
Abstract
Cardiovascular diseases form the most common cause of death worldwide, with atherosclerosis as main etiology. Atherosclerosis is marked by cholesterol rich lipoprotein deposition in the artery wall, evoking a pathogenic immune response. Characteristic for the disease is the pathogenic accumulation of macrophages in the atherosclerotic lesion, which become foam cells after ingestion of large quantities of lipoproteins. We hypothesized that, by inducing a CD8 T cell response towards lipoprotein derived apolipoprotein-B100 (ApoB100), lesional macrophages, that are likely to cross-present lipoprotein constituents, can specifically be eliminated. Based on in silico models for protein processing and MHC-I binding, 6 putative CD8 T cell epitopes derived from ApoB100 were synthesized. HLA-A2 binding was confirmed for all peptides by T2 cell binding assays and recall responses after vaccination with the peptides proved that 5 of 6 peptides could induce CD8 T cell responses. Induction of ApoB100 specific CD8 T cells did not impact plaque size and cellular composition in HLA-A2 and human ApoB100 transgenic LDLr−/− mice. No recall response could be detected in cultures of cells isolated from the aortic arch, which were observed in cell cultures of splenocytes and mesenteric lymph nodes, suggesting that the atherosclerotic environment impairs CD8 T cell activation.
Similar content being viewed by others


Introduction
Cardiovascular disease is the most common cause of death in the western world with atherosclerosis as the most common etiology1. Atherosclerosis is characterized by lipid deposition in the intima of medium to large-sized arteries, evoking immune infiltration in the vessel wall and inflammation. Among the immune cells attracted to the atherosclerotic lesions are CD8 T cells2. CD8 T cells in atherosclerotic lesions appear highly activated3 suggesting a pathogenic role for CD8 T cells atherosclerosis. Recent studies indeed support the notion of an overall pro-atherogenic role of CD8 T cells in atherosclerosis through the secretion of pro-inflammatory cytokines4,5,6. Nonetheless, atheroprotective effects of CD8 T-cells in the lesion have been reported as well7.
These conflicting reports may result from the fact that CD8 T cells form a heterogeneous population consisting of different subsets and cells recognizing different antigens. The largest CD8 T cell subset is the cytotoxic CD8 T cell or cytotoxic lymphocyte (CTL)8. The primary function of CTLs is to protect the host from intracellular pathogens9 and tumors10. Specific recognition of a target cell is established through positive interaction between the T cell receptor of the CTL, which is variable between individual CD8 T cells, and MHC-I complexed with a particular antigen derived peptide on the target cell. MHC-I/peptide complex recognition results in TNF-α and IFN-γ production by CTLs and leads to FAS ligand or perforin and granzyme B mediated cell death of the target cell11. The specificity of the TCR heavily impacts its pathogenicity, as depending on target antigen, vaccination induced CTLs can be atheroprotective12,13,14,15 or atherogenic16. Induction of CTL reactivity towards vascular cells like smooth muscle cells, enhanced vessel inflammation and atherosclerosis16. As suppressing apoptosis of macrophages was found to enhance atherosclerosis17,18 and the absence of MHCI molecules aggravates atherosclerosis19 we hypothesize that CD8 T cell mediated killing of macrophages is an essential process controlling progression of atherosclerosis. Intra-lesional TLR activation20 and presence of apoptotic bodies21, both potent inducers of cross presentation22,23, are likely to induce cross presentation of plaque antigens by lesion macrophages. Presumable cross-presentation of LDL derived apolipoprotein-B100 (ApoB100) epitopes on MHC-I by lesional phagocytes, suggests that inducing ApoB100 specific CD8 T cells could lead to killing of lesional macrophages and reduce atherosclerosis.
Since only a very small fraction of peptides binds to MHC-I24, we set out to identify CD8 T cell epitopes in ApoB100 to test this hypothesis. With in silico prediction models for HLA binding and antigen processing, human HLA-A2 restricted epitopes derived from human ApoB100 were predicted for translational relevancy. 6 ApoB100 derived peptides were selected and synthesized and binding of all peptides to HLA-A2 was confirmed with HLA-A2 assays in T2 cells25. Thereafter we performed vaccination studies using these peptides, inducing substantial levels of peptide specific memory CD8 T cells in HLA-A2 and human ApoB100 transgenic LDLr−/− mice. Although ApoB100 specific CTLs were induced by ApoB100 peptide vaccination, these CD8 T cells did not change cellular plaque composition, plaque collagen content, and plaque size, indicating that induction of ApoB100 specific CD8 T cells does not affect atherosclerosis.
Results
Predicted HLA-A2 restricted epitopes stabilize HLA-A2 and induce peptide specific CD8 T cell responses after DC vaccination
To target CD8 T cells towards plaque macrophages which are likely to cross-present plaque derived antigens, we predicted putative HLA-A2 restricted CD8 T cell epitopes in human ApoB100 using in silico models for immunoproteasomal processing and TAP binding26,27,28 and HLA-A2 binding models28,29,30,31,32,33,34,35. We synthesized 6 peptides with the highest putative HLA-A2 binding and processing score (Table 1). To establish the binding of the ApoB100 peptides to HLA-A2 in vitro, T2 cells were incubated overnight with ascending amounts of ApoB100 peptides and thereafter the expression of HLA-A2 on the T2 cells with flow cytometry was assessed (Supplementary Fig. S1). Because T2 cells are deficient for TAP1 and TAP2 expression, cytosolic peptide transport into the ER is reduced in T2 cells, reducing endogenous peptide loading onto MHC-I in the ER. MHC-I not complexed with a peptide is inefficiently transported to the cell membrane and is less stable, therefore leading to lowered expression of HLA-A2 on the cell membrane of T2 cells. Exogenous addition of peptides, forming MHC-I/peptide complexes on the cell surface, enhances MHC-I stability on the plasma membrane which increases expression of HLA-A2 on the T2 cell surface25. All 6 ApoB100 peptides increased HLA-A2 expression in a concentration dependent manner (Fig. 1A), confirming that these peptides bind to HLA-A2.

Predicted HLA-A2 restricted epitopes stabilize HLA-A2 and induces peptide specific CD8 T cell responses after DC vaccination. (A) Binding of in silico predicted CD8 T cell epitopes was assessed using T2 cells. T2 cell cultures (n = 3/peptide) were incubated overnight with a range of peptide concentrations or vehicle control (DMSO) and assessed for HLA-A2 expression by flow cytometry. (B) Next we assessed whether these ApoB100 derived peptides could induce an HLA-A2 restricted CD8 T cell response. HLA-A2tg mice (n = 2 per peptide), not expressing human ApoB100, were vaccinated with 2*106 HLA-A2tg bone marrow derived DCs pulsed with a single peptide (30 μM). A week after vaccination splenocytes were ex vivo incubated with the peptide against they were vaccinated. Graphs of peptide specific T cell responses a measured by flow cytometry through gating for CD44 and IFN-γ double positive T cells. Statistical analysis of A was performed with 2-way ANOVA and Bonferroni posttest, displayed as mean with SEM. For B, samples stimulated with peptide were compared to the unstimulated control of the same animal with contingency chi-square tests and Bonferroni posttest (corrected for 96 pairwise comparisons), individual samples are plotted. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To test the immunogenicity of the peptides we vaccinated HLA-A2 transgenic animals (HHD mice)36 with HHD bone marrow derived DCs (2*106 cells), which were overnight stimulated with LPS (100 ng/ml) and pulsed with a single peptide (10 µM). One week after vaccination, we assessed the induction of peptide-specific CTLs through flow cytometric measurement of IFN-γ positive antigen experienced (CD44+) CD4 and CD8 T cells (Supplementary Fig. S1), in splenocyte cultures which were incubated for 4 h with escalating concentrations of the ApoB100 peptide against which the particular animal was vaccinated, and Brefeldin A. For all peptides, except for ApoB2524–2532, the percentage of IFN-y positive antigen experienced CD8 T cells was increased over no peptide control, while the vaccinations did not affect CD4 T cell IFN-y production (Fig. 1B). This indicates that 5 of 6 peptides can induce HLA-A2 restricted CD8 T cell responses in vivo.
Peptide vaccination induces peptide responsive CD8 T cells in spleen and mediastinal lymph nodes but not in aorta
A suitable experimental model to assess the role of human ApoB100 specific T cells in the context of atherosclerosis was generated by crossbreeding HHD mice, transgenic for HLA-A2 and deficient in expression of murine MHC-I molecules36, with HuBL mice, expressing human ApoB100 and deficient for the LDLr37,38. HLA-A2 and human ApoB100 transgenic mice, which were LDLr deficient to allow atherosclerosis development, and either with normal expression (HuBL-A2m+) or devoid of murine MHC-I expression (HuBL-A2m−) were generated. Because immunization with the ApoB100 peptides in both mouse strains yielded similar immunological results and effect on plaque parameters in the atherosclerosis studies, mainly the results obtained with female HuBL-A2m− mice are shown with some complementary data obtained in male HuBL-A2m+ mice.
HuBL-A2m− mice received western type diet for 9 weeks starting from 15 weeks of age. Mice of the treatment group were i.v. vaccinated with HHD DCs (2*106 cells) pulsed with a mixture of the 6 ApoB100 derived peptides (10 μM/peptide) at the start of western type diet. Mice were i.v. boosted a week later with peptides (30 μM/peptide) adjuvanted with anti-CD40 (50 μg) and poly(I:C) (50 μg), previously shown to induce long lasting high levels of antigen-specific memory CD8 T cells39,40,41. Control animals received unpulsed control DCs (2*106 cells) at WTD initiation and a week later adjuvant without peptides (vehicle group), or received two PBS injections (PBS group). At the end of the experiment we determined whether vaccination induced peptide specific CD8 T cells. To that end, CD8 T cell recall responses were assessed with flow cytometry in cell cultures of the spleen (2*106 cells) and mediastinal lymph nodes (1*106 cells), which drain from the atherosclerosis prone aortic arch, incubated for 4 h with the ApoB100 derived peptides (10 μM/peptide) and Brefeldin A. The CD8 T cell percentage was significantly higher in the peptide-treated group compared to the PBS group and trended to be higher compared to the unpulsed DC/vehicle group in spleen (Fig. 2A) and blood (Supplementary Fig. S2) but not in mediastinal lymph nodes (Fig. 2D). Incubation of splenocytes with ApoB406–414, ApoB3070–3078 or ApoB4531–4539 enhanced the percentage of IFN-γ and TNF-α double positive CD8 T cells in the splenocytes cultures of the peptide-treated group, indicative of successful long-term induction of memory CD8 T cells for these peptides (Fig. 2B,C). In cell cultures from the mediastinal lymph nodes, incubated with a combination of all peptides, also a higher percentage of IFN-γ+ and TNF-α+ double positive CD8 T cells was observed in the peptide vaccinated group (Fig. 2E,F), although at a much lower percentage than in the spleen. In circulation, a strong increase in effector memory CD8 T cells and a reduction in naïve CD8 T cells were observed in the peptide-treated group compared to both control groups (Supplementary Fig. S2), suggesting that vaccination also resulted in peptide specific CD8 T-cells in circulation.

Peptide vaccination induces peptide responsive CD8 T cells. (A) Quantification of splenic CD8 T cell percentage. (B) Representative flow cytometry plots of splenic CD8 T cell activation by ApoB406–414 in the different treatment groups. (C) Quantification of IFN-γ and TNF-α double positive cells % from CD8 T cells. (D) Quantification of CD8 T cell percentage after ex vivo peptide stimulation mediastinal lymph nodes. (E) Representative flow cytometry plots of mediastinal lymph node CD8 T cell activation and (F) quantification of the IFN-γ and TNF-α double positive cell % from CD8 T cells after combined peptide stimulation. Statistical analysis was performed with 1-way ANOVA and Tukey’s multiple comparisons test. Depicted as mean with SEM, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Vaccination with ApoB100 derived CD8 epitopes does not affect atherosclerotic lesion size and composition
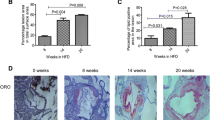
To assess the effect of ApoB100 specific CD8 T cell induction on atherosclerotic lesion development, we stained neutral lipids in aortic root sections with ORO (Fig. 3A), and quantified plaque size (control 4.18 ± 1.79*105, vehicle 4.55 ± 1.32*105, treated 5.57 ± 2.35*105) and vessel occlusion (Fig. 3B). To assess the stability of the plaques we stained aortic root sections with Masson’s Trichrome (Fig. 3C), and quantified the absolute area of plaque collagen and collagen content of the plaques (Fig. 3D). Finally, with immunohistochemical staining with MOMA-2 antibody (Fig. 3E), we quantified the absolute macrophage area and macrophage content of aortic root atherosclerotic plaques (Fig. 3F). None of the mentioned plaque parameters were significantly changed by induction of ApoB100 specific CD8 T cells. Also, cholesterol levels were not influenced by ApoB100 peptide vaccination (Supplementary Fig. S3).

Vaccination with ApoB100 derived CD8 epitopes does not affect lesion size and composition. (A) Representative microscopic images of ORO staining of aortic root tissue sections. (B) Quantification of average plaque size and average vessel occlusion. (C) Plaque stability was assessed by collagen content determination through analysis of Sirius red stained tissue slides. Representative microscopic images of aortic root tissue sections stained with Sirius red. (D) Quantification of collagen content of atherosclerotic lesions in the aortic root. (E) Representative microscopic images of tissue slides stained with MOMA-2 staining (F) and quantification of MOMA-2 surface area and plaque content. Statistical analysis was performed with 1-way ANOVA and Tukey’s multiple comparisons test. Plotted as mean with SEM.
ApoB100 peptide vaccination enhances CD8 T cell content but does not affect other immune cell populations in the aorta
Because the induced ApoB100 peptide specific CD8 T cells were hypothesized to reduce atherosclerosis through killing of plaque phagocytes cross-presenting the ApoB100 peptides, we assessed peptide reactivity and phenotype of CD8 T cells in the aortic arch, and the general immune content of the aortic arch.
To assess reactivity of CD8 T cells to the ApoB peptides, aortas from three mice from the same treatment group were pooled, digested, and cells were isolated and cultured for 4 h with all peptides combined (10 μM/peptide) and Brefeldin A, after which cells were assessed with flow cytometry (gating, Supplementary Fig. S4). The total percentage of CD8 T cells was significantly increased in the peptide treated group over PBS treated, and a trend towards increase (p = 0.0573) was observed compared to the vehicle treated (Fig. 4A). In contrast to the observed recall responses in the spleens and mediastinal lymph nodes from mice vaccinated with the ApoB100 peptides, we did not observe a recall response in the cell cultures from the aortic arch of the mice vaccinated with ApoB100 peptides (Fig. 4B,C).

Vaccination with ApoB100 peptides does enhance CD8 T cell levels but not reactivity towards ApoB100 peptides in the aortic root (A) Quantification of CD8 T cell percentage in aortic cultures. (B) Representative flow cytometry plots of aortic CD8 T cell activation and (C) quantification of IFN-γ and TNF-α double positive cell % from CD8 T cells after restimulation with a mix of all peptides. Statistical analysis was performed with 1-way ANOVA and Tukey’s multiple comparisons test. Depicted as mean with SEM, *P < 0.05.
In the atherosclerosis study using HuBL-A2m+ mice, the aortic CD8 T cell level, their maturation and activation was assessed with flow cytometry directly after isolation. In this study, a significant increase in CD8 T cell levels was found in ApoB peptide treated mice compared to vehicle treated mice, suggesting that migration of ApoB peptide specific T cells into the plaque occurred. We did not observe phenotypical differences in the aortic CD8 T cell population between vehicle and peptide treated animals (Supplementary Fig. S5). Besides an increase in CD8 T cells we did not detect significant changes in multiple myeloid and lymphoid cell populations in the aortic arch, shown for the aortic arch cultures of HuBL-A2m− mice (Supplementary Fig. S4), and myeloid and lymphoid cell populations directly after isolation from the aortic arch from HuBL-A2m+ mice (Supplementary Fig. S6).
As CD8 T cells require cell-cell contact for killing of target cells42, we also assessed whether CD8 T cells co-localized with macrophages, the most prominent APC population in the atherosclerotic lesion, in aortic root sections of female HuBL-A2m− mice. CD8 T cells were predominantly present in the cap structures of the atherosclerotic lesion possibly interacting with macrophages in the outer layers of the macrophage-rich area, and in addition we observed CD8 T cells in the adventitia. Macrophages were mostly present in the deeper layers of the plaque but in some tissue sections we also do observe CD8 T cells deeper in the plaque (Supplementary Fig. S7).
Thus, our data suggest that ApoB100 specific CD8 T cells do not have a major impact on the size and composition of the atherosclerotic lesion.
Discussion
The role of CD8 T cells in atherosclerosis is complex, with different roles for different CD8 T cell subsets and CD8 T cells with different antigen specificities. Recent studies suggest an overall pathogenic role for CTLs through secretion of pro-inflammatory cytokines4,5, which seems to correlate with observations of increased activated and cytokine producing CD8 T cells in peripheral blood of patients with coronary artery disease43,44,45. However, depending on target antigen, atherogenic and also atheroprotective12,13,14 antigen specific CD8 T cells responses have been reported. Because the suppression of macrophage apoptosis enhanced atherosclerosis17,18 and the absence of MHCI molecules aggravates atherosclerosis19, we reasoned that promoting CD8 T cell mediated killing of macrophages could act atheroprotective. As the inflammatory plaque environment is likely to promote cross-presentation of plaque antigens by lesional macrophages20,21,22,23, we hypothesized that we could specifically target CD8 T cells to kill lesional macrophages through vaccination with ApoB100 derived CD8 T cell epitopes, which was proposed to be the atheroprotective mechanism behind vaccination with ApoB100 derived peptide p21013,15.
To be applicable for vaccination in men, the CD8 epitopes have to be capable of binding human MHC-I. Therefore we predicted human HLA-A2 binding affinity and immunoproteasomal processing of peptide sequences present within ApoB100 with in silico prediction tools26,27,28,29,30,31,32,33,34,35. In line with a high accuracy of MHC-I binding models at predict MHC binders and non-binders46, all 6 predicted peptides were found to bind HLA-A2 in T2 cell binding assays. Subsequently we assessed the immunogenicity of the peptides in HLA-A2 transgenic HHD mice36. Vaccination of HHD mice with HHD DCs pulsed with a single ApoB100 derived peptide, led to robust peptide specific recall responses a week after vaccination for all peptides except for ApoB2524–2532, confirming immunogenicity for all ApoB100 derived peptides except ApoB2524–2532. Through utilization of mice and DCs without murine MHC-I expression, the observed CD8 T cell responses had to be restricted to HLA-A236.
The peptides used in this study were not previously assessed for their potential to modulate atherosclerosis. However, antibodies in the blood directed against LQWLKRVHANPLLIDVVTYLVALIPEPS, and FLIYITELLKKLQSTTVMNPYMKLAPGELT, containing the ApoB406–417 (LLIDVVTYLVAL), ApoB406–414 (LLIDVVTYL) and ApoB4531–4539 (FLIYITELL) peptides, were screened as a potential biomarker for acute myocardial infarction47. Antibodyies against LQWLKRVHANPLLIDVVTYLVALIPEPS and FLIYITELLKKLQSTTVMNPYMKLAPGELT were found in blood of patients suffering from acute myocardial infarction and individuals in the control group, and did not correlate with acute myocardial infarction47. Our vaccination approach is highly unlikely to have induced high antibody levels against the peptides, as priming occurred with peptide-pulsed MHC-I loaded DCs, followed by only a single booster with soluble peptide. Furthermore, a library screening of ApoB100 peptides for their capacity to induce T cell proliferation revealed that a peptide with the amino acid sequence HTFLIYITELLKKLQSTTVM, containing the ApoB4531–4539 (FLIYITELL) peptide that we used, could enhance proliferation of spleen derived T cells48.
To induce long lasting peptide specific CTLs in the atherosclerosis studies, HuBL-A2m+/− mice were primed with peptide pulsed HHD DCs, and then boosted after a week with peptide adjuvanted with an agonistic CD40 antibody and poly(I:C). In experimental cancer models this vaccination approach induces CTLs that are effective in penetrating and killing tumor cells, indicating that this vaccination approach yields migratory and functional CTLs39,40,41. Moreover multivalent vaccination enhanced the anti-tumor effect of this immunization approach41. For ApoB406–414, ApoB3070–3078, and ApoB4531–4539, robust recall responses were detected in the spleens of ApoB100 peptide vaccinated HuBL-A2m+/− mice, indicating successful vaccination. The vast majority of the ApoB100 specific CD8 T-cells made more than one cytokine (IFN-γ and TNF-α) suggesting they are functional CTLs49. Interestingly no CD8 T cell recall response could be detected for ApoB406–417 and ApoB2356–2364, suggesting thymic negative selection or peripheral tolerance induction towards ApoB406–417 and ApoB2356–2364 in the human ApoB100 expressing HuBL-A2m−/+ mice. Besides enhanced levels of ApoB100 peptide specific CD8 T cells in the spleen, we observed peptide specific CD8 T cells in the mediastinal lymph nodes, more effector CD8 T cells in the circulation and increased CD8 T cell levels in the aorta of ApoB100 vaccinated mice.
Despite induction of this relatively large antigen specific CD8 T cell response compared to studies in which p210 coupled to cationic BSA (p210-cBSA) was reported to reduce atherosclerosis through CD8 T cell induction13,15, we did not observe an atheroprotective effect of ApoB100 specific CD8 T cell induction. Although in silico prediction models suggest that p210 does not harbor CD8 T cell epitopes (murine H2Db and H2Kb MHC-I alleles), CD8 T cell involvement in the atheroprotective effect of p210-cBSA was shown by the transfer of atheroprotection by CD8 T cell transfer from p210-cBSA vaccinated donors to recipients, not observed in recipients from CD8 T cells of vehicle treated donors13. In our hands however, various vaccination protocols including vaccination of alum adjuvanted p210 coupled to PADRE (Pan-DR epitope), resulted in antibodies against p210 but did not affect the CD8 T cell population in human ApoB100 transgenic LDLr−/− (unpublished results). As FITC-p210 was more effectively taken up by DCs than unconjugated FITC13, this suggests that p210 possesses adjuvant properties. The LDLr binding sites of ApoB100, including p210, were also used in a construct with CD8 T cell epitope SIINFEKL to promote cross-presentation of SIINFEKL and induce SIINFEKL specific CD8 T cell activation50. As the antigen specificity of CD8 T cells after cBSA-p210 immunization was not assessed, this could imply that p210 acted as adjuvant for cBSA and enhanced uptake and (cross-) presentation of cationic BSA. As immunization with cationic BSA was previously reported to reduce atherosclerosis51, enhancing the immune response against cBSA could underlie the atheroprotective effect of cBSA-p210 vaccination.
As we observed strong recall responses towards 3 ApoB100 derived peptides, it is unlikely that the quality and quantity of the induced CD8 T cell response was insufficient to modulate atherosclerosis. As we observed a differential localization pattern of CD8 T cells and macrophages in the plaque it is possible that APCs and CD8 T cells not sufficiently co-localize to impact atherosclerosis. Alternatively it is possible that, although TLR induction and engulfment of apoptotic bodies induce cross presentation23,52, other lesional factors could have hampered cross-presentation, such as oxidized lipids which cause disturbance of lipid bodies53 which were found essential for cross presentation in DCs54,55. The observation that profoundly reduced cross priming capacity in batf3−/− chimeras does not affect atherosclerosis, suggests that cross presentation is not important in atherosclerosis56. In our restimulation assays however, lack of cross presentation and lack of co-localization is circumvented by exogenous addition of peptide which externally binds to MHC-I. Despite enhanced CD8 T cell levels in the aortas of ApoB peptide treated mice, we did not observe peptide responsiveness in the cultures with cells derived from the aortic root, suggesting that the atherosclerotic environment reduces the ability of CD8 T cells to respond to TCR stimuli. It is possible that this lack in responsiveness is CD8 T cell intrinsic, e.g. due to chronic antigen exposure in the plaque leading to CD8 T cell exhaustion57,58. In line with CD8 T cell exhaustion in atherosclerosis, upregulation of the co-inhibitory PD-1 expression was observed in atherosclerosis patients59. On the other hand, plaque cells could inhibit CD8 T cell activation, e.g. PD-L1 was found upregulated on macrophages in human lesions (Watanabe et al., 2017), which could provide a co-inhibitory signal to plaque CD8 T cells. The potential of plaque APCs to induce T cell activation is however largely unidentified, but would be very interesting to assess. Moreover it was recently shown that CD8 T-cells in the atherosclerotic lesion significantly downregulate cytokine production as a result of local adenosine signaling60. Therefore, it is possible that impaired antigen specific activation of plaque CD8 T cells, could have rendered induction of ApoB100 specific CD8 T cells ineffective.
MHC-I epitope elution from plaque material and single cell TCR sequencing of lesional CD8 T cells could greatly enhance understanding of CTL biology in atherosclerosis, especially since new bioinformatics avenues are opening up that allow linking of TCR sequences to antigens61,62,63. Identification of these antigens could help unravel atherogenic and atheroprotective CTL mediated immune responses, uncovering treatment options of atherosclerosis through vaccination of antigen specific CTLs.
In conclusion, here we assessed the effect of vaccination with ApoB100 derived CD8 T cell epitopes on atherosclerosis development. We could generate and maintain a very robust CD8 T cell response against 3 epitopes over 8 weeks, however in contrast to other studies which report CD8 T cell mediated protection from atherosclerosis resulting from vaccination with ApoB100 derived peptides13,15, boosting CD8 T cell immunity towards ApoB100 did not reduce atherosclerosis in our hands.
Materials and Methods
In silico HLA-A2 restricted human ApoB100 CD8 T cell epitope prediction
With use of the immune epitope database and analysis resource (iedb.org), putative human ApoB100 derived HLA-A2 restricted CD8 T cell epitopes were predicted. First top 1% predicted binders were selected using NetMHCpan29 which was reported to be the best prediction model for HLA-A264. Thereafter the Proteasomal cleavage/TAP transport/MHC class I combined predictor was used to rank the remaining peptides based on proteasomal cleavage and TAP transport26,27 and a consensus binding prediction28 combining Artificial neural network (ANN)31,32,33,34, Stabilized matrix method (SMM)30, and Scoring Matrices derived from Combinatorial Peptide Libraries (CombLib) models35. The 6 top peptides were synthesized (IHB, Leiden University Medical Centre).
HLA-A2 binding assay
10 mM peptide stocks were prepared in DMSO because of poor water solubility of the peptides and further diluted into complete IMDM. T2 cells, a kind gift from dr. Heemskerk (Leiden University Medical Centre, Leiden) were incubated with peptide concentrations ranging from 0.01–50 μM overnight and 3 μg/ml β-2 microglobulin (Sigma-Aldrich), and HLA-A2 expression was assessed with flow cytometry.
Animals
All animal work was approved by the Leiden University Animal Ethics Committee and the animal experiments were performed conform the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Human HLA-A2 transgenic (HHD), H-2Db−/− β2m−/− C57BL/6 mice36, a kind gift from dr. Lemonnier (Institut Pasteur, Paris), were crossbred with human ApoB100 transgenic LDLr−/− C57BL/6 mice37,38, a kind gift from dr. Nilsson (Lund University, Lund), to generate human ApoB100 and HLA-A2 transgenic H-2Db−/− β2m−/− LDLr−/− (without murine MHC-I expression, HuBL-A2m−) and hApoB100 and HLA-A2 transgenic H-2Db+/+− β2m+/+− LDLr−/− mice (with murine MHC-I expression, HuBL-A2m+). Expression of ApoB100 in blood plasma, was determined with the ApoB100 ELISA developer kit (Mabtech) according to manufacturer’s protocol. Expression of HLA-A2, H2Db and H2Kb on peripheral white blood cells was assessed with flow cytometry using HLA-A2-APC (BB7.2, Biolegend), H2Db-FITC (28-14-8, ThermoFisher) and H2Kb-BV421 (AF6-88.5, BD Biosciences) antibodies respectively. Blood for fenotyping purposes was obtained by lateral tail cut and collected in EDTA coated capillary tubes (Microvette®, Sarstedt). LDLr deficiency was assessed with PCR (Primers: forward-common; CAGTGCTCCTCATCTGACTTG, reverse-WT; CATCTCCCCGCAGTTTGTGT, reverse-KO; CGCCTTCTTGACGAGTTCTTCTG). All mice were kept in individual ventilated cages with aspen bedding, in groups of 2–4 mice per cage and were fed a regular chow diet or a ‘Western-type’ diet (WTD) containing 0.25% cholesterol and 15% cocoa butter (Special Diet Services, Witham, Essex, UK). All mice used in experiments were 12–20 weeks of age. Animals were randomized based on age, weight, plasma Apob100 levels, HLA-A2, H2Db and H2Kb expression. Diet and water were available ad libitum. At sacrifice, mice were anesthetized by a subcutaneous injection (120 μl) of a cocktail containing ketamine (40 mg/ml), atropine (50 μg/ml) and sedazine (6.25 mg/ml). Subsequently, the mice were euthanized and exsanguinated by femoral artery transection and perfusion with PBS through the left cardiac ventricle.
Immunization
Briefly, bone marrow was harvested from femurs and tibia from HHD mice. Bone marrow cells were cultured in complete IMDM (Lonza), IMDM supplemented with 8% FCS (GE Healthcare), 100 U/ml penicillin/streptomycin (Lonza), 2 mM Glutamax (Invitrogen) supplemented with 20 ng/ml GM-CSF (ImmunoTools) in non culture treated petri dishes at a concentration of 0.8*106 cells/ml (10 ml per plate). After 3 days 10 ml fresh complete IMDM with 20 ng/ml GM-CSF was added to the petridishes. On day 6, 10 ml medium was carefully aspirated avoiding removal of cells, and replaced with complete IMDM with 20 ng/ml GM-CSF. At day 9 non/slightly adherent DCs were harvested and cultured overnight with 25 μg/ml high molecular weight poly(I:C) (Invivogen) in complete IMDM in non culture treated petri dishes. DCs were harvested and pulsed with peptides (10 μM) or vehicle (DMSO) for 2 hours at 37 °C in complete IMDM in 50 ml falcon tubes (10*106 cells/ml) under constant agitation to avoid sticking to the plastic. DCs were washed twice with PBS and 2*106 cells were intravenously infused through the lateral tail vein.
In the atherosclerosis studies mice received an i.v. booster vaccination consisting of PBS (control group), or PBS with 5% DMSO and 50 μg high molecular weight Poly(I:C) (VacciGrade™, Invivogen) and 50 μg αCD40 (FGK4.5, BioXcell) (vehicle group), or PBS with 5% DMSO and 30 μM peptide (approximately 67 μg/peptide) and 50 μg high molecular weight Poly(I:C) and 50 μg αCD40 (vehicle group), in a total volume of 200 μl, a week after DC vaccination.
Primary cell preparation
For restimulation culture and flow cytometric analysis of lesional immune cells, aorta’s were isolated from just above the heart until at the height of the heart apex and cleaned from adipose tissue. Aorta’s were minced using scissors and digested in 0.5 ml PBS containing 400 U/ml collagenase type I (Sigma-Aldrich) and 120 U/ml collagenase type XI (Sigma-Aldrich) from Clostridium histolyticum, 60 U/ml type I-s hyaluronidase from bovine testes (Sigma-Aldrich) and 60 U/ml DNase 1 (Sigma-Aldrich) for 30 minutes at 37 °C under constant agitation. Aorta digests of 3 mice were pooled and mashed over 70μm strainers in RPMI to obtain single cell suspensions. Spleens and mediastinal lymph nodes were harvested and mashed over 70μm strainers to obtain single cell suspensions. At sacrifice, blood was obtained through terminal retro-orbital bleeding. Red blood cells from blood were lysed twice and splenocytes suspensions once, through incubation with 1 ml ACK lysis buffer (NH4Cl 150 mM, KHCO3 10 mM, Na2EDTA 0.1 mM) for 30 seconds at room temperature.
Evaluation of peptide specific immune responses
For measurement of antigen-specific CD8+ T cell responses, approximately 2.5*106 splenocytes were incubated with indicated concentrations of individual peptides in U bottom plates. For assessment of specific CD8 T cells in mediastinal lymph nodes, approximately 1.5*106 cells were incubated with 10 μM of all peptides combined in U bottom plates. For measurement of peptide specific CD8 T cell responses in aorta,cells from 3 aorta digests were combined, approximately 20*105 viable CD45+ cells (flow cytometric analysis), and incubated with with 10 μM of all peptides combined in V-bottom plates. Cells were incubated with peptides for 4 h together with Brefeldin A (BD Biosciences) and Monensin (BD Biosciences) in RPMI (Lonza), supplemented with 8% FCS, 100 U/ml penicillin/streptomycin and 2 mM Glutamax.
Flow cytometry
Extracellular staining of single cell suspensions was performed in PBS with 2% FCS and δCD16/32 antibody (93, Biolegend) and eBioscience™ Fixable Viability Dye eFluor™ 780 (ThermoFisher) to discriminate between living and dead cells at 4°C for 30 minutes. For intracellular cytokine staining, cells were incubated in Cytofix/Cytoperm™ Buffer (BD Biosciences) for 15 minutes at 4°C after extracellular staining, then washed twice with Perm/Wash Buffer (BD Biosciences) and stained in Perm/Wash Buffer for 45 minutes at 4°C. The following antibodies were purchased at BD biosciences: IL-17-FITC (TC11-18H10), F4/80-BV421 (BM8), CD4-V500 (RM-4-5), CD19PE-Cy7 (1D3). CD3 V500 (500A2). The following antibodies were purchased at Biolegend: CD8-BV510 (53-6.7), NK1.1-BV650 (PK136), CD4-PerCP (RM4-5), CD45-AF700 (30-F11), CD11b-PE/Dazzle 594 (M1/70), Thy1.2-PE-Cy7 (53-2.1), TNF-δ-PE (MP6-XT22), CD11c-FITC (N418), Ly6G-PerCP (1A8), Ly6C-APC (HK1.4). The following antibodies were purchases at ThermoFisher: IFN-Y-eFluor 450 (XMG1.2), IL-10-APC (JES5-16E3), CD11b –eVolve 605 (M1/70), MHCII-eVolve 655 (M5/114.15.2), CD19-PE (eBio1D3), CD8-PE-TR (5H10), CD62L-PerCP/Cy5.5 (MEL-14), CD44-APC (IM7). Compensation measurements were performed using UltraComp eBeads (ThermoFisher) and ArC Amine-Reactive Compensation Beads (ThermoFisher). Cells were analyzed with a Cytoflex S flow cytometer (Beckman Coulter) with Cytexpert 2.0 software (Beckman Coulter) and further analyzed using FlowJo software (Tree Star, inc.).
Histology
Hearts were cut in half and incubated in OCT medium for 30 minutes. After 30 minutes hearts were fast frozen on dry ice, and stored at −80°C before cryosections (10 μm) of the aortic root were collected on Superfrost Plus™ Adhesion Microscope Slides (ThermoFisher) at 70 μm intervals (7 slides/mice). Neutral fats were stained with Oil Red O to assess lesion size in five subsequent sections of the heart within the three aortic valve area. Lesion collagen content was determined with Masson trichrome staining (Sigma-Aldrich). Corresponding sections analyzed for plaque area and collagen content were immunohistochemically stained for macrophages with MOMA-2 antibody (Sanbio, 1:1000 dilution), CD8 T cells with CD8 antibody (Ly-2, 1:100 dilution, BD Pharmingen). Slides were blocked with 5% milk powder before primary antibody was added for 2 h at RT, after which primary antibody was incubated overnight at 4°C. Endogenous peroxidase activity was blocked by incubating slides in 0.3% Hydrogen peroxide for 30 minutes at RT. Then slides were incubated for 1 h at RT with a polyclonal Rabbit Anti-Rat Ig HRP (DAKO), after which VECTASTAIN ABC HRP Kit (Vector Laboratories) was used. Stained with NovaRed (Vector Laboratories).
Data availability
The data generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age–sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385, 117–171 (2015).
Gewaltig, J., Kummer, M., Koella, C., Cathomas, G. & Biedermann, B. C. Requirements for CD8 T-cell migration into the human arterial wall. Hum. Pathol. 39, 1756–1762 (2008).
Grivel, J.-C. et al. Activation of T Lymphocytes in Atherosclerotic Plaques. Arterioscler. Thromb. Vasc. Biol. 31, 2929–2937 (2011).
Kyaw, T. et al. Cytotoxic and Proinflammatory CD8+ T Lymphocytes Promote Development of Vulnerable Atherosclerotic Plaques in ApoE-Deficient Mice. Circulation 127, 1028–1039 (2013).
Cochain, C. et al. CD8 + T Cells Regulate Monopoiesis and Circulating Ly6C high Monocyte Levels in Atherosclerosis in MiceNovelty and Significance. Circ. Res. 117, 244–253 (2015).
van Duijn, J., Kuiper, J. & Slütter, B. The many faces of CD8+ T cells in atherosclerosis. Curr. Opin. Lipidol. 29, 1 (2018).
van Duijn, J. et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc. Res. 115, 729–738 (2019).
Mittrücker, H.-W., Visekruna, A. & Huber, M. Heterogeneity in the Differentiation and Function of CD8+ T Cells. Arch. Immunol. Ther. Exp. (Warsz). 62, 449–458 (2014).
Harty, J. T., Tvinnereim, A. R. & White, D. W. CD8+ T Cell Effector Mechanisms in Resistance to Infection. Annu. Rev. Immunol. 18, 275–308 (2000).
Reiser, J. & Banerjee, A. Effector, Memory, and Dysfunctional CD8(+) T Cell Fates in the Antitumor Immune Response. J. Immunol. Res. 2016, 8941260 (2016).
Halle, S., Halle, O. & Förster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 38, 432–443 (2017).
Hauer, A. D. et al. Vaccination Against VEGFR2 Attenuates Initiation and Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 27, 2050–2057 (2007).
Chyu, K.-Y. et al. CD8+ T Cells Mediate the Athero-Protective Effect of Immunization with an ApoB-100 Peptide. PLoS One 7, e30780 (2012).
van Wanrooij, E. J. A. et al. Vaccination against CD99 inhibits atherogenesis in low-density lipoprotein receptor-deficient mice. Cardiovasc. Res. 78, 590–596 (2008).
Dimayuga, P. C. et al. Identification of apoB‐100 Peptide‐Specific CD8+ T Cells in Atherosclerosis. J. Am. Heart Assoc. 6, e005318 (2017).
Ludewig, B. et al. Linking immune-mediated arterial inflammation and cholesterol-induced atherosclerosis in a transgenic mouse model. Proc. Natl. Acad. Sci. 97, 12752–12757 (2000).
Yamada, S. et al. Apoptosis Signal-Regulating Kinase 1 Deficiency Accelerates Hyperlipidemia-Induced Atheromatous Plaques via Suppression of Macrophage Apoptosis. Arterioscler. Thromb. Vasc. Biol. 31, 1555–1564 (2011).
Liu, J. et al. Reduced Macrophage Apoptosis Is Associated With Accelerated Atherosclerosis in Low-Density Lipoprotein Receptor-Null Mice. Arterioscler. Thromb. Vasc. Biol. 25, 174–9 (2004).
Fyfe, A. I., Qiao, J. H. & Lusis, A. J. Immune-deficient mice develop typical atherosclerotic fatty streaks when fed an atherogenic diet. J. Clin. Invest. 94, 2516–2520 (1994).
Curtiss, L. K. & Tobias, P. S. Emerging role of Toll-like receptors in atherosclerosis. J. Lipid Res. 50(Suppl), S340–5 (2009).
Van Vre, E. A., Ait-Oufella, H., Tedgui, A. & Mallat, Z. Apoptotic Cell Death and Efferocytosis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 887–893 (2012).
Corridoni, D. & Simmons, A. Innate immune receptors for cross-presentation: The expanding role of NLRs. Mol. Immunol. https://doi.org/10.1016/J.MOLIMM.2017.11.028 (2017).
Bellone, M. et al. Processing of engulfed apoptotic bodies yields T cell epitopes. J. Immunol. 159, 5391–9 (1997).
Yewdell, J. W. & Bennink, J. R. Immunodominance in major histocompatibility complex class I–restricted t lymphocyte responses. Annu. Rev. Immunol. 17, 51–88 (1999).
Stuber, G. et al. Assessment of major histocompatibility complex class I interaction with Epstein-Barr virus and human immunodeficiency virus peptides by elevation of membrane H-2 and HLA in peptide loading-deficient cells. Eur. J. Immunol. 22, 2697–2703 (1992).
Peters, B., Bulik, S., Tampe, R., Van Endert, P. M. & Holzhütter, H.-G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. 171, 1741–9 (2003).
Tenzer, S. et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. 62, 1025–37 (2005).
Moutaftsi, M. et al. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat. Biotechnol. 24, 817–819 (2006).
Nielsen, M. et al. NetMHCpan, a Method for Quantitative Predictions of Peptide Binding to Any HLA-A and -B Locus Protein of Known Sequence. PLoS One 2, e796 (2007).
Peters, B. & Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6, 132 (2005).
Buus, S. et al. Sensitive quantitative predictions of peptide-MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens 62, 378–84 (2003).
Lundegaard, C., Lund, O. & Nielsen, M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 24, 1397–1398 (2008).
Nielsen, M. et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017 (2003).
Lundegaard, C. et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 36, W509–12 (2008).
Sidney, J. et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 4, 2 (2008).
Pascolo, S. et al. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J. Exp. Med. 185, 2043–51 (1997).
Boren, J. et al. Identification of the low density lipoprotein receptor-binding site in apolipoprotein B100 and the modulation of its binding activity by the carboxyl terminus in familial defective apo-B100. J. Clin. Invest. 101, 1084–93 (1998).
Linton, M. F. et al. Transgenic mice expressing high plasma concentrations of human apolipoprotein B100 and lipoprotein(a). J. Clin. Invest. 92, 3029–37 (1993).
Barrios, K. & Celis, E. TriVax-HPV: an improved peptide-based therapeutic vaccination strategy against human papillomavirus-induced cancers. Cancer Immunol. Immunother. 61, 1307–1317 (2012).
Cho, H.-I. & Celis, E. Optimized Peptide Vaccines Eliciting Extensive CD8 T-Cell Responses with Therapeutic Antitumor Effects. Cancer Res. 69, 9012–9019 (2009).
Cho, H.-I., Jung, S.-H., Sohn, H.-J., Celis, E. & Kim, T.-G. An optimized peptide vaccine strategy capable of inducing multivalent CD8+ T cell responses with potent antitumor effects. Oncoimmunology 4, e1043504 (2015).
Halle, S. et al. In Vivo Killing Capacity of Cytotoxic T Cells Is Limited and Involves Dynamic Interactions and T Cell Cooperativity. Immunity 44, 233–245 (2016).
Bergström, I., Backteman, K., Lundberg, A., Ernerudh, J. & Jonasson, L. Persistent accumulation of interferon-γ-producing CD8+CD56+ T cells in blood from patients with coronary artery disease. Atherosclerosis 224, 515–520 (2012).
Hwang, Y. et al. Expansion of CD8+ T cells lacking the IL-6 receptor α chain in patients with coronary artery diseases (CAD). Atherosclerosis 249, 44–51 (2016).
Kolbus, D. et al. Association between CD8 + T-cell subsets and cardiovascular disease. J. Intern. Med. 274, 41–51 (2013).
Zhao, W. & Sher, X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 14, e1006457 (2018).
Zhang, X. et al. Detection of circulating IgG antibodies to apolipoprotein B100 in acute myocardial infarction. FEBS Open Bio 5, 712–6 (2015).
Gisterå, A. et al. Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J. Intern. Med. 281, 383–397 (2017).
Ghanekar, S. A. et al. Gamma interferon expression in CD8(+) T cells is a marker for circulating cytotoxic T lymphocytes that recognize an HLA A2-restricted epitope of human cytomegalovirus phosphoprotein pp65. Clin. Diagn. Lab. Immunol. 8, 628–31 (2001).
Sakamoto, N. & Rosenberg, A. S. Apolipoprotein B binding domains: evidence that they are cell-penetrating peptides that efficiently deliver antigenic peptide for cross-presentation of cytotoxic T. cells. 186, 5004–11 (2011).
Kolbus, D. et al. Immunization with cationized BSA inhibits progression of disease in ApoBec-1/LDL receptor deficient mice with manifest atherosclerosis. Immunobiology 216, 663–669 (2011).
Mäkelä, S. M., Osterlund, P. & Julkunen, I. TLR ligands induce synergistic interferon-β and interferon-λ1 gene expression in human monocyte-derived dendritic cells. Mol. Immunol. 48, 505–15 (2011).
Veglia, F. et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 8, 2122 (2017).
den Brok, M. H. et al. Saponin-based adjuvants induce cross-presentation in dendritic cells by intracellular lipid body formation. Nat. Commun. 7, 13324 (2016).
Bougnères, L. et al. A Role for Lipid Bodies in the Cross-presentation of Phagocytosed Antigens by MHC Class I in Dendritic Cells. Immunity 31, 232–244 (2009).
Legein, B. et al. Ablation of CD8α+ dendritic cell mediated cross-presentation does not impact atherosclerosis in hyperlipidemic mice. Sci. Rep. 5, 15414 (2015).
Day, C. L. et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354 (2006).
Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006).
Qiu, M.-K. et al. PD-1 and Tim-3 Pathways Regulate CD8+ T Cells Function in Atherosclerosis. PLoS One 10, e0128523 (2015).
Duijn, Jvan et al. CD39 identifies a microenvironment-specific anti-inflammatory CD8+ T-cell population in atherosclerotic lesions. Atherosclerosis 285, 71–78 (2019).
Shugay, M. et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427 (2018).
Dash, P. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017).
Glanville, J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017).
Shen, W.-J. et al. MHC binding prediction with KernelRLSpan and its variations. J. Immunol. Methods 406, 10–20 (2014).
Acknowledgements
F.H.S., H.D. and J.K. were funded by European Union’s Seventh Framework [grant number 603131], by contributions from Academic and SME/industrial partners. F.H.S. and J.K. are additionally supported by the Royal Netherlands Academy of Sciences for the GENIUS II project “Generating the best evidence-based pharmaceutical targets and drugs for atherosclerosis” (CVON2017-20). AF is supported by the Dutch Heart Foundation [grant number 2016T008 to AF].
Author information
Authors and Affiliations
Contributions
F.H.S. and J.K. devised the study, and designed the experiments. J.W.D. synthesized and purified the peptides. F.H.S., J.A., H.D., M.J.K., B.A.S., G.H.M.vP., I.B. and A.C.F., carried out the experiments. F.H.S. analyzed the data and wrote the manuscript. J.W.D., B.A.S. and J.K. contributed to revisions of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41598_2019_53642_MOESM1_ESM.pdf
Induction of HLA-A2 restricted CD8 T cell responses against ApoB100 peptides does not affect atherosclerosis in a humanized mouse model.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schaftenaar, F.H., Amersfoort, J., Douna, H. et al. Induction of HLA-A2 restricted CD8 T cell responses against ApoB100 peptides does not affect atherosclerosis in a humanized mouse model. Sci Rep 9, 17391 (2019). https://doi.org/10.1038/s41598-019-53642-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53642-z
- Springer Nature Limited