAReuk454FWD1 [CCA GCA SCY GCG GTA ATT CC] and TAReukREV3 [ACT TTC GTT CTT GAT YRA]). We also applied Illumina sequencing to mitochondrial 12S and 16S rRNA genes to detect fish taxa39. The mitochondrial 12S rRNA gene was amplified using a universal primer pair for Actinopterygii (Ac12Sf [ACT GGG ATT AGA TAC CCC ACT ATG] and Ac12Sr [GAG AGT GAC GGG CGG TGT]). The mitochondrial 16S rRNA gene was amplified using a universal primer pair for Actinopterygii (Ac16Sf [CCT TTT GCA TCA TGA TTT AGC] and Ac16Sr [CAG GTG GCT GCT TTT AGG C]). The DNA samples were amplified for sequencing in a two-step process. The forward primer was constructed (5′-3′) with the forward Illumina overhang adapter (TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG) added to the forward primer used. The reverse primer was constructed (5′-3′) with the reverse Illumina overhang adapter (GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G) added to the reverse primer used. Amplifications were performed in a 25 µL reaction with the Qiagen HotStar Taq master mix (Qiagen, Valencia, California, USA), 1 µL of each 5 µM primer, and 1 µL of template. Reactions were performed on the ABI Veriti thermocycler (Applied Biosytems, Carlsbad, California, USA) under the following thermal profile for 18S rRNA: an initial denaturation for 5 min at 95 °C, followed by 35 cycles of denaturation for 30 s at 94 °C, annealing for 1.5 min at 50 °C and ramp up at 0.5 °C per cycle (1.5 min) in the first 10 cycles and the following 25 cycles at 54 °C, and extension for 1 min at 72 °C, with a final extension of 10 min at 72 °C. In 12S and 16S rRNA analyses, we used the same thermal profile, but with a constant annealing temperature of 54 °C for all 35 cycles. The PCR product from the first stage amplification was added to a second PCR based on qualitatively determined concentrations. Primers for the second PCR were designed based on the Illumina Nextera PCR primers as follows: forward - AAT GAT ACG GCG ACC ACC GAG ATC TAC AC [i5index] TCG TCG GCA GCG TC and reverse - CAA GCA GAA GAC GGC ATA CGA GAT [i7index] GTC TCG TGG GCT CGG. The second stage amplification was run the same as the first except with only 10 cycle extensions. For the 18S rRNA gene, the first stage thermal profile used in 12S and 16S rRNA gene amplification was applied for these 10 cycles. Amplicons were visualized with eGels (Life Technologies, Grand Island, New York, USA) and products were pooled equimolar and each pool was size selected in two rounds using SPRIselect reagent (Beckman Coulter, Indianapolis, Indiana, USA) in a 0.75 ratio for both rounds. Size selected pools were then quantified using the Qubit 4 fluorometer (Life Technologies). The final library pool was analyzed on the MiSeq system (Illumina, San Diego, California, USA) using a Miseq reagent kit v3 (Illumina) with pair-end run setting of 2 × 300 flow cell at 10 pM. We ran 12S and 16S rRNA gene amplification on the same Miseq reagent kit, and 18S rRNA gene was run separately. All PCR reactions were run with no-template control; the negative control was also tagged and sequenced along with samples for contamination check purposes. Illumina sequencing was performed at RTL Genomics (Lubbock, Texas, USA).
To analyze the sequence data, forward and reverse reads were taken in FASTQ format and merged using PEAR Illumina paired-end read merger40. The formatted FASTQ files were then converted into FASTA-formatted files for subsequent analyses. DNA sequences were clustered into OTUs at 97% similarity using the CD-Hit-Est clustering algorithm41,42. OTUs consisting of less than five sequences were considered inconsequential and omitted from further analyses (12S rRNA gene sequences omitted: 2.06 ± 0.23% [mean ± SE]; 16S rRNA: 0.65 ± 0.08%; 18S rRNA: 1.28 ± 0.06%), with the remainder entering our identification protocol using BLAST.
Sequencing error of 12S and 16S rRNA genes was estimated to be 2% by comparing OTU centroid sequences identified as smalltooth sawfish by BLAST and their respective similarity percentages with a reference sequence (GenBank Accession: KP400584) for each gene via multiple sequence alignment using the MUSCLE program within MEGA7 software43 to manually check for chimeric sequences (Supplementary Fig. S2). This estimated sequencing error was used in our sequence identification protocol. Sequencing error was not estimated for 18S rRNA gene analysis due to its low associated taxonomic resolution, with the taxonomic class of the highest scoring BLAST similarity result accepted for each OTU. When class was not applicable, the next highest taxonomic level was used. Taxonomy follows Page and colleagues44 for fishes and Williams45 for crustaceans.
Mitochondrial 12S and 16S rRNA gene sequence identification protocol
Based on our estimation of sequencing error found in the host, BLAST results with a similarity score ≥98% were accepted for each prey OTU. If this criterion was met and ties were present, OTUs would be moved to the next step of our identification protocol. The remaining OTUs along with those with ties were aligned with ~40 bp species signature sequences identified for all fish species documented in the sample collection area12 that either had reference sequences available in GenBank or were sequenced for this study (Supplementary Table S1). This tag-sequencing strategy determined ~40 bp species signature sequences based on available data, with each sequence checked for specificity via BLAST. Exact matches were accepted as an accurate identification. If an exact match was not made with a tied OTU, the lowest shared taxonomic classification between the tied BLAST results was accepted (none were found higher than genus level, and all were Cynoscion sp.). All results were checked against available fisheries data22, with any dubious identifications relegated to the lowest viable taxonomic classification (none higher than genus level and all were Menticirrhus sp.).
Sanger sequencing for mitochondrial 12S and 16S rRNA genes of potential fish prey
Twenty-four fish species known from our sampling area were sequenced (Fig. 1; Supplementary Table S3). Sixteen species were sequenced for the mitochondrial 12S rRNA gene and 23 species for the mitochondrial 16S rRNA gene using the same primer sets used in the high-throughput sequencing39 except for a minor modification in Ac16Sr(C-) [CAG GTG GCT GCT TTT AGG C]). Preparation of sequencing samples was carried out as described previously11.
Data deposit
High-throughput mitochondrial 12S and 16S rRNA, and 18S rRNA gene sequences of smalltooth sawfish fecal samples were deposited in the National Center for Biotechnology Information sequence read archive under accession number SAMN10130720. Mitochondrial 12S and 16S rRNA gene fish sequences were deposited in GenBank under accession numbers MH715297-312 and MH715980-6002, respectively.
References
Carlson, J., Wiley, T. & Smith, K. Pristis pectinata. The IUCN Red List of Threatened Species 2013: e.T18175A141791261, https://doi.org/10.2305/IUCN.UK.2013-1.RLTS.T18175A141791261.en (accessed 5 August 2019) (2013).
Wueringer, B. E., Squire, L. & Collin, S. P. The biology of extinct and extant sawfish (Batoidea: Sclerorhynchidae and Pristidae). Rev. Fish Biol. Fish. 19, 445–464 (2009).
Brame, A. B. et al. Biology, ecology, and status of the smalltooth sawfish Pristis pectinata in the USA. Endang. Species Res. 39, 9–23 (2019).
Blewett, D. A., Hensley, R. A. & Stevens, P. W. Feeding habits of common snook, Centropomus undecimalis, in Charlotte Harbor, Florida. Gulf. Caribb. Res. 18, 1–14 (2015).
Bangley, C. W., Rulifson, R. A. & Overton, A. S. Evaluating the Efficiency of flushed stomach-tube lavage for collecting stomach contents from dogfish sharks. Southeast. Nat. 12, 523–533 (2013).
Dias, E., Morais, P., Leopold, M., Campos, J. & Antunes, C. Natural born indicators: Great cormorant Phalacrocorax carbo (Aves: Phalacrocoracidae) as monitors of river discharge influence on estuarine ichthyofauna. J. Sea Res. 73, 101–108 (2012).
Sinclair, E. H. & Zeppelin, T. K. Seasonal and spatial differences in diet in the western stock of steller sea lions (Eumetopias Jubatus). J. Mammal. 83, 973–990 (2005).
Gagliardi, A., Martinoli, A., Preatoni, D., Wauters, L. A. & Tosi, G. From mass of body elements to fish biomass: A direct method to quantify food intake of fish eating birds. Hydrobiologia. 583, 213–222 (2007).
Mariano-jelicich, R. & Favero, M. Assessing the diet of the black skimmer through different methodologies: is the analysis of pellets reliable? Waterbirds. 29, 81–87 (2006).
Barrett, R. T. Food web interactions in the southwestern Barents Sea: Black-legged kittiwakes Rissa tridactyla respond negatively to an increase in herring Clupea harengus. Mar. Ecol. Prog. Ser. 349, 269–276 (2007).
Poulakis, G. R. et al. Sympatric elasmobranchs and fecal samples provide insight into the trophic ecology of the smalltooth sawfish. Endang. Species Res. 32, 491–506 (2017).
Poulakis, G. R., Matheson, R. E. Jr., Mitchell, M. E., Blewett, D. A. & Idelberger, C. F. Fishes of the Charlotte Harbor estuarine system, Florida. Gulf Mex. Sci. 2, 117–150 (2004).
King, R. A., Read, D. S., Traugott, M. & Symondson, W. O. C. Molecular analysis of predation: A review of best practice for DNA-based approaches. Mol. Ecol. 17, 947–963 (2008).
Pompanon, F. et al. Who is eating what: Diet assessment using next generation sequencing. Mol. Ecol. 21, 1931–1950 (2012).
Symondson, W. O. C. & Harwood, J. D. Special issue on molecular detection of trophic interactions: Unpicking the tangled bank. Mol. Ecol. 23, 947–693 (2014).
Traugott, M., Kamenova, S., Ruess, L., Seeber, J. & Plantegenest, M. Empirically characterising trophic networks: what emerging dna-based methods, stable isotope and fatty acid analyses can offer. Adv. Ecol. Res. 49, 177–224 (2013).
Hindell, M. A., Gales, N. J., Jarman, S. N., Casper, R. M. & Deagle, B. E. Detecting prey from DNA in predator scats: A comparison with morphological analysis, using Arctocephalus seals fed a known diet. J. Exp. Mar. Bio. Ecol. 347, 144–154 (2007).
Deagle, B. E. et al. Analysis of Australian fur seal diet by pyrosequencing prey DNA in faeces. Mol. Ecol. 18, 2022–2038 (2009).
Deagle, B. E. et al. Molecular scatology as a tool to study diet: Analysis of prey DNA in scats from captive Steller sea lions. Mol. Ecol. 14, 1831–1842 (2005).
Bowser, A. K., Diamond, A. W. & Addison, J. A. From puffins to plankton: A DNA-based analysis of a seabird food chain in the northern Gulf of Maine. PLoS One 8, 1–17 (2013).
Leray, M., Meyer, C. P. & Mills, S. C. Metabarcoding dietary analysis of coral dwelling predatory fish demonstrates the minor contribution of coral mutualists to their highly partitioned, generalist diet. PeerJ 3, e1047 (2015).
Idelberger, C. F. & Greenwood, M. F. D. Seasonal variation in fish assemblages within the estuarine portions of the Myakka and Peace Rivers, southwest Florida. Gulf Mex. Sci. 23, 224–240 (2005).
Hardy, N. et al. Assessing the trophic ecology of top predators across a recolonisation frontier using DNA metabarcoding of diets. Mar. Ecol. Prog. Ser. 573, 237–254 (2017).
Johnson, J. H., DiRado, J. A., Mackey, G. & Abbet, R. Comparative diets and foraging strategies of subyearling Atlantic salmon, brown trout, and rainbow trout during winter. J. Appl. Ichthyol. 33, 1158–1165 (2017).
Olin, J. A. et al. Variable δ15N diet-tissue discrimination factors among sharks: implications for trophic position, diet and food web models. PLoS One 8, 1–12 (2013).
Hussey, N. E., Chapman, D. D., Donnelly, E., Abercrombie, D. L. & Fisk, A. T. Fin-icky samples: An assessment of shark fin as a source material for stable isotope analysis. Limnol. Oceanogr. Methods. 9, 524–532 (2011).
Hussey, N. E. et al. Stable isotopes and elasmobranchs: tissue types, methods, applications and assumptions. J. Fish Biol. 80, 1449–1484 (2012).
Hussey, N. E. et al. Size-based analysis of diet and trophic position of the white shark Carcharodon carcharias in South African waters in global perspectives on the biology and life history of the white shark, M. Domeier, Eds, pp. 27–48 (CRC Press, 2012).
Malpica-Cruz, L., Herzka, S. Z., Sosa-Nishizaki, O. & Lazo, J. P. Tissue-specific isotope trophic discrimination factors and turnover rates in a marine elasmobranch: empirical and modeling results. Can. J. Fish. Aquat. Sci. 69, 551–564 (2012).
Poulakis, G. R., Stevens, P. W., Timmers, A. A., Wiley, T. R. & Simpfendorfer, C. A. Abiotic affinities and spatiotemporal distribution of the endangered smalltooth sawfish, Pristis pectinata, in a south-western Florida nursery. Mar. Freshw. Res. 62, 1165–1177 (2011).
Poulakis, G. R., Blewett, D. A. & Mitchell, M. E. The effects of season and proximity to fringing mangroves on seagrass-associated fish communities in Charlotte Harbor, Florida. Gulf Mex. Sci. 21, 171–184 (2003).
Poulakis, G. R., Stevens, P. W., Timmers, A. A., Stafford, C. J. & Simpfendorfer, C. A. Movements of juvenile endangered smalltooth sawfish, Pristis pectinata, in an estuarine river system: Use of non-main-stem river habitats and lagged responses to freshwater inflow-related changes. Environ. Biol. Fishes. 96, 763–778 (2013).
Huston, C. A. et al. Diel movements of juvenile smalltooth sawfish: Implications for defining the size of a nursery hotspot. Endang. Species Res. 34, 311–322 (2017).
Bethea, D. M., Buckel, J. A. & Carlson, J. K. Foraging ecology of the early life stages of four sympatric shark species. Mar. Ecol. Prog. Ser. 268, 245–264 (2004).
Newman, S. P., Handy, R. D. & Gruber, S. H. Ontogenetic diet shifts and prey selection in nursery bound lemon sharks, Negaprion brevirostris, indicate a flexible foraging tactic. Environ. Biol. Fishes. 95, 115–126 (2012).
Scharer, R. M., Patterson, W. F., Carlson, J. K. & Poulakis, G. R. Age and growth of endangered smalltooth sawfish (Pristis pectinata) verified with LA-ICP-MS analysis of vertebrae. PLoS One 7, e47850 (2012).
Wueringer, B. E., Squire, L., Kajiura, S. M., Hart, N. S. & Collin, S. P. The function of the sawfish’s saw. Curr. Biol. 22, R150–R151 (2012).
Poulakis, G. R. & Grubbs, R. D. Biology and ecology of sawfishes: global status of research and future outlook. Endang. Species Res. 39, 77–90 (2019).
Evans, N. T. et al. Quantification of mesocosm fish and amphibian species diversity via environmental DNA metabarcoding. Mol. Ecol. Resour. 16, 29–41 (2016).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina paired-end read merger. Bioinformatics 30, 614–620 (2014).
Huang, Y., Niu, B., Gao, Y., Fu, L. & Li, W. CD-HIT suite: A web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682 (2010).
Li, W. & Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Page, L. et al. Common and scientific names of fishes from the United States, Canada, and Mexico. 7 th ed. (American Fisheries Society, Bethesda, 2013).
Williams, A. Shrimps, lobsters, and crabs of the Atlantic coast of the eastern United States, Maine to Florida (Smithsonian Institution, Washington, D.C., 1984).
Acknowledgements
Our smalltooth sawfish research is ongoing and has been supported primarily by funding from the U.S. Department of Commerce (DOC), National Oceanic and Atmospheric Administration’s (NOAA) National Marine Fisheries Service through Section 6 (Cooperation with the States) of the U.S. Endangered Species Act under the following grant awards to the Florida Fish and Wildlife Conservation Commission from NOAA: NA10NMF4720032 and NA13NMF4720047. Part of this research was also supported by the Save Our Seas Foundation, Marco Island Shell Club, the Florida Gulf Coast University Whitaker Center for STEM Education, and the Blair Foundation. Statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the views or policies of the funders. This research was conducted under Endangered Species Permit numbers 1475 and 15802 issued to GRP by NOAA Fisheries.
Author information
Authors and Affiliations
Contributions
T.L.H. and H.U. conducted laboratory analyses, G.R.P. and R.M.S. conducted sample collection, and all authors (T.L.H., H.U., G.R.P., R.M.S. and S.G.T.) contributed to data interpretation and manuscript writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hancock, T.L., Poulakis, G.R., Scharer, R.M. et al. High-resolution molecular identification of smalltooth sawfish prey. Sci Rep 9, 18307 (2019). https://doi.org/10.1038/s41598-019-53931-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53931-7
- Springer Nature Limited