
Abstract
Phasevarions (phase-variable regulons) are emerging as an important area of bacterial gene regulation. Many bacterial pathogens contain phasevarions, with gene expression controlled by the phase-variable expression of DNA methyltransferases via epigenetic mechanisms. Non-typeable Haemophilus influenzae (NTHi) contains the phase-variable methyltransferase modA, of which multiple allelic variants exist (modA1-21). We have previously demonstrated 5 of 21 these modA alleles are overrepresented in NTHi strains isolated from children with middle ear infections. In this study we investigated the modA allele distribution in NTHi strains isolated from patients with chronic obstructive pulmonary disease, COPD. We demonstrate that the distribution of modA alleles in a large panel of COPD isolates is different to the distribution seen in middle ear infections, suggesting different modA alleles may provide distinct advantages in the differing niches of the middle ear and COPD airways. We also identified two new phase-variable modA alleles – modA15 and modA18 – and demonstrate that these alleles methylate distinct DNA sequences and control unique phasevarions. The modA15 and modA18 alleles have only been observed in COPD isolates, indicating that these two alleles may be markers for isolates likely to cause exacerbations of COPD.
Similar content being viewed by others

Introduction
Non-typeable Haemophilus influenzae (NTHi) is a major human-adapted pathogen that is the etiological agent of a number of acute and chronic diseases of the human respiratory tract1,2, and for invasive infections such as septicaemia and meningitis3,4,5. NTHi is intimately involved with the pathogenesis of chronic obstructive pulmonary disease (COPD). Approximately 65 million people worldwide suffer from COPD, which is the fourth most common cause of death globally6. NTHi is known to colonise patients with COPD and persist for extended periods of time. This results in increased inflammation and resulting tissue damage, and further loss of pulmonary function7. A sudden worsening of COPD symptoms is known as an exacerbation. Half of all COPD exacerbations are the result of bacterial infection, with NTHi being the most common bacterial pathogen isolated from COPD patients experiencing an exacerbation8. Individual NTHi strains show a high level of genotypic and phenotypic heterogeneity9. Previous work has shown that a significant number (33%) of COPD exacerbations occur upon acquiring a new strain of NTHi10, and that the lung microbiome changes both during and after COPD exacerbations11. There is also a demonstrated link to particular genomic features associated with strains of NTHi isolated from patients experiencing a COPD exacerbation12, which is strongly indicative that COPD exacerbations are not only due to host factors, but are triggered by bacterial factors.
We have previously reported the presence of phase-variable regulons (phasevarions)13 in a range of human adapted bacterial pathogens such as Streptococcus pneumoniae14, Neisseria gonorrhoeae and Neisseria meningitidis15, Helicobacter pylori16, Haemophilus influenzae17, and Moraxella catarrhalis18. Phasevarions result in differential expression of a regulon of genes via epigenetic mechanisms through the phase-variation (ON-OFF switching or variation in specificity) of DNA methyltransferases associated with restriction-modification (R-M) systems. In all cases described, phasevarions control the expression of genes required for pathobiology. In many cases phasevarion switching also leads to differential expression of vaccine candidates in multiple bacterial pathogens such as S. pneumoniae14, N. gonorrhoeae and N. meningitidis15, and NTHi19. The presence of phasevarions in several major human pathogens makes identification of stably expressed antigens problematic, as the genes subject to regulation by phasevarions do not contain any easily identifiable features. Currently, the only way to identify genes in a phasevarion is by detailed study of gene and/or protein expression in the alternate phasevarion states. The identification of surface antigens regulated by phasevarions is critical to find stably expressed vaccine antigens and also to understand their role in pathobiology.
In work carried out with a large panel of NTHi isolates taken from children with middle ear infection, or otitis media (OM), we have previously demonstrated that ~65% of these NTHi isolates contain one of just five phase-variable Type III N6 adenine DNA methyltransferases, ModA2, 4, 5, 9, or 10, among the 21 known modA alleles. Each modA allele methylates a distinct DNA sequence and controls a different set of genes, i.e., a different phasevarion19. The genes encoding these alleles are highly conserved at their 5′ and 3′ regions (95% DNA identity), but contain a highly variable central region20, encoding the Target Recognition Domain (TRD). The TRD dictates the sequence recognised and methylated by the ModA protein. A different TRD means a different sequence is methylated, and consequently a different phasevarion of regulated genes is controlled. We hypothesised that strains of NTHi isolated from a different host micro-environment, i.e., the airways of COPD patients, may contain a different set of modA alleles when compared to those isolated from the middle ear of children with OM19, as the environment of the COPD lung differs significantly from the OM middle ear. Here we describe the modA alleles present in a large collection (n = 269) of NTHi strains isolated from the sputum of people with COPD21 to investigate the modA allele distribution, and characterise gene expression differences resulting from phase-variation of two previous unstudied modA alleles, modA15 and modA18.
Methods
Bacterial strains and growth conditions
NTHi strains 10P129H1 (modA15) and 84P36H1 (modA18) were isolated from COPD patients in Buffalo, USA, as part of a previous study21. The institutional review boards of the University at Buffalo and the Veterans Affairs Western New York Healthcare System approved collection of samples, as detailed previously21; study participants provided written informed consent before enrollment, as detailed previously21. All methods were performed in accordance with the relevant guidelines and regulations of the University at Buffalo and the Veterans Affairs Western New York Healthcare System, as detailed previously21. NTHi were grown in BHI broth (Oxoid) supplemented (sBHI) with hemin (1% v/v) and NAD (2 µg/ml), or on sBHI agar (as broth but with 1% w/v bacteriological agar; Oxoid). Liquid cultures were grown aerobically at 37 °C with shaking at 150 rpm. Plates were grown at 37 °C supplemented with 5% (v/v) CO2. Natural modA15 and modA18 ON and OFF variants in NTHi strains 10P129H1 and 84P36H1, respectively, were identified by fragment length analysis of the modA repeat tract of multiple single colonies using the fluorescently labeled (6-Carboxyfluorescein; FAM) forward primer Him1F (5′-FAM-ATGGCGGACAAAGCACCGAAGG-3′) and the reverse primer Him3 (5′-CAAAAAGCCGGTCAATTTCATCAAA-3′)22, and fragments were analyzed by the Australian Genome Research Facility (AGRF, Brisbane, Australia). Isolates containing ≥90% ON or OFF were considered to be natural ON or OFF respectively, as determined by fragment length analysis, and were used in subsequent studies. Fluorescently labelled fragments were measured using a 3130xl Genetic Analyser and GeneScan system (Applied Biosystems), and peaks analysed using Peakscanner version 1.0 (Applied Biosystems). We have previously described the modA15 and modA18 ON/OFF pairs23 and shown by Western blot that expression only occurs in the ON strain of each strain pair23.
Analysis of modA allelic diversity in COPD isolates
Whole genome shot-gun sequencing had previously been carried out for all 269 NTHi strains isolated from COPD patients21. These genome sequences were used in a BLAST search using prototype modA alleles20, and the modA sequence isolated from each strain. Alignments were carried out using Clustal-W, and each allele manually checked in a nucleotide sequence alignment against the twenty-one known modA alleles. To compare the distribution of modA alleles in our COPD collection with the distribution of modA alleles in our previously published OM collection19, we used a one-tailed z-score test to compare individual modA allele proportions between the two populations.
Preparation of outer membrane proteins (OMPs) from NTHi
NTHi modA15 and modA18 ON/OFF pairs were grown in sBHI broth (50 ml) at 37 °C overnight with shaking at 100 rpm. Cells were pelleted at 4500 rpm for 15 mins at 4 °C, resuspended in 4 ml 10 mM HEPES-NaOH pH 7.5, and OMPs were prepared as detailed previously24. Briefly, cells were lysed by sonication, and debris pelleted as above. Sarkosyl was added to the clarified supernatant to a final concentration of 1%, and incubated at 25 °C for 30 mins. Supernatants were then centrifuged at 35,000 rpm for 90 mins. Pellets were resuspended in 10 mM HEPES-NaOH pH 7.5, sarkosyl added to a final concentration of 1%, and incubation and centrifugation steps repeated twice more. Final pellets containing the OMP-enriched fraction were resuspended in 100 μl of 10 mM HEPES-NaOH pH 7.5, and the protein concentration quantified using the BCA protein assay kit according to manufacturer’s instructions (Thermo Scientific). Each of the OMP preparations (5 µg) were run on the Novex Bis-Tris pre-cast gel system with MOPS running buffer according to the manufacturer’s instructions (Life Technologies). Ammoniacal silver staining was carried out to visualize proteins25 and observe if any proteins were differentially expressed due to modA phase-variation. Differences were identified visually, and confirmed by processing each band with imageJ using a band that was observed as the same intensity in the respective ON vs OFF lane (loading control; LC). Only differences that were confirmed by ImageJ as >2-fold intensity difference relative to the loading control were considered differentially expressed. Difference in intensity is presented as the fold difference ON vs OFF normalised to loading control.
Single-Molecule, Real-Time (SMRT) sequencing and methylome analysis
We previously sequenced and submitted annotated genomes of NTHi strains 10P129H1 (modA15) and 84P36H1 (modA18)23. Briefly, DNA was sequenced at the Yale Center for Genome Analysis (YCGA) using the PacBio RS II platform with P6-C4 chemistry and a library size of 10 kb, with one strain per SMRT cell, and assembled de novo using the hierarchical genome assembly process (HGAP)26. Polishing for a pure-PacBio assembly was carried out using the Quiver algorithm26 from the SMRT Analysis software suite (version 2.3.0 – http://www.pacb.com/devnet/) with default parameters. Consensus sequences were submitted to NCBI for annotation with the Prokaryotic Genome Annotation Pipeline (PGAP), and annotated sequences submitted to GenBank (accession numbers CP029620 [strain 10P129H1] and CP029621 [strain 84P36H1]). Methylome analysis was carried out as described previously27,28 by YCGA. Each genome was sequenced to sufficient depth to allow methylome analysis of m6A methylation (cytosine methylation was not searched for). Coverage of each strain was as follows - strain 10P129H1 - ModA15 ON coverage of 393.5 fold, and ModA15 OFF coverage of 435.5 fold; strain 84P36H1 – ModA18 ON coverage of 435.5 fold, ModA18 OFF coverage of 380.0 fold.
RNA Seq analysis
Triplicate biological replicates of total RNA were prepared using Trizol (Thermo Fisher) according to manufacturer’s instructions from mid-log cultures of NTHi modA15 and modA18 ON/OFF pairs (OD600 = 0.5) as previously used for modA ON vs OFF expression analysis19. RNA Seq data sets used have been recently announced29. RNA quality was assessed by AGRF using an Agilent Bioanalyser. All RNA preps had an RNA integrity number (RIN) of above 8.0, indicating high quality RNA. Libraries were prepared using the Illumina Ribo-Zero Gold protocol. Briefly, RNA was fragmented, and randomly primed first strand cDNA synthesis carried out using SuperScript II Reverse Transcriptase (Invitrogen) according to manufacturer’s protocols. Following second-strand cDNA synthesis, fragments were adenylated at the 3′ end, and polyT containing sequencing adapters ligated. Libraries were then amplified via PCR (13 cycles). Library quality was assessed using an Agilent Bioanalyser DNA 1000 chip. qPCR quantification was used to assess individual libraries before normalizing (2 nM) and pooling using the Illumina cBot system with TruSeq PE Cluster Kit v3 reagents. Sequencing (150 bp paired end runs) was performed on the Illumina NovaSeq system with TruSeq SBS Kit v3 reagents (average number of sequence reads for each triplicate sample is as follows – modA15 ON - 37,726,839; modA15 OFF - 32,023,168; modA18 ON - 42,069,056; modA18 OFF - 34,004,813). Sequence quality was assessed according to the standard protocols of AGRF. Unfiltered sequence reads were aligned against the respective reference genomes (CP029620 [10P129H1; modA15]; CP029621 [84P36H1; modA18]) using Bowtie2 aligner (v2.3.3.1) using standard settings. Default software settings were used throughout unless otherwise stated. Transcripts were assembled with Stringtie v1.3.3 (http://ccb.jhu.edu/software/stringtie/) utilising the reads alignment and reference annotation based assembly option. This methodology generated assemblies for known and potentially novel transcripts. The Gencode annotation containing both coding and non-coding annotation for each genome was used as a guide (http://www.gencodegenes.org/). Raw gene count values were analysed with edgeR (https://bioconductor.org/packages/release/ bioc/html/edgeR.html) to compute differential gene expression values. Counts were summarised at the gene level using the featureCounts v1.5.3 utility of the subread package (http://subread.sourceforge.net/). Gene expression differences between respective modA ON and OFF were expressed as logFC (log2-fold change of expression). Analysis generated logCPM values (average log count per million for the gene across all samples), F values (quasi-likelihood F-statistic for the gene across all samples), p-values for the test of statistically different expression, and the FDR (False discovery rate/Adjusted p-value for multiple hypothesis testing). Annotated genomes for each strain were used as the reference genome CP029620 (10P129H1; modA15 ON/OFF pair) and CP029621 (84P36H1; modA18 ON/OFF pair).
Results and Discussion
An NTHi collection isolated from COPD patients contains different proportions of modA alleles compared to those isolated from OM patients
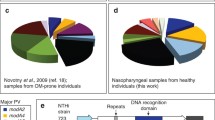
Using sequenced genomes for 269 NTHi isolates from patients presenting with COPD21 we carried out BLAST analysis with our previously, well-defined modA allele sequences19,20 to determine the modA allele distribution within this population. The collection contains 168 independent strains consisting of 67 cleared strains and the first and last isolates of 101 persistent strains (n = 202) collected longitudinally over a fifteen year period (April 1994–March 2009)21. Cleared strains are defined as those isolated once at a single monthly visit to the clinic, then not isolated again on subsequent visits21. For modA allele distribution, we only counted the cleared strains (n = 67) and the first isolate of each persistent strain pair (n = 101), meaning we analysed the modA allele distribution in 168 unique strains. We could not detect any significant homology to any modA allele in seven of these strains, indicating that these isolates do not contain a modA gene. We have not observed this before in any other data sets, i.e., all previously sequenced NTHi always contain a modA gene. There was insufficient sequence coverage to accurately determine the modA allele present in 3 strains. Therefore 158 strains in total were analysed for their modA allele distribution. In order to determine if there is a different proportion of modA alleles associated with COPD isolates, we compared this collection to our previously characterised OM collection19. The majority (82.5%) of these isolates where collected in the USA19,22,30,31, with the remaining 17.5% from Finland32. Three major OM-associated phase-variable modA alleles, modA2, 4, and 5, were all represented in COPD isolates at very similar levels to those seen in NTHi isolated from our previous studies19 (Fig. 1; modA2 [19.6% COPD vs 17.2% OM], modA4 [11.4% COPD vs 12.4% OM] and modA5 ([11.4% COPD vs 9.5% OM]; p value of >0.05 for all three alleles using a one-tailed z-score test). However, the two other major modA alleles seen in children with OM, modA9 and modA1019 were present in a much reduced proportion of strains in our COPD population; modA9 was not present in any strain isolated from the COPD airway (vs 4.7% of OM isolates; a statistically significant p value of 0.0028 using a one-tailed z-score test), and modA10 was present in only 3 COPD isolates (1.9%) despite being the second most prevalent phase-variable modA allele present in our OM isolate collection (15.3% OM; a statistically significant p value of <0.00001 using a one-tailed z-score test)19. This implies that the genes being differentially regulated by modA9 and modA10 provide an advantage, and are therefore selected for, in the OM middle ear, but not in COPD airways. It is even possible that the phasevarions controlled by modA9 and modA10 are disadvantageous in the context of COPD airways. Alternatively, and as a caveat to our statistical analysis, the different distribution of modA alleles in COPD and OM isolates could be due to geographic and/or temporal differences in the two strain collections: COPD isolates were collected at the Buffalo Veterans Affairs Medical Center, New York, USA, between 1994–200921; OM strains comprise a number of different sub-collections collected in multiple locations across the globe, and have been detailed previously19. Therefore, the precise role, if any, of modA9 and modA10 in COPD would require extensive experimental confirmation, and this is hindered by the lack of any non-COPD isolates from the Buffalo area to determine if the lack of modA9 in isolates taken from the COPD lung are due to selection against this allele in COPD, or due to lack of this allele in general in strains from this area.

Distribution of the modA allele in a collection of NTHi isolated from patients with COPD. We analysed 269 unique strains taken from COPD patients and previously genome sequenced21 consisting of 67 cleared strains, and 101 persistent strain pairs (202 strains in total). BLAST search and sequence alignment for each strain was carried out in order to determine the modA allele present in each strain. We only analysed the 67 cleared strains, and the first isolate of each strain pair (n = 168). Of these 168 strains, 7 strains did not contain a modA allele, and 3 strains did not contain sufficient sequence to determine the modA allele present. (A) The modA allele distribution in 158 strains where we could unequivocally identify the modA allele sequence. modA15, modA18, and modA22 are highlighted with a *. PV = phase-variable. The % of each modA allele present in the collection is given in the key; (B) sequence alignment of newly identified/studied modA alleles modA15, modA18, and modA22 (newly identified as part of this study). Alignments were carried out using ClustalW, and visualized in JalView overview feature. Nucleotides are represented as vertical blue bars (dark blue >80% identity; light blue >50% identity; white <50% identity or gap.
During our analysis we characterised phasevarions controlled by two previously described modA alleles - modA15 and modA18 - and discovered one new modA allele, which we have designated modA22. Although modA15 and modA18 had been identified previously20, their methylation specificity and the genes that they control, i.e., their respective phasevarions, had never been defined. Analysis of the collection showed that five strains contained modA15 (3.16%) and seven strains contained modA18 (4.43%). All modA genes in the study contain a AGCC[n] DNA repeat tract of variable length, which mediates phase-variable expression. We picked prototype strains containing each allele - strain 10P129H1 contained modA15 and strain 84P36H1 contained modA18. We have previously sequenced and annotated the genomes of these strains (Accession numbers CP029620 (strain 10P129H1) and CP029621 (strain 84P36H1)23. Fragment length analysis showed that the AGCC[n] repeat tracts of these strains was phase-variable, and we were able to isolate ON and OFF enriched populations23.
The new modA allele identified in our COPD collection, which we have designated modA22, was not phase-variable. All strains containing a modA22 allele (11 strains; 6.96%) contained three AGTC repeats in place of a variable AGCC[n] repeat tract. Simple sequence repeat (SSR) tracts of this length phase-vary at very low rates33,34,35, and our own analysis showed only a single peak when carrying out fragment length analysis across this region of DNA, with no evidence of any sub-populations showing different SSR tract lengths (data not shown).
ModA15 and ModA18 methylate different target sequences to currently characterised ModA alleles
We used methylome analysis in order to determine the methyltransferase specificity of ModA15 and ModA18. Using our previously described methodology and rationale19,36 we compared the methylomes from the ModA15 and ModA18 ON/OFF pairs to determine the specificity of each ModA methyltransferase. This analysis showed that ModA15 methylates the sequence 5′-G(m6A)ANTCNNCG-3′, and ModA18 methylates the sequence 5′-CTS(m6A)AGNNNNCG-3′ (Table 1). Both these sequences are unusual for Type III methyltransferases, as the majority recognise and methylate five base-pair non-palindromic sequences37,38. The ModA15 recognition sequence, 5′-G(m6A)ANTCNNCG-3′, occurs just 364 times in the genome of NTHi strain 10P129H1, and the ModA18 recognition sequence 5′-CTS(m6A)AGNNNNCG-3′ appears only 94 times in the genome of NTHi strain 84P36H1. Unlike other ModA alleles that methylate >95% of their recognition sites19,36, we could only detect methylation at 15.4% (56/364) of ModA15 sites, and at 48.9% (46/94) of ModA18 sites in the respective modA ON genomes. No methylation of these sequences was detected in the respective modA OFF genomes of each strain (Table 1). Our previous Western blot analysis of the ON/OFF pairs used for SMRT sequencing shows that ModA is expressed in each ON strain23 so the low methylation coverage was unexpected. However, these methyltransferases may recognise a hierarchy of sequences, with the identity of the non-specific bases (non-specific bases or ‘N’ refers to any of the four DNA bases A, C, G, or T) in each consensus sequence playing a role in the specificity/methylation at that particular site. This relaxed specificity has been observed before with ModA11 in Neisseria meningitidis36, with between 100% and 4.6% methylation detected at specific sites, dependent on the non-specific bases within the 5′-NCGY(m6)AGN-3′ consensus recognition sequence. In both ModA15 and Mod18 ON/OFF pairs we could detect Dam methylation at its characterised 5′-G(m6A)ATC-3′ site, but at only approximately 65% of all GATC sites in all four genomes (Table 1). We have previously seen at least 95% of GATC sites methylated in other genomes where we have carried out methylome analysis19,36. Perhaps the low level of methylation seen with both strains, including with Dam, is a result of either relaxed specificity, or a deficiency in the metabolism of these strains, but this would require extensive experimental confirmation, and is beyond the scope of the current study. An interesting follow up study could be to determine if competition with other methyltransferases, or other DNA binding proteins, exists at ModA15 and ModA18 sites to explain low levels of methylation, or if there is a general, as yet unknown, deficiency in ability to methylate in these strains.
Phase-variation of ModA15 and ModA18 results in outer-membrane protein expression differences
In order to investigate if phase-variation of modA15 and modA18 resulted in differences in protein expression, we prepared outer-membrane protein (OMP) fractions from our modA15 and modA18 ON/OFF pairs. Following ammoniacal silver staining of 5 µg of each OMP preparation, clear differences in the expression level of several proteins is visible when comparing modA15 ON vs OFF, and modA18 ON vs OFF (Fig. 2). Differences in size of a number of comparable protein bands can also been seen, implying that modA15 and modA18 phase-variation leads to differences in expression of allelic variants of these proteins, or in differences in post-translational modifications of these proteins. As described in previously characterised modA phasevarions19, this demonstrates that phase-variation of modA15 and modA18 results in gene and protein expression differences, and that these modA alleles control phasevarions. Differences in expression of outer-membrane proteins are likely to influence the interaction of the bacterial population and the human host. As the outer-membrane of NTHi is the main interface with the host, this could have implications on pathobiology. Phase variation of OMPs could also impact vaccine development, as alterations in the expression level and/or modification state of proteins targeted by vaccines could render those vaccines ineffective if the target is no longer expressed or is modified so as to prevent recognition of epitopes.

Silver stained protein gel showing outer-membrane protein (OMP) expression differences resulting from modA15 and modA18 phase-variation. OMPs were prepared using a sarkosyl OMP preparation protocol. 5 μg of each OMP fraction was separated on SDS PAGE and visualized using ammoniacal silver staining. Differences in protein expression between modA15 ON vs OFF, and modA18 ON vs OFF are highlighted with arrows. Differences were identified visually, and confirmed by processing each band with imageJ using a band that was observed as the same intensity in the respective ON vs OFF lane (loading control; LC). Only differences that were confirmed by ImageJ as >2-fold intensity difference ON vs OFF relative to the loading control are highlighted. Difference in intensity as fold difference ON vs OFF normalised to loading control. modA15 ON vs modA15 OFF: (i) 0.11 fold; (ii) 4.4 fold; (iii) 6.3 fold. modA18 ON vs modA18 OFF: (iv) 0.46 fold; (v) 0.40 fold. In addition, a difference in size of a protein of equivalent expression is highlighted with * in modA18 ON vs OFF. The full gel image is provided in Supplementary Fig. 1.
RNA Seq analysis reveals phase-variation of modA15 and modA18 results in whole cell gene expression differences
In order to determine the effects of modA15 and modA18 phase-variation on whole cell gene expression differences, we carried out whole cell RNA Seq analysis using a single ON/OFF pair of strains for each modA allele. RNA was prepared from the same cultures used to carry out Western blotting to show that each methyltransferase is only expressed in the ON strain, and confirmed by fragment length analysis, as described previously23. Our analysis showed that 4 genes were up-regulated and 40 genes down-regulated in modA15 ON relative to OFF, and that 8 genes were up-regulated and 2 genes down-regulated in modA18 ON relative to OFF (Table 2). Given the unusual recognition sequences of these two ModA alleles, and their rarity in the genomes of the strains containing them, the small number of differentially expressed genes is not surprising. A comparison of the genome sequence of each strain pair showed there were minimal differences between each strain pair except within the relevant modA AGCC[n] repeat tract.
In the ModA15 phasevarion, the DNA binding transcriptional regulator Fis [DLJ98_05145] was up-regulated in ON relative to OFF. Therefore, in addition to the direct regulatory effects of ModA15 methylation, i.e., regulation of genes by differential methylation of individual gene promoter regions, the differential regulation of Fis could be thought of as indirect gene regulation by modA phase-variation39 (regulation of a regulator – the gene expression changes resulting from Fis being differentially regulated are indirectly due to ModA15, as modA15 phase-variation regulates the expression of the regulator Fis). Fis has been shown to modulate virulence gene expression in a number of bacterial pathogens, including Salmonella enterica and Dickeya dadantii40, so differential regulation of Fis by ModA15 could play a key role in NTHi pathobiology. The transcriptional repressor LacI (DLJ98_05540) is differentially regulated in the ModA15 phasevarion, showing a 2-fold decrease in expression in ModA15 ON. Lower expression of LacI in ModA15 ON may impart more metabolic flexibility in vivo due to increased expression of genes involved in carbon metabolism. A number of metabolic genes are down-regulated in the ModA15 phasevarion when ModA15 is ON; these include genes involved in nucleotide metabolism (e.g., D-ribose pyranase [DLJ98_05515], hypoxanthine phosphoribosyltransferase [DLJ98_04115]) central carbon metabolism (C4-dicarboxylate ABC transporter permease [DLJ98_04415], malate dehydrogenase [DLJ98_03795]) and phosphate metabolism (acid phosphatase AphA [DLJ98_05485], phosphonate ABC transporter substrate-binding protein [DLJ98_10115]). This suggests that a sub-population of cells where ModA15 is OFF may be better equipped to colonise different host niches compared to sub-populations of cells where ModA15 is ON, and indicates that dynamic interplay between ModA15 ON and ModA15 OFF sub-populations could occur during colonisation and disease.
A number of genes regulated in the ModA18 phasevarion could have effects on NTHi pathobiology; for example, ModA18 ON shows increased expression of the regulatory protein GemA (DLK00_05500), that is widespread in the Pasteurellaceae, but has an as yet undefined regulon. The anaerobic dimethyl sulphoxide reductase (DmsBCD; DLK00_02880 - DLK00_02890) is also up-regulated in ModA18 ON. Dimethyl sulphoxide reductase has been shown to be important in virulence in Actinobacillus pleuropneumoniae41.
Our demonstration of the presence of two new phase-variable modA alleles controlling phasevarions in NTHi adds an extra level of complexity to vaccine development in this organism. The identification of what could be considered COPD-associated modA alleles, modA15 and modA18, means that characterisation of the complete phasevarions regulated by these two alleles could aid development of more effective treatment options for COPD patients colonised by NTHi, as well as directing and informing overall vaccine development against NTHi.
References
Haggard, M. Otitis media: prospects for prevention. Vaccine 26(Suppl 7), G20–24 (2008).
Johnson, R. H. Community-acquired pneumonia: etiology, diagnosis, and treatment. Clinical therapeutics 10, 568–573 (1988).
van Wessel, K. et al. Nontypeable Haemophilus influenzae Invasive Disease in the Netherlands: A Retrospective Surveillance Study 2001–2008. Clinical Infectious Diseases 53, e1–e7, https://doi.org/10.1093/cid/cir268 (2011).
Wan Sai Cheong, J. et al. Trends in the epidemiology of invasive Haemophilus influenzae disease in Queensland, Australia from 2000 to 2013: what is the impact of an increase in invasive non-typable H. influenzae (NTHi)? Epidemiology and infection, 1–8, https://doi.org/10.1017/s0950268815000345 (2015).
Whittaker, R. et al. Epidemiology of Invasive Haemophilus influenzae Disease, Europe, 2007–2014. Emerging infectious diseases 23, 396–404, https://doi.org/10.3201/eid2303.161552 (2017).
Sethi, S. & Murphy, T. F. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. The New England journal of medicine 359, 2355–2365, https://doi.org/10.1056/NEJMra0800353 (2008).
Desai, H. et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Annals of the American Thoracic Society 11, 303–309, https://doi.org/10.1513/AnnalsATS.201310-350OC (2014).
Murphy, T. F., Brauer, A. L., Schiffmacher, A. T. & Sethi, S. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine 170, 266–272, https://doi.org/10.1164/rccm.200403-354OC (2004).
Van Eldere, J., Slack, M. P., Ladhani, S. & Cripps, A. W. Non-typeable Haemophilus influenzae, an under-recognised pathogen. The Lancet. Infectious diseases 14, 1281–1292, https://doi.org/10.1016/s1473-3099(14)70734-0 (2014).
Sethi, S., Evans, N., Grant, B. J. & Murphy, T. F. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. The New England journal of medicine 347, 465–471, https://doi.org/10.1056/NEJMoa012561 (2002).
Huang, Y. J. et al. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. Journal of clinical microbiology 52, 2813–2823, https://doi.org/10.1128/jcm.00035-14 (2014).
Fernaays, M. M., Lesse, A. J., Sethi, S., Cai, X. & Murphy, T. F. Differential genome contents of nontypeable Haemophilus influenzae strains from adults with chronic obstructive pulmonary disease. Infect Immun 74, 3366–3374, https://doi.org/10.1128/iai.01904-05 (2006).
Atack, J. M., Tan, A., Bakaletz, L. O., Jennings, M. P. & Seib, K. L. Phasevarions of Bacterial Pathogens: Methylomics Sheds New Light on Old Enemies. Trends in Microbiology 26, 715–726, https://doi.org/10.1016/j.tim.2018.01.008 (2018).
Manso, A. S. et al. A random six-phase switch regulates pneumococcal virulence via global epigenetic changes. Nat. Commun. 5, https://doi.org/10.1038/ncomms6055 (2014).
Srikhanta, Y. N. et al. Phasevarions mediate random switching of gene expression in pathogenic. Neisseria. PLoS Pathog. 5, e1000400 (2009).
Srikhanta, Y. N. et al. Phasevarion mediated epigenetic gene regulation in Helicobacter pylori. PLoS One 6, e27569 (2011).
Srikhanta, Y. N., Maguire, T. L., Stacey, K. J., Grimmond, S. M. & Jennings, M. P. The phasevarion: A genetic system controlling coordinated, random switching of expression of multiple genes. Proc. Natl. Acad. Sci. USA 102, 5547–5551, https://doi.org/10.1073/pnas.0501169102 (2005).
Blakeway, L. V. et al. ModM DNA methyltransferase methylome analysis reveals a potential role for Moraxella catarrhalis phasevarions in otitis media. FASEB J. 28, 5197–5207, https://doi.org/10.1096/fj.14-256578 (2014).
Atack, J. M. et al. A biphasic epigenetic switch controls immunoevasion, virulence and niche adaptation in non-typeable Haemophilus influenzae. Nat. Commun. 6, https://doi.org/10.1038/ncomms8828 (2015).
Gawthorne, J. A., Beatson, S. A., Srikhanta, Y. N., Fox, K. L. & Jennings, M. P. Origin of the diversity in DNA recognition domains in phasevarion associated modA genes of pathogenic Neisseria and Haemophilus influenzae. PLoS One 7, e32337, https://doi.org/10.1371/journal.pone.0032337 (2012).
Pettigrew, M. M. et al. Haemophilus influenzae genome evolution during persistence in the human airways in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA, https://doi.org/10.1073/pnas.1719654115 (2018).
Fox, K. L. et al. Haemophilus influenzae phasevarions have evolved from type III DNA restriction systems into epigenetic regulators of gene expression. Nucleic Acids Res. 35, 5242–5252, https://doi.org/10.1093/nar/gkm571 (2007).
Atack, J. M., Murphy, T. F., Bakaletz, L. O., Seib, K. L. & Jennings, M. P. Closed Complete Genome Sequences of Two Nontypeable Haemophilus influenzae Strains Containing Novel modA Alleles from the Sputum of Patients with Chronic Obstructive Pulmonary Disease. Microbiology Resource Announcements 7, https://doi.org/10.1128/mra.00821-18 (2018).
Murphy, T. F., Dudas, K. C., Mylotte, J. M. & Apicella, M. A. A subtyping system for nontypable Haemophilus influenzae based on outer-membrane proteins. J. Infect. Dis. 147, 838–846 (1983).
Oakley, B. R., Kirsch, D. R. & Morris, N. R. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem 105, 361–363 (1980).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10, 563–569, https://doi.org/10.1038/nmeth.2474 (2013).
Clark, T. A. et al. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 40, e29, https://doi.org/10.1093/nar/gkr1146 (2012).
Murray, I. A. et al. The methylomes of six bacteria. Nucleic Acids Res. 40, 11450–11462, https://doi.org/10.1093/nar/gks891 (2012).
Atack, J. M., Murphy, T. F., Pettigrew, M. M., Seib, K. L. & Jennings, M. P. Transcriptome Sequencing Data Sets for Determining Gene Expression Changes Mediated by Phase-Variable DNA Methyltransferases in Nontypeable Haemophilus influenzae Strains Isolated from Patients with Chronic Obstructive Pulmonary Disease. Microbiol Resour Announc 8, https://doi.org/10.1128/mra.00526-19 (2019).
Novotny, L. A. et al. Epitope mapping immunodominant regions of the PilA protein of nontypeable Haemophilus influenzae (NTHI) to facilitate the design of two novel chimeric vaccine candidates. Vaccine 28, 279–289, https://doi.org/10.1016/j.vaccine.2009.08.017 (2009).
Fox, K. L. et al. Selection for phase variation of LOS biosynthetic genes frequently occurs in progression of non-typeable Haemophilus influenzae infection from the nasopharynx to the middle ear of human patients. PLoS One 9, e90505, https://doi.org/10.1371/journal.pone.0090505 (2014).
Meats, E. et al. Characterization of encapsulated and noncapsulated Haemophilus influenzae and determination of phylogenetic relationships by multilocus sequence typing. Journal of clinical microbiology 41, 1623–1636, https://doi.org/10.1128/jcm.41.4.1623-1636.2003 (2003).
Bayliss, C. D. et al. Phase variable genes of Campylobacter jejuni exhibit high mutation rates and specific mutational patterns but mutability is not the major determinant of population structure during host colonization. Nucleic Acids Res. 40, 5876–5889, https://doi.org/10.1093/nar/gks246 (2012).
Cox, E. C. Bacterial mutator genes and the control of spontaneous mutation. Annu. Rev. Genet. 10, 135–156, https://doi.org/10.1146/annurev.ge.10.120176.001031 (1976).
Farabaugh, P. J., Schmeissner, U., Hofer, M. & Miller, J. H. Genetic studies of the lac repressor. J. Mol. Biol. 126, 847–863, https://doi.org/10.1016/0022-2836(78)90023-2 (1978).
Seib, K. L. et al. Specificity of the ModA11, ModA12 and ModD1 epigenetic regulator N6-adenine DNA methyltransferases of Neisseria meningitidis. Nucleic Acids Res. 43, 4150–4162, https://doi.org/10.1093/nar/gkv219 (2015).
Hadi, S. M., Bachi, B., Iida, S. & Bickle, T. A. DNA restriction–modification enzymes of phage P1 and plasmid p15B. Subunit functions and structural homologies. J. Mol. Biol. 165, 19–34 (1983).
Roberts, R. J., Vincze, T., Posfai, J. & Macelis, D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299, https://doi.org/10.1093/nar/gku1046 (2015).
Phillips, Z. N., Husna, A. U., Jennings, M. P., Seib, K. L. & Atack, J. M. Phasevarions of bacterial pathogens - phase-variable epigenetic regulators evolving from restriction-modification systems. Microbiology, https://doi.org/10.1099/mic.0.000805 (2019).
Duprey, A., Reverchon, S. & Nasser, W. Bacterial virulence and Fis: adapting regulatory networks to the host environment. Trends Microbiol 22, 92–99, https://doi.org/10.1016/j.tim.2013.11.008 (2014).
Baltes, N., Hennig-Pauka, I., Jacobsen, I., Gruber, A. D. & Gerlach, G. F. Identification of dimethyl sulfoxide reductase in Actinobacillus pleuropneumoniae and its role in infection. Infect. Immun. 71, 6784–6792 (2003).
Acknowledgements
We thank the Yale Centre for Genomic Analysis (YCGA; USA) for expert technical assistance in carrying out SMRT sequencing and methylome analysis. We thank the Australian Genome Research Facility (AGRF) for expert technical assistance in carrying out RNA Seq. This work was supported by the Australian National Health and Medical Research Council (NHMRC) Project Grant 1099279 to K.L.S. and J.M.A., Career Development Fellowship 1045235 to K.L.S., Program Grant 1071659 and Principal Research Fellowship 1138466 to M.P.J.; Garnett Passe and Rodney Williams Grant-in-Aid (Supplementation) to K.L.S. and J.M.A.; Australian Research Council (ARC) discovery grants 180100976 to JMA and 170104691 to M.P.J.; and National Institute of Health (NIH, USA) R01 AI19641 to T.F.M. and M.M.P.
Author information
Authors and Affiliations
Contributions
J.M.A., M.P.J., T.F.M. and K.L.S. conceived the research project and designed the experiments. J.M.A. performed the experiments and analysed the experimental results. J.M.A. prepared the manuscript. T.F.M. collected samples from COPD patients, and curates the COPD isolates collection. M.M.P. provided modA sequence data. All authors edited and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Atack, J.M., Murphy, T.F., Pettigrew, M.M. et al. Non-typeable Haemophilus influenzae isolates from patients with chronic obstructive pulmonary disease contain new phase-variable modA methyltransferase alleles controlling phasevarions. Sci Rep 9, 15963 (2019). https://doi.org/10.1038/s41598-019-52429-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52429-6
- Springer Nature Limited
This article is cited by
-
Reduction in Rubicon by cigarette smoke is associated with impaired phagocytosis and occurs through lysosomal degradation pathway
Clinical and Experimental Medicine (2023)