
Abstract
Inflammation is a complex physiological process that poses a serious threat to people’s health. However, the potential molecular mechanisms of inflammation are still not clear. Moreover, there is lack of effective anti-inflammatory drugs that meet the clinical requirement. Procyanidin A1 (PCA1) is a monomer component isolated from Procyanidin and shows various pharmacological activities. This study further demonstrated the regulatory role of PCA1 on lipopolysaccharide (LPS)-stimulated inflammatory response and oxidative stress in RAW264.7 cells. Our data showed that PCA1 dramatically attenuated the production of pro-inflammatory cytokines such as NO, iNOS, IL-6, and TNF-α in RAW264.7 cells administrated with LPS. PCA1 blocked IκB-α degradation, inhibited IKKα/β and IκBα phosphorylation, and suppressed nuclear translocation of p65 in RAW264.7 cells induced by LPS. PCA1 also suppressed the phosphorylation of JNK1/2, p38, and ERK1/2 in LPS-stimulated RAW264.7 cells. In addition, PCA1 increased the expression of HO-1, reduced the expression of Keap1, and promoted Nrf2 into the nuclear in LPS-stimulated RAW264.7 cells. Cellular thermal shift assay indicated that PCA1 bond to TLR4. Meanwhile, PCA1 inhibited the production of intracellular ROS and alleviated the depletion of mitochondrial membrane potential in vitro. Collectively, our data indicated that PCA1 exhibited a significant anti-inflammatory effect, suggesting that it is a potential agent for the treatment of inflammatory diseases.
Similar content being viewed by others


Introduction
A large number of studies have shown that inflammation is a defensive body response after stimulation by all kinds of ligands. However, in some cases, inflammation is potentially harmful. If inflammation cannot be effectively controlled, it will lead to excessive production of pro-inflammatory factors and a series of pathological change, such as diabetes, pneumonia, hepatitis, and nephritis1,2. There are overwhelming studies on inflammation recently, among which the most popular in vitro model is RAW264.7 cell induced by LPS3. RAW264.7, a monocyte/macrophage-like cell line, is also the most commonly used in vitro study on screening anti-inflammatory activity from natural compounds. LPS is a common inducer from Escherichia coli O111:B44,5. LPS can upregulate a large number of inflammatory mediators such as nitric oxide (NO), cyclooxygenase-2 (COX-2), tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), etc. in RAW264.7 cells during the inflammatory reaction6.
As an important transcriptional regulation factor, Nuclear factor (NF-κB) participates in regulating the body of chronic inflammation and produce certain physiological changes7. The NF-κB family members including p50, p52, Rel (p65), c-Rel, and RelB, contain one N-terminal Rel homology region that mediates polymerization, DNA binding, and c-terminal nuclear localization sequence8. The most abundant and active form of NF-κB is p65-p50 heterodimer. At rest, the IκB protein binds to the nuclear NF-κB subunit and stays in the cytoplasm9. Specifically, under stimulation, IKK is activated and leads to degradation of the phosphorylation-dependent proteasome IκB, and then releases NF-κB subunit from the cytoplasm to the nucleus. The NF-κB subunit binds to the κB element and initiates the expression of target genes related to the inflammatory response10. MAPKs pathway also plays an important role in the inflammatory process. The family members of MAPKs mainly include extracellular signal-regulated kinase (ERK), stress-activated protein kinase (JNK), and p38 mitogen-activated protein kinase (p38MAPK)11. In general, LPS stimulation promotes the phosphorylation of ERK, JNK, and p38 to further stimulate nuclear translocation of the nuclear transfer-factor AP-1and induction of some inflammatory factors expression11. Therefore, inhibition of the NF-κB and MAPKs signaling pathway can be a good therapy method on some inflammatory diseases.
Oxidative stress also plays an important role in the inflammation involved in chronic diseases. Nuclear factor erythroid 2-related factor (Nrf2) can regulate the antioxidant proteins expression and protect the body from oxidative damage caused by injury and inflammation12. Normally, Nrf2 binds with its suppressor Keap1 in the cytoplasm. While in the activation state, Nrf2 translocates into the nucleus where it binds to ARE and induces the expression of downstream target genes such as HO-113. Therefore, regulation of Nrf2 nucleus translocation and increasing the activity of HO-1 can appropriately inhibit the occurrence of inflammatory reactions14.
Procyanidin, a polyphenolic compound existing in most plants, exhibits anti-inflammatory, anti-oxidant, anti-apoptotic and other pharmacological effects15,16. Procyanidin A1 (PCA1), as one of the procyanidins that were isolated and identified, drew attention to further research. Previous studies showed that PCA1 has immune modulation, cholesterol modulation, and anti-oxidative effects17,18. However, the role of PCA1 in the inflammatory response and oxidative stress is still lack of research evidence. To clarify the role of PCA1 in regulating inflammation more clearly, it is necessary to do further investigation on the anti-inflammatory and anti-oxidative mechanisms of PCA1. Thus, in this study, using LPS-stimulated RAW264.7 cells, we investigated the anti-inflammatory activity and the molecular mechanism of PCA1.
Results
PCA1 diminished LPS-stimulated inflammatory reaction in RAW264.7 cells
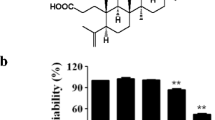
The cytotoxicity of PCA1 (Fig. 1A) was determined by MTT assay. As shown in Fig. 1B, the results indicated that PCA1 (20, 40, 80 μM) displayed no significant toxicity in RAW264.7 cells compared with normal control (NC). Nitrite level, NO release, iNOS, and COX-2 are major indicators of inflammatory response19. Our study demonstrated that PCA1 pretreatment suppressed LPS-stimulated nitrite level and iNOS protein expression in a dose-dependent manner. From Fig. 1C, the nitrite level of cells treated with LPS sharply increased. However, the pretreatment with PCA1 in 20 μM exhibits 19.5% inhibition on nitrite level, 37.4% in 40 μM, 47.6% in 80 μM, compared with LPS group (Fig. 1C,D), respectively. In addition, as shown in Fig. 1E,F, the flow cytometry assay indicated that PCA1 diminished NO production in 20 μM. However, we found that PCA1 has no effect on the expression of COX-2 protein (Fig. 1D). As shown in Fig. 1E,F, using the flow cytometry assay, PCA1 suppressed NO release in LPS-stimulated RAW264.7 cells in accordance with the results of the nitrite level and iNOS protein expression.

PCA1 diminished LPS-stimulated inflammatory response in RAW264.7 cells. (A) Chemical structures of Procyanidin A1 (PCA1). (B) The cytotoxicity of PCA1 in RAW264.7 cells was determined by MTT assay after 24 h treatment. (C) RAW264.7 cells treated with PCA1 (20, 40, 80 μM) for 1 h and then induced with LPS (1 μg/ml) for 18 h. The medium was collected to determine the nitrite level using Griess Regent. (D) RAW264.7 cells treated with PCA1 (20, 40, 80 μM) for 1 h and then induced with LPS (1 μg/ml) for 18 h. iNOS and COX-2 expressions were detected by Western blotting. The grouping of gels/blots cropped from different parts of the same gel (targets vs loading control). The full blots are shown in Supplementary Information. (E) RAW264.7 cells were the treatment of PCA1 (20, 40, 80 μM) for 2 h before the co-treatment of LPS (1 μg/ml) for 6 h. Then the cells were marked with DAF-FM for 1 h, which were assessed by flow cytometry. (F) Statistical analysis of the NO per group. **p < 0.01, and ***p < 0.001 versus the LPS induced group.
PCA1 reduced LPS-induced the expression pro-inflammatory cytokines in RAW264.7 cells
According to previous study, RAW264.7 cells release large amounts of pro-inflammatory cytokines such as TNF-α, IL-6, IL-1β, etc. due to LPS-mediated the inflammatory reaction20. Ca2+, another inflammatory signal, will lead to influx in cell during inflammatory process21,22. Therefore, we explored whether PCA1 could exhibit inflammatory effect on the release of pro-inflammatory cytokines. Our ELISA assay results indicated that PCA1 attenuated TNF-α and IL-6 levels in a dose-dependent manner (Fig. 2A,B). PCA1 can inhibit TNF-α and IL-6 more than 50% under the concentration of 40 μM. Calcium production was determined by Fluo-3/AM (1 μM) for 1 h. Our results showed that 20 μM administration with PCA1 decreased calcium production significantly compared with the control group (Fig. 2C,D). Above all, these findings showed that PCA1 diminished inflammatory responses in LPS-stimulated RAW264.7 cells.

PCA1 reduced LPS-induced pro-inflammatory cytokines in RAW264.7 cells. (A,B) RAW264.7 cells treated with PCA1 (0, 20, 40, 80 μM) for 1 h and then induced with LPS (1 μg/ml) for 18 h. The TNF-α and IL-6 level was detected using ELISA kits. (C) RAW264.7 cells were pretreated with PCA1 (0, 20, 40, 80 μM) for 1 h before co-incubation with LPS (1 μg/ml) for 8 h. Then the cells were marked with Fluo-3/AM for 1 h and assessed by flow cytometry. (D) Statistical analysis of the NO per group. *p < 0.05,**p < 0.01, and ***p < 0.001 versus the LPS group.
PCA1 disrupted LPS-induced NF-κB nuclear translocation
There has been reported that LPS can induce translocation of NF-κB/p65 from the cytoplasm to the nucleus23. And the nucleus translocation of NF-κB can regulate the release of large amounts of inflammatory mediators such as TNF-α, IL-6, IL-1β, NO, and iNOS24. As shown in Fig. 3A, we found that LPS-induced RAW264.7 cells increased p-p65 protein expression, while the pre-treatment with PCA1 significantly attenuated p-p65 protein expression. Furthermore, LPS promoted NF-κB nucleus translocation. However, PCA1 also prevented the nucleus translocation of NF-κB with the concentration of 80 μM (Fig. 3B). Meanwhile, our immunofluorescence results showed that LPS increased NF-κB nucleus translocation, which was decreased by PCA1 (Fig. 3C). Based on the results above, we have a conclusion that PCA1 suppressed NF-κB nuclear translocation.

PCA1 hindered LPS-induced NF-κB nuclear translocation. (A) RAW264.7 cells treated with PCA1 for 2 h were induced by LPS (1 μg/ml) for 2 h. p-p65 and p65 protein level was detected by Western blotting. (B) RAW264.7 cells were pretreated with PCA1 (80 μM) for 1 h and then stimulated with LPS (1 μg/ml) for 1 h. The localization of p65 in the cytoplasm and nucleus were detected by Western blotting. (C) RAW264.7 cells treated with for PCA1 for 2 h were included with LPS for 2 h. p65 translocation was determined using immunofluorescence analysis. The grouping of gels/blots cropped from different parts of the same gel (targets vs loading control) or different gels (phosphorylation). The full blots are shown in Supplementary Information. ***p < 0.001 versus the LPS group.
The effect of PCA1 on NF-κB and MAPKs pathways in RAW264.7 cells
PCA1 plays a vital role in NF-κB pathway, which remarkably inhibited NF-κB activity and translocation25. As shown in Fig. 4A,C, the phosphorylation of IκBα induced by LPS was inhibited, while IκBα protein level was increased by pretreatment with PCA1 in a dose-dependent manner in RAW264.7 cells. Then PCA1 also inhibited IKKα/β phosphorylation induced by LPS with no impact of the expression of IKKα and IKKβ.

Impact of PCA1 on the NF-κB and MAPK pathway in RAW264.7 cells. (A,B) RAW264.7 cells treated with PCA1 for 2 h were induced by LPS. The NF-κB and MAPKs signaling pathway expression proteins were detected using Western blotting. (C,D) Statistical analysis of the IκB-α, IKK-α/β, IKK-α, IKK-β, p-JNK1/2, JNK1/2, p-ERK1/2, ERK1/2, p-p38, and p38 per group. The grouping of gels/blots cropped from different parts of the same gel (targets vs loading control) or different gels (phosphorylation). The full blots are shown in Supplementary Information. *p < 0.05, **p < 0.01, and ***p < 0.001 versus the LPS group.
The proteins of MAPKs family (JNK1/2, ERK1/2, and p38MAPK) involved in anti-inflammatory responses, also played an important role in inflammatory diseases26. As shown in Fig. 4B,D, PCA1 remarkably suppressed the protein level of p-JNK1/2, p-ERK1/2, and p-p38MAPK with no impact on the expression of total JNK1/2, ERK1/2, and p38MAPK. Collectively, this study demonstrated that PCA1 exhibited anti-inflammatory activity through NF-κB and MAPKs signaling pathway.
PCA1 inhibited ROS production and reduced mitochondrial membrane potential (MMP)
ROS, a vital signaling factor, plays a critical role in inflammatory disorder. An increase of ROS level can be induced by LPS, meanwhile, inflammation can also cause accumulation of ROS to further promote oxidative stress27. In our study, ROS generation was examined by DCFH2-DA. As shown in Fig. 5, PCA1 significantly suppressed LPS-induced ROS generation in RAW264.7 cells. In addition to the flow cytometry assay, immunofluorescent assay and intracellular ROS kit were employed to detect the intracellular ROS generation. The results indicated that PCA1 decreased intracellular ROS level (Fig. 5A–D). LPS disrupts the stability of mitochondrial membrane potential (MMP), which is not conducive to maintaining normal physiological functions of cells28. As shown in Fig. 6, the JC-1 monomer increased by LPS-stimulation (green staining), but the pretreatment with PCA1 decreased the JC-1 monomer and restored it to aggregates (red fluorescence). Taken together, PCA1 inhibited ROS production and aggrandized MMP in RAW264,7 cells.

PCA1 inhibited ROS generation. (A) RAW264.7 cells treated with for PCA1 (20, 40, 80 μM) for 1 h were incubated with LPS for 8 h. Cells were stained with DCFH2-DA (1 μM) for 30 min. The fluorescence intensity was determined by flow cytometry at the FITC channel. (B) Statistical analysis of the ROS per group. (C) RAW264.7 cells were seeded into 96-well plates and cultured overnight. Cells pretreated with PCA1 (0, 20, 40, 80 μM) for 1 h were stimulated with LPS (1 μg/ml) for 8 h. The ROS level was determined by a ROS kit (Sigma MAK143) according to the manufacturer’s instructions. (D) ROS level in cells was stained with DCFH2-DA. The images were captured by fluorescence microscopy. ***p < 0.001 versus the LPS group.

PCA1 reduced mitochondrial membrane potential (MMP). RAW264.7 cells treated with for PCA1 for 1 h were incubated with LPS for 8 h. The probe of JC-1 (5 μg/mL) was employed to detect cells for 20 min. Images were captured by fluorescence microscopy.
Impact of PCA1 on Keap1-Nrf2 signaling pathway in RAW264.7 cells
The recent study demonstrated that LPS activates the Nrf2 signaling pathway in RWA264.729,30. In the study, the Nrf2 pathway was investigated. As shown in Fig. 7A,C,D, LPS stimulation induced upregulation of Keap1 expression, while pretreatment with PCA1 slightly downregulated Keap1 protein expression. Pretreatment with PCA1 increased Nrf2 and HO-1 protein expressions in RWA264.7 cell. The pretreatment with PCA1 increased Nrf2 protein level in the nucleus but decreased the protein level in the cytoplasm, compared with NC and LPS stimulation group (Fig. 7B).

Impact of PCA1 on Keap1-Nrf2 signaling pathway in RAW264.7 cells. (A) RAW264.7 cells treated with for PCA1 for 1 h were incubated with LPS for 8 h. the expression of Keap1, Nrf2, and HO-1 were detected using Western blotting. (B) RAW264.7 cells treated with for PCA1 for 2 h were co-cultured with LPS for 2 h. The protein expression of Nrf2 in cytoplasm and nucleus were detected by Western blotting. (C–E) Statistical analysis of the Keap1, Nrf2, and HO-1 content per group. The grouping of gels/blots cropped from different parts of the same gel (targets vs loading control) or different gels (phosphorylation). The full blots are shown in Supplementary Information. *p < 0.05, **p < 0.01, and ***p < 0.001 versus the LPS induced group.
Toll-like receptor participated in PCA1’s anti-inflammatory process
TLR4 that is specifically found on multiple cells makes an important difference in the inflammatory process31. In presence of LPS and MD2, TLR4 dimerizes, modulating the NF-κB, MAPK, and other signaling cascades, eliciting pathogen-specific innate immune responses via pro-inflammatory cytokine release23. To evaluate the PCA1’s effect on TLR4, the cellular thermal shift assay was employed to detect whether PCA1 binds to TLR4. Our results indicated that PCA1 attenuated LPS-induced TLR4 expression and PCA1 bond to TLR4 (Fig. 8A,B).

PCA1 binds to TLR4. (A) RAW264.7 cells treated with for PCA1 (0, 20, 40, 80 μM) for 2 h were incubated with LPS for 2 h. The expression of TLR4 were detected using Western blotting. (B) Treated with PCA1 (80 μM) for 2 h, cells were lysed by RIPA and total proteins were collected. Then, proteins were equally divided into 6 parts, which were heated at 44, 48, 52, 56, 60, and 64 °C for 3 min, respectively. The corresponding loading buffer were added into the proteins, which were analysed by western blotting. The grouping of gels/blots cropped from different parts of the same gel (targets vs loading control). The full blots are shown in Supplementary Information. *p < 0.05, **p < 0.01, and ***p < 0.001 versus the LPS-induced/compared group.
Discussion
The inflammatory reaction is a common pathological reaction in life, which exists in various tissues and organs32. Inflammation is related to the occurrence and development process of many diseases including stroke, arthritis, neurodegenerative disease, and cardiovascular disease33. Therefore, the inhibition of inflammation plays a key role in inflammation-related diseases therapy. In previous studies, we found that PCA1 is one of the monomers isolated from procyanidins, which could reduce the inflammatory response34. This study aimed to elucidate the molecular mechanism of the anti-inflammatory effect of PCA1.
Macrophages play vital roles in regulating inflammatory responses25. LPS, one of the main components of the outer membrane of gram-negative bacteria, can bind to the TLR4 receptor and promote inflammation35. Thus, the RAW264.7 cell inflammatory model induced by LPS was used in this study. Under the stimulation of LPS, macrophages can release a variety of inflammatory factors, such as TNF-α, IL-6, NO, and IL-1β36. Therefore, blocking the occurrence of these inflammatory mediators in inflammatory responses may inhibit the development of inflammation. In our study, PCA1 attenuated NO, iNOS, TNF-α, and IL-6 production, but no effect on COX-2 level (Figs 1 and 2). These results suggested that PCA1 has an anti-inflammatory activity.
Calcium plays a crucial role in the stimulated macrophages, which is considered as a second signal messenger on multiple signaling pathways37. Stimulation of RAW264.7 cells by LPS leads to an increase level of intracellular calcium. Previous studies have shown that calcium are involved in the transcriptional activation of inflammatory cytokines and chemokines besides IL-6, TNF-α, IL-1β, and NO38. Our study illustrated that PCA1 decreased the production of calcium (Fig. 3).
Abundant evidence has shown that LPs binding with TLR4 can activate many inflammatory pathways, such as NF-κB pathway and MAPKs pathaway39. NF-κB signaling pathway plays a necessary role in the pathological process of the inflammatory response. NF-κB is a multipotent transcription factor of the protein family. It can promote transcriptional expression through binding to κB DNA elements40. Normally, NF-κB and IκB binds together and forms a complex in the cytoplasm. However, provoking the degradation of IκB and nucleus translocation of NF-κB subunit p65 from the cytoplasm is induced by LPS stimulation of inflammation41. The upregulation of p65 in the nuclear promotes the release of a large number of cytokines and chemokines10. Our data indicated that PCA1 dramatically attenuated LPS-mediated upregulation of phosphorylation of IKKα/β, IκB and p65, degradation of IκB, and the translocation of p65 from the cytoplasm and into the nucleus (Fig. 3). It is reported that TLR4 and MAPKs signaling pathway are also actively involved in inflammatory responses42. Enhancing the phosphorylation of JNK1/2, P38, ERK1/2 in LPS-induced RAW264.7 cells43. We have shown that PCA1 markedly inhibited the phosphorylation of JNK1/2, P38, ERK1/2 without changing the activation of total JNK1/2, P38, ERK1/2 protein level (Fig. 4). In addition, our results showed that PCA1 binds to TLR4 (Fig. 8). Taken collectively, PCA1 exhibited anti-inflammatory response by mediating NF-κB and MAPKs pathways.
ROS generation mediates a variety of inflammatory signaling pathways which promotes inflammation reaction44. MMP is also an important inflammatory response signal as well as ROS45. These suggested that either the inhibition of ROS or the promotion of MMP can also be the most important therapeutic target of inflammatory diseases46. In this study, PCA1 significantly reduced ROS production and restored mitochondrial membrane potential (Figs 5 and 6).
HO-1, an antioxidant enzyme in the stress response, catalyzing the degradation of heme, biliverdin, and carbon monoxide (CO), also plays an important role in inflammation and oxidation responses47. Chronic inflammation has been reported in HO-1 deficient models48. As the main protein regulating HO-1 expression, Nrf2 is distributed in the cytoplasm together with Keap1 normally. However, when Keap1 was degraded under oxidative stress, Nrf2 was released and migrated to the nucleus, then induced HO-1 expression49. Our results showed that pretreatment with PCA1 decreased Keap1 expression, promoted Nrf2 nuclear translocation and induction of HO-1 in vitro (Fig. 7).
In conclusion, we proved that PCA1 exhibited anti-inflammatory and anti-oxidant effects on RAW264.7 cells via the NF-κB and MAPK signaling pathway (Fig. 9). Thus, PCA1 is a potential agent for the research and development of the treatment with inflammatory diseases.

The schematic of the anti-inflammatory activities of procyanidin A1 in vitro. Procyanidin A1 (PCA1) exerts anti-inflammatory activity, which decreases calcium exclusion, reactive oxygen species (ROS) generation, and the release of pro-inflammatory cytokines through NF-kB, MAPK, and Nrf2/HO-1pathways. LPS, lipopolysaccharides; TNF-α, tumor necrosis factor-α; NO, nitric oxide; COX-2, cyclooxygenase-2; iNOS, inducible nitric oxide synthase; NF-κB, nuclear factor-κB; IL-6, interleukin-6; MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase; ERK, extracellular regulated protein kinase; ROS, reactive oxygen species; Nrf2, nuclear factor erythroid 2-related factor; HO-1, heme oxygenase-1.
Materials and Methods
Chemical and reagents
Procyanidin A1 was purchased from PUSH BIO-TECHNOLOGY, (Chengdu, China). Lipopolysaccharides from Escherichia coli O111:B4, Griess reagent (modifiziert-G4410), 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH2-DA) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified eagle medium (DMEM), Fluo-3/AM, NO detector DAF-FM, fetal bovine serum (FBS) were purchased from Life Technologies/Gibco Laboratories (Grand Island, NY, USA). ELISA kits were obtained from Neobioscience (Shenzhen, China). Antibodies against COX-2 (#4842), NF-κB/p65 (#8242T), p-NF-κB/p65 (#3033), IKKα (#2682), IKKβ (#8943T), p-IKKα/β (#2078), IκBα (#4814), p-IκBα (#2859), JNK1/2 (#9252T), p-JNK1/2 (#4668T), ERK1/2 (#4695T), p-ERK1/2 (#4370T), p38MAPK (#9212), p-p38MAPK (#4511), Keap-1 (#4678), Nrf2 (#12721), HO-1 (#70081), PARP (#9532) and GAPDH (#5174) were purchased from Cell Signaling (Beverly, MA, USA).
Cell culture
RAW264.7 (murine macrophage) cell line was from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cell line was cultured in DMEM High glucose normal culture medium including 10% FBS, 100 U/mL penicillin and 100 mg/mL streptomycin in a humidified incubator, where containing 5% CO2 and maintaining at 37 °C.
Cell viability assay
Cell viability was measured by MTT assay. Probably 1 × 105 RAW264.7 cells were seeded in 96-well plates and cultured overnight, then treated with PCA1 (0, 20, 40, 80 μM) for 24 h, following MTT (5 mg/ml) was added to each well and cultured for 3 h. Then removing culture media, the crystal violet was dissolved with DMSO (100 μL/well), which was determined at 570 nm using microplate reader50.
Assessment of nitrite level and cytokine release
RAW264.7 cells were seeded in 12-well plates with a density of 2 × 105 cells/well for 24 h. Cells were pretreated with PCA1 for 1 h, and then inducted an inflammatory response by LPS (1 μg/ml) for 18 h. Collection of culture media for the detection of nitrite and inflammatory cytokines with Griess Regent and ELISA kit, respectively.
Western blotting analysis
RAW264.7 cells were seeded into a dish or 6-well plates and fostered overnight. PCA1 (20, 40, 80 μM) was pretreated at indicated time points, after LPS induction for a certain period time. Total cell proteins were extracted using RIPA (1% PMSF and 1% cocktail). According to the manufacturer’s instruction, the extraction of cytoplasmic and nuclear protein with the kit (Beyond time, Shanghai, China) was used to obtain the cytoplasmic and nuclear proteins. BCA protein kit (Waltham, MA, USA) was employed to determine protein concentrations. The denatured protein was separated by 8% or 10% SDS-PAGE gels and transferred them to PVDF membrane (Millipore, Billerica, MA, USA). After blocking the PVDF membrane with 5% nonfat milk for 1 h, the PVDF membrane was incubated with primary antibodies (1:1000) for more than 12 h at 4 °C. After washing with TBST and incubated with secondary antibody (1:5000) for 2 h at room temperature, the membrane was exposure by ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA, USA).GAPDH was used as a housekeeping protein.
Flow cytometry assay
RAW264.7 cells were seeded in 24-well plates with a density of 1.5 × 105 cells per well and cultured overnight. The Cells were pretreated with PCA1 for 1 h, and then treated with or without LPS (1 μg/ml). Different probes were used to detect the corresponding indicators, including NO detector DAF-FM (5 μM, 1 h)51, ROS detector DCFH2-DA (10 μM, 30 min), Ca2+ detector Fluo-3/AM (1 μM, 1 h) or MMP detector JC-1 (10 μg/ml, 30 min). After probes incubation, cells were collected and tested by flow cytometry (Becton-Dickinson, Franklin Lakes, NJ, USA).
Immunofluorescence assay
RAW264.7 cells were seeded in confocal dishes with a density of 15 × 105 cells/ml overnight. Then these cells were pretreated with PCA1 (80 μM) for 4 h and LPS-stimulated (1 μg/ml) for 2 h. After fixed, punched, blocked, the cells in the dished were incubated with NF-κB p65 antibody (1:100) overnight at 4 °C. Finally, cells were incubated with Alexa Fluor 594 secondary antibody for 1 h. Nuclei were revealed by Hoechst 33,342 staining. Fluorescence images were collected under a confocal microscope system (Leica, Wetzlar, Germany).
Fluorescence assay
RAW264.7 cells were seeded in 96-well plates with a density of 4 × 105 cells/ml overnight. Cells were pretreated with PCA1 (80 μM) for 1 h and then incubated with or without LPS for another 8 h. Staining with JC-1 (10 μg/ml) and DCFH2-DA (100 μM) for 30 min. Fluorescence images were captured by fluorescence microscopy.
Cellular thermal shift assay
RAW264.7 cells were seeded into a dish (2 × 106 cells) overnight. Treated with PCA1 (80 μM) for 2 h, cells were lysed by RIPA and total proteins were collected. Then, proteins were equally divided into 6 parts, which were heated at 44, 48, 52, 56, 60, and 64 °C for 3 min, respectively. The corresponding loading buffer were added into the proteins, which were analysed by western blotting.
Statistical analysis
Date are presented as means ± SD or means ± S.E.M. All experiments are repeated at least three times. Data are normally distributed and analyzed by one-way-ANOVA or Student’s t-test by GraphPad Prism 6.0 software. Results are considered as statistical significance when p < 0.05.
References
Travelli, C., Colombo, G., Mola, S., Genazzani, A. A. & Porta, C. NAMPT: A pleiotropic modulator of monocytes and macrophages. Pharmacological research (2018).
Wang, C.-C., Lin, H.-L., Wey, S.-P. & Jan, T.-R. Areca-nut extract modulates antigen-specific immunity and augments inflammation in ovalbumin-sensitized mice. Immunopharmacology and immunotoxicology 33, 315–322 (2011).
Nakanishi-Matsui, M., Yano, S., Matsumoto, N. & Futai, M. Lipopolysaccharide induces multinuclear cell from RAW264. 7 line with increased phagocytosis activity. Biochemical and biophysical research communications 425, 144–149 (2012).
Andrade, L. Sesquiterpenes from Essential Oils and Anti-Inflammatory Activity. Natural product communications 10, 1767–1774 (2015).
Yoon, H. J., Moon, M. E., Park, H. S., Im, S. Y. & Kim, Y. H. Chitosan oligosaccharide (COS) inhibits LPS-induced inflammatory effects in RAW 264.7 macrophage cells. Biochemical and biophysical research communications 358, 954–959 (2007).
Cagiola, M. et al. In vitro down regulation of proinflammatory cytokines induced by LPS tolerance in pig CD14+ cells. Veterinary immunology and immunopathology 112, 316–320 (2006).
Doyle, S. L. & O’Neill, L. A. Toll-like receptors: from the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochemical pharmacology 72, 1102–1113 (2006).
Hoesel, B. & Schmid, J. A. The complexity of NF-κB signaling in inflammation and cancer. Molecular cancer 12, 86 (2013).
Kim, D. et al. PKCalpha-LSD1-NF-kappaB-Signaling Cascade Is Crucial for Epigenetic Control of the Inflammatory Response. Mol Cell 69, 398–411 e396 (2018).
Gao, H. et al. Isoacteoside, a dihydroxyphenylethyl glycoside, exhibits anti‐inflammatory effects through blocking toll‐like receptor 4 dimerization. British journal of pharmacology 174, 2880–2896 (2017).
Chang, L. & Karin, M. Mammalian MAP kinase signalling cascades. Nature 410, 37 (2001).
Bak, M.-J., Truong, V. L., Kang, H.-S., Jun, M. & Jeong, W.-S. Anti-inflammatory effect of procyanidins from wild grape (Vitis amurensis) seeds in LPS-induced RAW 264.7 cells. Oxidative medicine and cellular longevity 2013 (2013).
Chen, S., Zou, L., Li, L. & Wu, T. The protective effect of glycyrrhetinic acid on carbon tetrachloride-induced chronic liver fibrosis in mice via upregulation of Nrf2. PloS one 8, e53662 (2013).
Kilic, U. et al. Melatonin suppresses cisplatin-induced nephrotoxicity via activation of Nrf-2/HO-1 pathway. Nutrition & metabolism 10, 7 (2013).
Chacón, M. R. et al. Grape-seed procyanidins modulate inflammation on human differentiated adipocytes in vitro. Cytokine 47, 137–142 (2009).
Terra, X. et al. Grape-seed procyanidins act as antiinflammatory agents in endotoxin-stimulated RAW 264.7 macrophages by inhibiting NFkB signaling pathway. Journal of agricultural and food chemistry 55, 4357–4365 (2007).
Wang, A. et al. Procyanidins mitigate osteoarthritis pathogenesis by, at least in part, suppressing vascular endothelial growth factor signaling. International journal of molecular sciences 17, 2065 (2016).
Takano, F. et al. Aqueous extract of peanut skin and its main constituent procyanidin A1 suppress serum IgE and IgG1 levels in mice-immunized with ovalbumin. Biological and Pharmaceutical Bulletin 30, 922–927 (2007).
Ronchetti, D. et al. Modulation of iNOS expression by a nitric oxide-releasing derivative of the natural antioxidant ferulic acid in activated RAW 264.7 macrophages. European journal of pharmacology 532, 162–169 (2006).
Waseem, T., Duxbury, M., Ito, H., Ashley, S. W. & Robinson, M. K. Exogenous ghrelin modulates release of pro-inflammatory and anti-inflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Surgery 143, 334–342 (2008).
Yaron, J. R. et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis 6, e1954 (2015).
Giorgi, C., Marchi, S. & Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nature reviews Molecular cell biology, 1 (2018).
Kawai, T. & Akira, S. Toll-like receptor downstream signaling. Arthritis Res. Ther. 7, 12–19 (2005).
Ying, X. et al. Piperine inhibits LPS induced expression of inflammatory mediators in RAW 264.7 cells. Cellular immunology 285, 49–54 (2013).
Yuan, R. et al. Dihydrotanshinone exhibits an anti-inflammatory effect in vitro and in vivo through blocking TLR4 dimerization. Pharmacological Research 142, 102–114 (2019).
Gao, H. et al. Isoacteoside, a dihydroxyphenylethyl glycoside, exhibits anti-inflammatory effects through blocking toll-like receptor 4 dimerization. Br J Pharmacol 174, 2880–2896 (2017).
Luciani, A. et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nature cell biology 12, 863 (2010).
Murphy, M. P. & Hartley, R. C. Mitochondria as a therapeutic target for common pathologies. Nature Reviews Drug Discovery (2018).
Vo, V. A. et al. Phosphorylation of Akt mediates anti-inflammatory activity of 1-p-coumaroyl β-D-glucoside against lipopolysaccharide-induced inflammation in RAW264. 7 cells. The Korean. Journal of Physiology & Pharmacology 18, 79–86 (2014).
Hsu, W.-H., Lee, B.-H., Huang, Y.-C., Hsu, Y.-W. & Pan, T.-M. Ankaflavin, a novel Nrf-2 activator for attenuating allergic airway inflammation. Free Radical Biology and Medicine 53, 1643–1651 (2012).
Hennessy, E. J., Parker, A. E. & O’Neill, L. A. Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307 (2010).
Gan, W. Q., Man, S., Senthilselvan, A. & Sin, D. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax 59, 574–580 (2004).
Zhang, Z.-G., Li, Y., Ng, C. T. & Song, Y.-Q. Inflammation in Alzheimer’s disease and molecular genetics: recent update. Archivum immunologiae et therapiae experimentalis 63, 333–344 (2015).
Martinez‐Micaelo, N., González‐Abuín, N., Ardevol, A., Pinent, M. & Blay, M. T. Procyanidins and inflammation: Molecular targets and health implications. Biofactors 38, 257–265 (2012).
Wu, H. et al. Nuciferine Ameliorates Inflammatory Responses by Inhibiting the TLR4-Mediated Pathway in Lipopolysaccharide-Induced Acute Lung Injury. Frontiers in Pharmacology 8 (2017).
Borges, P. V. et al. Protective effect of gedunin on TLR-mediated inflammation by modulation of inflammasome activation and cytokine production: evidence of a multitarget compound. Pharmacological research 115, 65–77 (2017).
Ribeiro, C. M. P. The role of intracellular calcium signals in inflammatory responses of polarised cystic fibrosis human airway epithelia. Drugs in R & D 7, 17–31 (2006).
Schulz, R., Nava, E. & Moncada, S. Induction and potential biological relevance of a Ca2+‐independent nitric oxide synthase in the myocardium. British journal of pharmacology 105, 575–580 (1992).
Guo, S. et al. Magnoflorine Ameliorates Lipopolysaccharide-Induced Acute Lung Injury via Suppressing NF-κB and MAPK Activation. Frontiers in Pharmacology 9 (2018).
Wen, D. et al. A selective small molecule IκB kinase β inhibitor blocks nuclear factor κB-mediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes, and mast cells. Journal of Pharmacology and Experimental Therapeutics 317, 989–1001 (2006).
Karin, M. & Delhase, M. In Seminars in immunology. 85–98 (Elsevier) (2000).
Arthur, J. S. C. & Ley, S. C. Mitogen-activated protein kinases in innate immunity. Nature Reviews Immunology 13, 679 (2013).
Kaminska, B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1754, 253–262 (2005).
Newsholme, P. et al. Diabetes associated cell stress and dysfunction: role of mitochondrial and non‐mitochondrial ROS production and activity. The Journal of physiology 583, 9–24 (2007).
Uchikura, K. et al. Lipopolysaccharides induced increases in Fas ligand expression by Kupffer cells via mechanisms dependent on reactive oxygen species. American Journal of Physiology-Gastrointestinal and Liver Physiology 287, G620–G626 (2004).
Chung, Y. M., Bae, Y. S. & Lee, S. Y. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radical Biology and Medicine 34, 434–442 (2003).
Jin, C. H. et al. Isoegomaketone inhibits lipopolysaccharide-induced nitric oxide production in RAW 264.7 macrophages through the heme oxygenase-1 induction and inhibition of the interferon-β-STAT-1 pathway. Journal of agricultural and food chemistry 58, 860–867 (2009).
Poss, K. D. & Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proceedings of the National Academy of Sciences 94, 10925–10930 (1997).
Kansanen, E., Kuosmanen, S. M., Leinonen, H. & Levonen, A.-L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox biology 1, 45–49 (2013).
Gao, H. et al. Total tanshinones-induced apoptosis and autophagy via reactive oxygen species in lung cancer 95D cells. The American journal of Chinese medicine 43, 1265–1279 (2015).
Gao, H. et al. Simultaneous purification of dihydrotanshinone, tanshinone I, cryptotanshinone, and tanshinone IIA from Salvia miltiorrhiza and their anti-inflammatory activities investigation. Scientific reports 8, 8460 (2018).
Acknowledgements
We would like to appreciate the support from Guangxi Science and Technology Base and Talent Special Project (2018AD19034), Guangxi Natural Science Foundation (2018JJB140265), Ph.D. Fund of Guangxi University of Chinese Medicine (B170023), the Fund of Creating the First-class Subject in Chinese Medicine of Guangxi University of Chinese Medicine (0501802819), and the Project of Guangxi Overseas “100 persons plan” High-level Expert.
Author information
Authors and Affiliations
Contributions
H. Gao, L. Zhao, and J. Feng designed the research. Q. Wang, S. Han, and X. Li conducted most of the experiments. J. Li help to finish flow cytometry and immunofluorescence experiments. Q. Wang and H. Gao wrote the manuscript. M. Tang, B. Dong, and S. Yang revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, S., Gao, H., Chen, S. et al. Procyanidin A1 Alleviates Inflammatory Response induced by LPS through NF-κB, MAPK, and Nrf2/HO-1 Pathways in RAW264.7 cells. Sci Rep 9, 15087 (2019). https://doi.org/10.1038/s41598-019-51614-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51614-x
- Springer Nature Limited
This article is cited by
-
Anti-inflammatory effects of TP1 in LPS-induced Raw264.7 macrophages
Applied Biological Chemistry (2024)
-
Yohimbine Treatment Alleviates Cardiac Inflammation/Injury and Improves Cardiac Hemodynamics by Modulating Pro-Inflammatory and Oxidative Stress Indicators
Inflammation (2024)
-
Litchi procyanidins inhibit colon cancer proliferation and metastasis by triggering gut-lung axis immunotherapy
Cell Death & Disease (2023)
-
Changes in Intracellular and Extracellular Metabolites of Mixed Lactobacillus Strains Enhance Inhibition of Pathogenic Bacterial Growth and Lipopolysaccharide-Induced Alleviation of RAW264.7 Cellular Inflammation
Probiotics and Antimicrobial Proteins (2023)
-
Procyanidin alleviates ferroptosis and inflammation of LPS-induced RAW264.7 cell via the Nrf2/HO-1 pathway
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)