
Abstract
The human skin is colonized by a wide array of microorganisms playing a role in skin disorders. Studying the skin microbiome provides unique obstacles such as low microbial biomass. The objective of this study was to establish methodology for skin microbiome analyses, focusing on sampling technique and DNA extraction. Skin swabs and scrapes were collected from 9 healthy adult subjects, and DNA extracted using 12 commercial kits. All 165 samples were sequenced using the 16S rRNA gene. Comparing the populations captured by eSwabs and scrapes, 99.3% of sequences overlapped. Using eSwabs yielded higher consistency. The success rate of library preparation applying different DNA extraction kits ranged from 39% to 100%. Some kits had higher Shannon alpha-diversity. Metagenomic shotgun analyses were performed on a subset of samples (N = 12). These data indicate that a reduction of human DNA from 90% to 57% is feasible without lowering the success of 16S rRNA library preparation and without introducing taxonomic bias. Using swabs is a reliable technique to investigate the skin microbiome. DNA extraction methodology is crucial for success of sequencing and adds a substantial amount of variation in microbiome analyses. Reduction of host DNA is recommended for interventional studies applying metagenomics.
Similar content being viewed by others
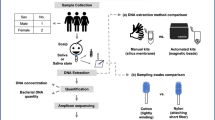

Introduction
The human skin is colonized by millions of bacteria, fungi and vira composing the skin microbiome. It has long been recognized that microbes are important players in skin diseases. Recently the relationship between host and skin microbes has experienced a renaissance of research activity after the rise of high-throughput DNA sequencing in 20061 and subsequently increased access and continued decrease in cost. Most protocols for characterization of human microbial communities have been developed for gut microbiome studies. The skin harbors completely different niches and unique methodological challenges such as high contamination risk2 mainly due to low microbial biomass combined with a risk of adding contaminating microbes during handling of samples, e.g. from extraction reagents3. Difficulties in acquiring sufficient bacterial DNA for microbiome analysis is also an obvious challenge in skin microbiome studies4,5.
The most common approach to characterize the skin microbiome is amplicon sequencing of the small subunit ribosomal RNA (16S rRNA in prokaryotes) gene. This gene is ideal for community fingerprinting since it is highly conserved and possesses conserved and variable regions. Groundbreaking studies of the skin microbiome have been made with amplicon sequencing. Grice and colleagues have shown that physiologically comparable skin sites (sebaceous, moist and dry) harbor similar bacterial communities and that sampling with swabs, scrapes and punch biopsies captures the same dominant microbial components6,7. Optimal methodologies for conducting skin microbiome research have received increased focus (reviewed in Kong et al.8) and it is becoming apparent that multiple steps in the analyses pipeline influence the results. Concerning sampling strategy different methodologies differ not only in sampling depth and discomfort but also in biomass yield and human DNA contribution. Compared to swabs, scrapes potentially increase the biomass collected which is useful in studies with rare taxa9. When isolating DNA for sequencing, most scientists conducting skin microbiome research use commercial kits relying on protocols which often differ in their strategy for disrupting bacterial cells. Enzymatic treatment, thermal disruption and/or mechanical lysis are commonly used. Bead size and material are likely to influence and select certain microbial populations.
Over the past few years, whole metagenomic shotgun sequencing has become a common method for assembling genomes. This method has increased resolution and higher internal consistency10 compared to 16S rRNA gene sequencing and can identify bacteria to strain-level. Shotgun sequencing captures all the genetic material in a sample without a targeted amplicon step, allowing comparisons on kingdom abundances. One such study of the skin has shown that site specificity also applies to fungi, but not to eukaryotic vira11. The same study showed that healthy adults maintain their skin communities for up to two years. However, since the human genome is about one thousand times larger than bacterial genomes, the total DNA pool can easily become dominated by host DNA, which might obscure small microbial differences e.g. induced in interventional studies. Depleting host DNA in samples for shotgun sequencing may be a crucial approach to improve data and different approaches have recently been compared in saliva samples12.
While more thorough investigations of the effect of sampling procedures have been conducted for samples with high bacterial load and low amount of human DNA13,14, a comparable effort focusing on the skin microbiome is lacking. With a very large amount of human DNA and a particular microbiome composition, this environment presents different challenges. Here we present a comparison of two sampling strategies and 12 commercially available DNA extraction kits for investigating the skin and nasal microbiome. We amplified the 16S rRNA gene and sequenced hypervariable regions 3–4. Nasal samples were also sequenced with shotgun metagenomics to evaluate the usefulness of host DNA depletion applied in two kits by use of lysis of human cells and subsequent addition of nucleases.
Results
Success of 16S rRNA gene sequencing varied with DNA extraction method
Nine healthy volunteers (5 women, 4 men), aged 26 to 64 years, were included in the study. In total, 220 samples were collected, including 144 skin samples from eSwabs and scrapes (Fig. S1, Table S1), 36 nasal eSwabs, 16 E. coli positive controls, and 24 negative controls containing either preservation medium from eSwabs or buffer from the kits. All 220 samples were analysed with 16S rRNA gene sequencing. Quality filtering removed samples with loading mistakes or less than 5000 reads, leaving 165 samples including 137 skin and nares samples used for figures. In total, 4,017,433 reads were produced in the 137 samples. The minimal number of reads in a sample was 11,360 and the median was 27,947. Furthermore, operational taxonomic units (OTUs) present in less than 10% of the samples were removed, leaving 4814 OTUs in the 137 samples (Table S2, a complete OTU table).
We applied 12 different DNA extraction kits (Table 1) with different success rates of library preparation (Table 2). Kits number 2, 10, 11 and 12 performed poorly with rates of successful libraries ranging from 39–79%. Success in library preparation was overall independent of DNA concentrations in the samples (Table 2). However, the worst performing kit, number 11, with a success rate of 39%, also had a very low average DNA concentration of 0.09 ng/µl.
The eSwab is a preferable method
We used eSwabs collecting material from the surface of the skin and scrapes collecting skin cells and microbes from the outermost part of the epidermis. There were no differences in concentrations of isolated DNA or success of library preparation between eSwabs and scrapes (data not shown). Furthermore, of the total 4814 OTUs, 4325 (89.8%) were identified with both sampling methods and these OTUs represented 3,989,311 sequence reads of the total 4,017,433 reads (99.3%) (Fig. 1a). Unique OTUs were identified using both eSwabs and scrapes. To further evaluate potential differences between microbial populations, Shannon alpha-diversity and Chao1 richness were compared (Fig. 1b). Differences between eSwabs and scrapes were found in Shannon alpha-diversity (mean scrape: 3.2; mean swab: 3.8; unadjusted p-value: 0.04), and in Chao1 richness, with lower richness in scrapes (mean scrape: 743; mean swab: 1180; unadjusted p-value: 0.00008). Notably, scrapes captured more Pseudomonas than eSwabs (Fig. S2). Redoing this analysis after removing all Pseudomonadales, this conclusion still holds for the richness, albeit with a higher p-value (Shannon’s p-value: 0.08; Chao1: p = 0.016). The eSwab seems to be a more consistent method than scrapes, with better Pearson’s product-moment coefficients at all taxonomic levels compared to scrapes (Figs. 1c and S3–5). Since the nare samples were only collected by eSwabs, we also made the scatter plots without those samples to rule out that inclusion of those made the scrapes seem worse off (Fig. 1c). Excluding Pseudomonadales improves Pearson’s r at all taxonomic levels, but scrapes still perform consistently worse than swabs.

Comparison of skin sampling method. (a) A Venn diagram illustrating overlap of OTUs with ≥98% similarity and percent of sequence reads overlapping in parenthesis. (b) Violin plots illustrating Shannon alpha-diversity and Chao1 richness according to sampling method. (c) Scatter plots comparing the proportion of reads from a pair of samples from the same clade at the genus taxonomic level. Each sample is a pair of samples from the same skin site in the same individual, extracted with different kits. Pearson’s product moment and Spearman’s rank correlation were calculated for each plot.
DNA extraction method captured different microbial communities
The spread observed on the scatter plots (Figs. 1 and S3–5) suggests that different extraction methods capture somewhat different communities since each pair of samples on the plots comes from the same skin site in the same individual but were extracted with different kits. If the performance of all kits were the same, the points would be close to the y = x diagonal. The 12 different DNA extraction kits applied (Table 1) had some influence on Shannon alpha-diversity and Chao1 richness. Especially kit number 8 differs from most of the other kits, with high Shannon alpha-diversity and Chao1 richness (Fig. 2). Kit number 7 had this tendency as well. Notably, both kit number 7 and 8 applied a 3-mm stainless steel ball and bead beating in a Tissuelyser for mechanical disruption of bacterial cell walls and membranes (Table 1). However, kit number 10 also applied this bead beating protocol and did not yield higher Shannon alpha-diversity or Chao1 richness. Kit number 4 and 5 (same kits with different lot numbers, Table 1) had low Shannon-diversity and Chao1 richness (Fig. 2) and were dominated by Enterobacteriales (Fig. 3c), which might indicate contamination. Kits 5 and 6 also presented a large relative abundance of Pseudomonadales, also suggesting kit contamination. Our negative controls show that the main background is Pseudomonas (average 72% in all kit negative controls) and E. coli is present in most negative controls as well (average 3%) (Fig. S6). Two kits, number 8 and 10, have diverse profiles in their negative controls. Kit number 10 has a large representation (17%) of Burkholderia.

Influence of DNA extraction kit on microbiome diversity and richness. (a) Violin plots illustrating Shannon alpha-diversity and Chao1 richness according to DNA extraction kit. (b) Tables with p-values from Kruskal-Wallis-tests corrected for multiple testing by the Benjamini-Hochberg procedure, bold and underlined when statistical significance.

Variation by skin site. (a) A heatmap of Bray-Curtis distances between samples, with metadata plotted on the axis above and color code to the right. 0 indicates that samples share the same OTU and 1 that they are totally different. (b) Violin plots illustrating Shannon alpha-diversity and Chao1 richness according to skin site, * when statistical significance in a Kruskal-Wallis-test corrected for multiple testing by the Benjamini-Hochberg procedure (p < 0.05). (c) Bar charts depicting relative abundances of bacteria at the order taxonomic level. Samples are sorted by skin site and number of kit used is assigned above the charts. Individual subject numbers is indicated by the colour bar at the bottom of the figure.
The heatmap (Fig. 3a) of Bray-Curtis dissimilarities did not indicate clustering according to DNA extraction kit. The subject had more influence on clustering than kit (Figs. 3a and 4).

Reduction of host DNA does not influence microbial communities. (a) A MetaPhlAn2 clustered heatmap showing the distribution of microbes in the 12 samples, each representing one nasal sample from the kits applied (Table S2). Kit number is annotated along the x-axis and detected species-level names on the y-axis on the right side. (b) Percent viral DNA (x-axis) in the samples from each kit (y-axis). (c) Scatter plots illustrating Shannon alpha-diversity and Chao1 richness.
Variation by skin location
Evaluation of beta-diversity using Bray-Curtis Dissimilarity reveals clustering by skin site (Fig. 3a) first and then by subject. Whether samples were taken from the antecubital and popliteal fossae or volar forearm had a relatively minor impact on Shannon-alpha Diversity, Chao1 richness (Fig. 3b) and the composition of the skin microbiome (Fig. 3c). Nasal samples differed significantly from skin samples, with lower Shannon-alpha Diversity, Chao1 richness and a microbiome dominated by Staphylococcus (primarily S. epidermidis) and Corynebacterium. The skin also contained Staphylococcus and Corynebacterium (Fig. S2). The experimental procedures used here did not allow us to assess the impact of Cutibacterium (see Discussion for details).
Reduction of host DNA content for shotgun sequencing
To investigate the efficacy of host DNA removal, one nasal sample from each of the 12 DNA extraction kits were also shotgun sequenced (Table S3). The samples were from subjects 2 (kit number 1, 2, 4 and 5), 5 (kit number 3, 6, 10 and 11) and 8 (kit number 7, 8, 9 and 12). A detailed description of the sequencing procedure and basic sequencing data quality measures is available in the Methods section.
Kit number 9 reduced the percent of human DNA from app. 90% to 57% (Table 2). Kit number 12 did not succeed in depleting or reducing human DNA. Importantly, using kit number 9 does not seem to introduce taxonomic biases as the sample clusters with samples from other kits (Fig. 4a) and the microbial profile is similar to samples from other kits.
The sample from kit number 7 gave the most distinct microbial profile (Fig. 4c) with a predominance of Cutibacterium acnes and unclassified Escherichia (Fig. 4a), where the latter might indicate contamination. As with 16S, the other samples were dominated by Staphylococcus and Corynebacterium. The sample from kit number 8 clusters with the one from kit number 7 and lacks a high abundance of Corynebacterium propinquum and C.- accolens (Fig. 4a).
The proportion of viral DNA (Fig. 4b) was influenced by the subject sampled. All samples from subject 2 contained viral DNA. Additionally, viral DNA was found in the sample extracted with kit number 8 from subject 8.
Discussion
We compared sampling of skin using eSwabs and scrapes with subsequent 16S ribosomal RNA gene sequencing. A very large overlap was found, both in OTU identified and in OTU counts, indicating that these methods can be used interchangeably. This is in line with data from Chng et al. comparing a modified cup scrub, swab and tape-strip10 data from Ogai et al. comparing swabs and tape strips15 and data from Grice et al. comparing swabs, scrapes and punch biopsies7. Grice et al. argue that microbiota from swabs and scrapes represent a history of skin differentiation, implying that the microorganisms from deeper layers transit to the surface with differentiating skin cells. With this perspective, the outermost microbiome (live or dead) can very well indicate which processes and physiological roles the microorganism deeper in the skin16 may have. However, this might be too much of a simplification, as we also identify unique OTUs using each sampling method and a difference in Chao1 richness. A study applying repeated tape stripping for removal of the stratum corneum layers also show some significant differences in microbial composition between superficial and deeper layers of the stratum corneum with an increase in the relative abundance of Firmicutes (Staphylococcus) in the deeper layers and a decrease of Actinobacteria (Cutibacterium)17. We do not find such striking differences in microbial composition between samples of the outermost skin taken with eSwabs and scrapes going deeper in the stratum corneum. This could be due to differences in methodology, as our scrapes also capture the outermost microbiota which swabbing of specific layers after sequential tape stripping does not, but it might also be an artefact of the primers used, which do not appropriately amplify Cutibacterium. When sampling superficially by swabs one might overlook specific microbiota and potential interactions between microbiota and live human cells deeper in the skin. However, in our hands, data collected by eSwabs were more consistent. This technique is also less invasive and therefore more useful for certain purposes.
Many samples contained trace amounts of chloroplasts. Two of them, however, (kit 10, subject 4 and 6, flexure) contained large numbers of chloroplasts (15–25%). Since they do not consistently appear in all of kit 10’s samples, we suspect that these subjects may have considerable physical contact with plants in their everyday life or made regular use of plant-based cosmetics.
Choice of DNA extraction kit affects the observed microbial profiles, but not more than inter-individual variation. It is difficult to assess which kit comes closest to the biological truth and a limitation of this study is a lack of a proper mock community as positive control. However, kit number 4 and 5 seemed to be dominated by contaminating bacteria and are not recommended for examination of the skin microbiome.
Other factors one should consider when choosing a protocol for DNA extraction is success in sequencing and convenience of usage in a specific setting. Kits number 2, 10, 11 and 12 performed poorly with rates of successful libraries for 16S rRNA gene sequencing ranging from 39–79%. We would avoid these kits. When taking samples from patients in the clinic it is of priority that they can be stored immediately. Kit number 12 had considerable hands-on time before a storage is possible. In general, less total hands-on time is also preferable.
Kit number 9 reduced the content of human DNA from nasal samples from 90% to 57%. We were worried that adding a nuclease for reduction of host DNA would destroy free microbial DNA as well and skew the picture of microbial communities compared to extraction kits without this step. Fortunately, no taxonomic skewing was observed in 16S or shotgun data. The good correspondence between the 16S and shotgun taxonomy profile using this kit also demonstrates that reagent contamination after DNA extraction was not an issue here. Another concern was that reduction of total DNA in the samples would increase the risk of failure in library preparation, as the DNA concentrations in skin swabs generally are low, around 5 ng in total. Again, this was not a problem. Had it been a problem it would probably be advisable to sequence deeper to discover the effects of interventions rather than to reduce human DNA.
One drawback of using kit number 9 was that the total hands-on time was substantial. One recently published study compared this specific kit (Qiagen QIAamp DNA Microbiome Kit) with other methods of depleting host DNA in saliva12. It was found that treatment involving osmotic lysis of human cells and subsequent treatment with propidium monoazide is very efficient in removing host-derived sequences with a small taxonomic bias compared to untreated samples. Furthermore, this treatment is much cheaper than using the kit and requires fewer washing steps and less hand-on time12. Future studies should test this treatment on skin samples.
Whether samples were taken from popliteal and antecubital flexures or volar forearms had no impact on Shannon-alpha Diversity, Chao1 richness (Fig. 3b) or the composition of the skin microbiome (Fig. 3c). This is contradictory to the pioneer work performed by Grice et al.6 and Findley et al.9 showing that moist, dry and sebaceous areas have distinct microbial profiles. This is a general picture, and the actual differences found in these studies between the specific volar forearm and flexures are modest. Also, factors such as body composition and posture, clothing and weather can affect the moistness of the flexures. A dry flexure might be relatively similar to a volar forearm. Nasal samples differed significantly from skin samples with lower Shannon-alpha Diversity, Chao1 richness and a microbiome dominated by Staphylococcus (primarily S. epidermidis) and Corynebacterium. Choosing to investigate more distinct skin areas or including more areas and using metagenomics would possibly enable us to see a general pattern in differences between moist, dry and sebaceous areas.
We interpret the spread on the scatter plots as extraction methods capturing different communities. However, as we sampled non-overlapping skin areas, local differences in communities might also contribute to this spread. There are known variations in transepidermal water loss within the volar forearm, with higher values near the wrist compared to other sites of the forearm18. Also, recent studies show that sebum and hydration levels are predictors of microbiome composition19 and that the specific composition of epidermal lipids strongly affects bacterial colonization20. It is however not possible to circumvent this issue when comparing multiple factors, as in our study.
In addition to false negatives, DNA extraction kits can contribute with false positives, especially in environments with relatively low bacterial abundance, as the human skin. Indeed, kit 4 and 5, which had the lowest DNA extraction yield for skin samples (Table 2), also had the highest amount of Pseudomonadaceae. This family has been described to be abundant in human skin before7, but this observation has not been reproduced. Furthermore, Pseudomonadaceae have been found in high abundance in the “kit-ome”, i.e. the background of bacterial DNA present in DNA extraction kits and PCR reagents2. No thermal disruption was applied in kit number 4 and 5 (Table 1) which could result in the low amounts of isolated microbial DNA from skin samples and higher amplification of contaminating DNA. However, our main conclusions hold even when excluding all Pseudomonadales and Enterobacteriales.
After sampling and DNA extraction, another major source of bias in amplicon-based studies is primer choice. The primer pair used here was suboptimal for skin microbiome studies, since it specifically excludes Cutibacterium, as evidenced by this clade being found in shotgun, but not in amplicon samples. However, since this bias was kept constant for all samples investigated, they can still be compared. Still, future studies on the human skin microbiome will benefit from using a shotgun approach when possible (see e.g.10), or another primer pair for the 16S region. In this case, two approaches are possible, either selecting a different region of this gene21,22 or simply modifying the reverse primer to amplify the V4 region of Cutibacterium spp.23.
Conclusion
Swabs and scrapes can be used interchangeably to investigate the skin microbiome. Swabs may be preferable as they are more consistent and less invasive. DNA extraction methodology is crucial for success of sequencing and adds a substantial amount of variation in microbiome analyses. However, clustering of data was more influenced by subject than kit. Using the QIAamp DNA Microbiome Kit from Qiagen, host DNA is reduced without introducing taxonomic biases, which is recommended for interventional studies applying metagenomics.
Methods
The study was approved by the ethics committee of the Capital Region of Denmark (H-16020971). All participants signed a written informed consent form prior inclusion and any sampling. All methods were performed in accordance with relevant guidelines and regulations.
Study participants
Nine healthy Caucasian volunteers were recruited from Hospital office staff in September 2016. Inclusion criteria were age 18 or older. Exclusion criteria were current or previous eczema, pregnancy, breastfeeding, scar tissue on sampling areas, active infections and use of antibiotics or probiotics within the past four weeks. The volunteers were instructed not to shower, use chlorinated pools, sauna, steam bath, sun tanning and topicals (e.g. moisturizers) two days before sampling at the Department of Dermatology and Allergy at Herlev and Gentofte Hospital.
Sampling
Skin samples were collected from non-overlapping areas on the dry volar forearms and moist antecubital and popliteal fossae. The fossae were defined as the region from the flexure + /− 4 cm and the volar forearm as starting after the antecubital fossa to 4 cm from the wrist. One side of the body was randomized to sampling with eSwabs (8 samples in total) and the other with scrape (8 samples in total). Four nasal eSwabs were collected from each subject as well, giving a total of 20 samples from each subject (Fig. 1S and Table S1). For sample collection, no prior cleaning or preparation of the skin surface was done. A fresh pair of gloves were worn for each sample. The flocked swab was premoistened in either preservation medium or enzymatic lysis buffer. A timer was set at 30 sec. for rubbing the skin area. Superficial skin scrapings were obtained by taking 20 strokes in different directions at the skin with a disposable scalpel. Sample material was placed into 2 ml LoBind Eppendorf tubes containing buffer from the kit (according to manufacturer).
E. coli ATCC 8739–0483E7 Epower pellets (SSI Diagnostica, CFU per pellet approximately 5 × 107) were suspended (according to manufacturer) in either preservation medium, buffer from the kit or enzymatic lysis buffer. Samples were either stored at −20 °C, −80 °C or processed immediately, according to DNA extraction protocol.
DNA extraction
DNA was extracted using 12 different commercial kits (kit number 4 and 5 were similar, but had different lot numbers) according to manufactures’ protocols: 1. BiOstic Bacteremia DNA Isolation Kit (MO BIO, lot no.: BC16C25), 2. Microbial DNA Isolation Kit (MO BIO, lot no.: U16E2), 3. PowerLyzer UltraClean Microbial DNA Isolation Kit (MO BIO, lot no.: PL16C29), 4. UltraClean Tissue & Cells DNA Isolation Kit (MO BIO, lot no.: U15I14), 5. UltraClean Tissue & Cells DNA Isolation Kit (MO BIO, lot no.: U16D18), 6. PowerSoil DNA Isolation kit (MO BIO, lot no.: PS16C29), 7. MasterPure yeast DNA purification kit (Epicentre, lot no.: 0020027874), 8. QIAamp DNA Investigator Kit (QIAGEN, lot no.: 154018987), 9. QIAamp DNA Microbiome Kit (QIAGEN, lot no.: 154026306), 10. PureLink Genomic DNA Kit (Invitrogen, lot no.: 17462207), 11. PureLink Microbiome DNA purification (Invitrogen, lot. No.: 1761498), 12. MolYsis Complete5 Ultra-deep Microbiome prep kit (Molzym, lot no.: S22qKG020025). These kits were chosen because they were applied in published skin microbiome studies and/or recommended by the manufacturer to be useful for skin microbiome analysis. The combinations of kits, locations and subjects are described in Fig. S1 and Table S1.
16S rRNA gene sequencing
DNA concentration was determined with Quant-iT ds DNA broad range kit (Thermo Fisher Scientific, Waltham, MA, USA). The V3-V4 hypervariable region of the 16S rRNA gene was amplified using universal primers F341 and R80524. The PCR was performed according to the 2-step PCR protocol as described in Hugerth et al. 2018 with 23 µL of DNA solution as input25. The product was then cleaned with AMPure XP beads (Beckman Coulter, Brea, CA, USA) before being submitted to a 13 cycle barcoding reaction with Nextera XT index kit V2 (Illumina, San Diego, CA, USA) according to instructions from the manufacturer. The amplicons were sequenced on Illumina’s MiSeq platform with 2 × 300 bp reads and a cutoff value of 5000 reads was applied. A blank (negative) PCR control was amplified and sequenced with each plate.
Operational taxonomic unit picking
After amplicon sequencing, we used Cutadapt v.1.1626 to remove read pairs not carrying both primers or with an average Phred score < 15. Read pairs were then merged using Vsearch v.2.6.227 and excluding non-merging reads, merged pairs containing any ambiguous bases, with more than 3 expected errors over the full length or with a length <380 bp or >520 bp. We then used the unoise algorithm from Usearch v.10.0.24028 to denoise reads. For quantification, all merged reads were mapped back to the centroid sequences requiring at least 98% identity over the full length of the query. To differentiate between S. aureus and S. epidermidis an additional run was made with 99.8% identity clustering. Taxonomy was assigned based on the SILVA database v12829 using the algorithm described by Hugerth et al. (2018). Plant-chloroplast and mitochondrial OTUs were removed. All calculations on 16S rRNA gene data are OTU-based.
Metagenomic sequencing and analysis
Shotgun metagenomic sequencing was performed on 12 nasal samples (Table S3, accession number table). Sequencing libraries were constructed using Rubicon ThruPLEX DNA-seq, with an average fragment size of 365 base pairs (min: 304, max: 441, stdev: 42.2). Clustering was done by ‘cBot’ and samples were sequenced on an Illumina HiSeq2500 (HiSeq Control Software 2.2.58/RTA 1.18.64) with a 2 × 126 setup using ‘HiSeq SBS Kit v4’ chemistry. The Bcl to FastQ conversion was performed using bcl2fastq_v2.19.1.403 from the CASAVA software suite. The samples were sequenced in 2 lanes with 259 and 261 million clusters each, respectively, producing an average of 40.8 million reads (min: 30.8, max: 55.2, stdev: 8.1) with on average 93.0 of bases with Q-scores greater than 30. Metagenomic sequencing data was analyzed using StaG-mwc30,31 version 0.2.0-dev. Reads were preprocessed with BBDuk32 37.99 using the default settings defined in StaG-mwc. Host contamination was removed with BBMap32 v37.99 by mapping reads to a masked version of hg19 (http://seqanswers.com/forums/showthread.php?t = 42552) using the default settings defined in StaG-mwc. Taxonomic profiling was performed using MetaPhlAn233 v2.7.7 using the default settings defined in StaG-mwc, with the addition of ‘-t rel_ab_w_read_stats‘ to produce estimated read counts per taxa used for downstream calculations.
Statistical analyses
Intra-sample diversity and richness were calculated using Shannon’s entropy and Chao1 richness, respectively. Inter-sample diversity was estimated as Bray-Curtis divergence. Pairwise comparisons were performed with the Kruskal-Wallis test. All pairwise statistical comparisons were corrected for multiple testing using the Benjamini-Hochberg procedure unless otherwise stated. All calculations for 16S data were performed in R v.3.4.3, with the additional packages Vegan v.2.4-534, Fossil v.0.3.735 and Vioplot v.0.2 (an R package based on the original work by Hintze et al.36). Calculations and visualizations of shotgun metagenomic data was performed in a Jupyter notebook (Jupyter v4.4.0) using SciKit-bio v0.5.4, matplotlib v3.0.037, seaborn v0.9.038, pandas v0.22.039 in Python v3.6.6.
Data availability
The sequence files and metadata for each sample in this study is publicly available at NCBI (Submission number: SUB4053477). Code used for the analysis of amplicon sequencing data is available at this repository: https://github.com/ctmrbio/Amplicon_workflows. The workflow used for the analysis of the shotgun metagenomics data is available at this repository (version 0.2.0-dev): https://github.com/boulund/stag-mwc (commit: ea3781d). Jupyter notebooks used to produce plots are available at https://doi.org/10.6084/m9.figshare.8319842.v1.
References
Sogin, M. L. et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences 103, 12115–12120 (2006).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC biology 12, 87, https://doi.org/10.1186/s12915-014-0087-z (2014).
de Goffau, M. C. et al. Recognizing the reagent microbiome. Nat Microbiol 3, 851–853, https://doi.org/10.1038/s41564-018-0202-y (2018).
Hannigan, G. D. et al. Culture-Independent Pilot Study of Microbiota Colonizing Open Fractures and Association with Severity, Mechanism, Location, and Complication from Presentation to Early Outpatient Follow-Up. J. Orthop. Res. 32, 597–605, https://doi.org/10.1002/jor.22578 (2014).
Altunbulakli, C. et al. Relations between epidermal barrier dysregulation and Staphylococcus species-dominated microbiome dysbiosis in patients with atopic dermatitis. J Allergy Clin Immunol, https://doi.org/10.1016/j.jaci.2018.07.005 (2018).
Grice, E. A. et al. Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 (2009).
Grice, E. A. et al. A diversity profile of the human skin microbiota. Genome Res. 18, 1043–1050, https://doi.org/10.1101/gr.075549.107 (2008).
Kong, H. H. et al. Performing Skin Microbiome Research: A Method to the Madness. J Invest Dermatol 137, 561–568, https://doi.org/10.1016/j.jid.2016.10.033 (2017).
Findley, K. et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 498, 367–370, https://doi.org/10.1038/nature12171 (2013).
Chng, K. R. et al. Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nature Microbiology 1, https://doi.org/10.1038/nmicrobiol.2016.106 (2016).
Oh, J., Byrd, A. L., Park, M., Kong, H. H. & Segre, J. A. Temporal Stability of the Human Skin Microbiome. Cell 165, 854–866, https://doi.org/10.1016/j.cell.2016.04.008 (2016).
Marotz, C. A. et al. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 6, 42, https://doi.org/10.1186/s40168-018-0426-3 (2018).
Costea, P. I. et al. Towards standards for human fecal sample processing in metagenomic studies. Nature biotechnology 35, 1069–1076, https://doi.org/10.1038/nbt.3960 (2017).
Sinha, R. et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nature biotechnology 35, 1077–1086, https://doi.org/10.1038/nbt.3981 (2017).
Ogai, K. et al. A Comparison of Techniques for Collecting Skin Microbiome Samples: Swabbing Versus Tape-Stripping. Front Microbiol 9, 2362, https://doi.org/10.3389/fmicb.2018.02362 (2018).
Nakatsuji, T. et al. The microbiome extends to subepidermal compartments of normal skin. Nature Communications 4, https://doi.org/10.1038/ncomms2441 (2013).
Zeeuwen, P. L. J. M. et al. Microbiome dynamics of human epidermis following skin barrier disruption. Genome Biol. 13, https://doi.org/10.1186/gb-2012-13-11-r101 (2012)
Tupker, R. A. et al. Guidelines on sodium lauryl sulfate (SLS) exposure tests. A report from the Standardization Group of the European Society of Contact Dermatitis. Contact Dermatitis 37, 53–69 (1997).
Mukherjee, S. et al. Sebum and Hydration Levels in Specific Regions of Human Face Significantly Predict the Nature and Diversity of Facial Skin Microbiome. Sci Rep 6, 36062, https://doi.org/10.1038/srep36062 (2016).
Baurecht, H. et al. Epidermal lipid composition, barrier integrity, and eczematous inflammation are associated with skin microbiome configuration. J Allergy Clin Immunol 141, 1668–1676.e1616, https://doi.org/10.1016/j.jaci.2018.01.019 (2018).
Meisel, J. S. et al. Skin Microbiome Surveys Are Strongly Influenced by Experimental Design. J Invest Dermatol 136, 947–956, https://doi.org/10.1016/j.jid.2016.01.016 (2016).
Kuczynski, J. et al. Experimental and analytical tools for studying the human microbiome. Nature reviews. Genetics 13, 47–58, https://doi.org/10.1038/nrg3129 (2011).
Zeeuwen, P. et al. Reply to Meisel et al. J Invest Dermatol 137, 961–962, https://doi.org/10.1016/j.jid.2016.11.013 (2017).
Hugerth, L. W. et al. DegePrime, a program for degenerate primer design for broad-taxonomic-range PCR in microbial ecology studies. Appl Environ Microbiol 80, 5116–5123, https://doi.org/10.1128/aem.01403-14 (2014).
Hugerth, L. W. et al. A comprehensive automated pipeline for human microbiome sampling, 16S rRNA gene sequencing and bioinformatics processing. bioRxiv, 286526. Preprint at, https://www.biorxiv.org/content/biorxiv/early/2018/03/21/286526.full.pdf (2018).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12 (2011).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Edgar, R. C. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv, 081257. Preprint at, https://www.biorxiv.org/content/biorxiv/early/2016/10/15/081257.full.pdf (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research 41, D590–D596 (2012).
Boulund, F. StaG-mwc v0.3.0-beta. Zenodo, https://doi.org/10.5281/zenodo.1483891 (2018).
Boulund, F. & Olsson, L. StaG Metagenomic Workflow Collaboration (mwc). GitHub, https://github.com/ctmrbio/stag-mwc (2018).
Bushnell, B. BBMap: a fast, accurate, splice-aware aligner. Lawrence Berkeley National Lab.(LBNL), Berkeley, CA (United States). Poster at, https://jgi.doe.gov/wp-content/uploads/2013/11/BB_User-Meeting-2014-poster-FINAL.pdf (2014).
Truong, D. T. et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature methods 12, 902 (2015).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 1.17-2. R Development Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing (2010).
Vavrek, M. J. Fossil: palaeoecological and palaeogeographical analysis tools. Palaeontologia Electronica 14, 16 (2011).
Hintze, J. L. & Nelson, R. D. Violin plots: a box plot-density trace synergism. The American Statistician 52, 181–184 (1998).
Hunter, J. D. Matplotlib: A 2D graphics environment. Computing in science & engineering 9, 90 (2007).
Waskom, M. et al. mwaskom/seaborn: v0.9.0. Zenodo, https://doi.org/10.5281/zenodo.1313201 (2018).
McKinney, W. pandas: a foundational Python library for data analysis and statistics. Python for High Performance and Scientific Computing 14 (2011).
Acknowledgements
The authors would like to acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure, NGI, and Uppmax (Sweden) for providing assistance in massive parallel sequencing and computational infrastructure. A grant from Danish Environmental Protection Agency is supporting salary of the scientists at the National Allergy Research Centre. Running costs were supported by Aage Bangs Fond.
Author information
Authors and Affiliations
Contributions
R.D.B. and J.D.J. designed the experiment. R.D.B. collected samples, extracted DNA and wrote the manuscript with input from all authors. M.S. made libraries for amplicon sequencing. L.W.H., F.B., R.D.B. and L.E. analyzed, interpreted and visualized data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors declare that they have no financial competing interests. J.D.J. is head of the cosmetic counsel (unpaid position) advisory to the Danish Environmental Protection Agency.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bjerre, R.D., Hugerth, L.W., Boulund, F. et al. Effects of sampling strategy and DNA extraction on human skin microbiome investigations. Sci Rep 9, 17287 (2019). https://doi.org/10.1038/s41598-019-53599-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53599-z
- Springer Nature Limited
This article is cited by
-
Recovery of microbial DNA by agar-containing solution from extremely low-biomass specimens including skin
Scientific Reports (2023)
-
Human microbiome transfer in the built environment differs based on occupants, objects, and buildings
Scientific Reports (2023)
-
Optimization of DNA extraction and sampling methods for successful forensic microbiome analyses of the skin and saliva
International Journal of Legal Medicine (2023)
-
Skin Microbiota and the Cosmetic Industry
Microbial Ecology (2023)
-
Controlling skin microbiome as a new bacteriotherapy for inflammatory skin diseases
Inflammation and Regeneration (2022)