
Abstract
The genetic diversity of the FMD viruses collected from the outbreaks during the second half of the 20th Century in Sri Lanka was assessed in the present study. We sequenced the VP1 genomic region of the samples collected during FMDV epidemics caused by serotype O in Sri Lanka during 1962 and 1997. For comparison, we sequenced the VP1 of the related viral isolates collected from other Asian countries. We analyzed the VP1 sequences of the viral strains using the UPGMA method with uncorrected pairwise distances. Nucleotide divergence (ND) thresholds of 15%–20% and 5%–<15% were used to differentiate topotypes and lineages, respectively. We calibrated the divergence times and lineage-specific substitution rates using Bayesian-skyline models. Based on the ND estimations and phylogenetic relationships, we identified and named two new topotypes [CEYLON 1 (CEY-1) and WEST, CENTRAL AND SOUTH ASIA 1 (WCSA-1)] and six new lineages (Syr-62, Srl-77, Tur-69, May-78, Tai-87 and Bur-77) of serotype O. We believe that the novel topotypes and lineages named may have disappeared although they have similar substitution rates for epizootic outbreaks. Because the amino acid selection analysis revealed that the two topotypes and six lineages identified were under purifying selection during the outbreaks.
Similar content being viewed by others
Introduction
Foot-and-mouth disease (FMD) is considered as one of the most contagious and acute diseases in ruminants and swine1,2. FMD imposes a massive threat to the livestock industry3,4. The identification of the correct FMD virus (FMDV) causing a particular FMD incident/epidemic plays a crucial role in taking measures to control and eradicate the disease5. There are seven FMDV serotypes (A, C, O, Asia 1, SAT 1, SAT 2, and SAT 3) described according to their antigenic and immunological properties. According to the antigenic and genetic attributes, scientists divided the distribution of FMDV into seven different pools where some serotypes exhibit a tendency to be restricted within specific pools. Among the seven serotypes described, serotype O is the most prevalent and distributed form of FMDV spreading across six pools out of the seven described4,6.
Samuel and Knowles, (2001)1 divided serotype O into eight geographically and genetically distinct topotypes, viz, MIDDLE EAST-SOUTH ASIA (ME-SA), SOUTHEAST ASIA (SEA), INDONESIA-1 (ISA-2), INDONESIA-2 (ISA-2), CATHAY, WEST AFRICA (WA), EAST AFRICA (EA) and EUROPE-SOUTH AMERICA (Euro-SA). Later EA was renamed EA-1 when three further East African topotypes (EA-2, EA-3, and EA- 4) were described7. The topotype and lineage level differentiation is mainly carried out using Nucleotide Divergence (ND) of the VP1 genomic region of FMDV genome1. The topotypes and lineages are defined at the VP1 nucleotide identity (NI) thresholds of 80–85% and 90–95%, respectively1,4,8.
The VP1 structural protein together with proteins VP2, VP3, and VP4 form the capsid of FMDV. The VP1 capsid protein consists of 211 amino acids (coded by 633 nucleotides). The genotyping, molecular epidemiology and evolutionary studies and development of vaccines against FMDV are widely carried out using the coding region of the VP1 capsid protein due to its high variability among different topotypes and lineages1,5,9,10,11,12. The highest variability within VP1 genomic region among different topotypes and lineages is located in the critical antigenic sites of VP1 ranging from 140–160 (G-H loop) and 200–213 (C-terminus region)13. The G-H loop contains an RGD tripeptide which mediates host cell binding of FMDV via an integrin host cell receptor14. Due to the high variability (mutation by substitutions, and insertions and deletions)15,16,17, the serotype specificity and the importance in host cell binding18, the VP1 genomic region is widely used for genotyping. The VP1 genomic region is also used to understand the patterns of distributions and molecular phylogenetics of FMDV6.
FMD was first reported in Sri Lanka in the mid-19th century19,20. Although numerous outbreaks occurred since then, most of the causative strains were not identified. Abeyratne et al.21 characterized the viral isolates collected from FMD outbreaks from 1997–2014 and introduced a novel endemic lineage, Srl-97, present in Sri Lanka. However, no study has systematically described the outbreaks and the reported cases before 1997 in Sri Lanka leaving a considerable knowledge gap in FMDV evolutionary dynamics within the country. We believe that some of the causative strains could be new to the FMD research community and it is essential to characterize past outbreaks and report the novel lineages and topotypes even if they have become extinct. Thus we have sequenced the VP1 region of the FMDV samples isolated during 1962–1990 in Sri Lanka. For comparison purposes, we carried out VP1 sequencing of the viral isolates which might be related to the FMDV strains of Sri Lanka. Thereby, in the present study, we introduce novel topotypes and lineages of serotype O through detailed molecular phylogenetic and deduced amino acid (AA) analyses.
Materials and Methods
Reverse transcription PCR (RT-PCR) and sequencing
Vesicular epithelium samples collected by the veterinary authorities from infected animals in Sri Lanka (n = 22) and Southeast Asian and Middle Eastern countries (n = 34) during 1962–1990 were submitted to the FAO World Reference Laboratory for FMD at Pirbright. Viruses were isolated on primary bovine thyroid cells or on the IB-RS-2 pig kidney cell line (explained in Mao et al.22 and Chapman and Ramshaw23 respectively). Total RNA was extracted from virus isolates using the RNeasy kit (Qiagen, Crawley, West Sussex, UK) as described in Knowles et al.8. To amplify the VP1 region of the FMDV genome two one-step RT-PCRs were performed using serotype O specific forward primers, O-1C244F and O-1C272F each with the reverse primer, EUR-2B52R using conditions previously described8 (Knowles et al., 2016). Sequencing of each amplicon was performed using the BigDye® Terminator v3.1 Cycle Sequencing kit and run on an ABI 3730 DNA Analyzer. The VP1 sequences generated in this study are deposited in NCBI, GenBank repository (Table S1). The Pirbright Institute where all the viral experiments were conducted is a United Kingdom Accreditation Service (UKAS) Laboratory. The testing and analysis were conducted according to the strictest bio-containment and safety procedures given in https://www.pirbright.ac.uk/node/37681#panel-9334.
Phylogenetic analysis
For the differentiation of the FMDV topotypes, we created an alignment using ClustalW algorithm24 with all the identified topotypes of serotype O in MEGA 725. We manually checked the total alignment for the addition of unnecessary gaps and interference to the reading frame. As the high amount of missing data can delude the analytical power, we excluded the sequences having less than 500 nucleotides to increase the robustness of the analysis. We analyzed the alignment using Unweighted Pair Group Method with Arithmetic Mean (UPGMA) and uncorrected pairwise distance methods. The tree was then plotted using FigTree v1.4.326.
We subjected the data matrix of the alignment to a model selection analysis to find out the best fit model. We implemented Akaike Information Criterion (AIC)27, Bayesian Information Criterion (BIC) and Decision Theory (DT) in jModel Test 2.1.1028 for the model selection using CIPRES Science Portal29. A rapid boostrap algorithm30, using 1000 pseudo-replicates, was implemented using the RAxML software31. The best ML tree was selected using – (minus) log-likelihood values, and we imprinted bootstrap values to the tree topology given in the best ML tree.
Molecular dating to infer divergence times
The nucleotide alignment file was imported into the BEAUti 2 software to create an XML file. The GTR site model32 with nucleotide substitution parameters given in the model selection in jModel Test was used. We set the model to the relaxed lognormal clock by considering the substitution rates along each branch. Since we dealt with several topotypes and lineages which have different diversification rates, we used the uncorrelated relaxed lognormal clock as it allows the formation of each clade using one lognormal distribution33. We used the software BEAST 2 (2.4.8)34 to create the divergence time tree. BEAST allows utilizing a range of stochastic branching models and permits to use the best model that fits the specific data set generated in the present study. Since some of the lineages of the virus that we sequenced could have gone extinct and no longer detected, it is more reliable to use mechanical species level process when drawing the tree topology. We used birth-death process which accounts for both speciation as well as extinction35. Since we used the time-stamped sequences to infer the divergence time, we used Birth Death Skyline Serial model as the branching model36. We kept the reproductive number (R0) to its default value and maximum boundary up to 10 as the rate of spreading of an epidemic is rarely above 10. Usually, FMD hosts are known to gain immunity in one week after the infection, thus we set infectious-rate-prior to 52 per year37. The RNA viruses have a mutation rate of 0.001 substitutions per year per site. Thus we used the mean clock rate of 1 × 10−3 with a lognormal distribution. We ran 50 million cycles of Markov Chain Monte Carlos (MCMC) chains to sample from the posterior distribution. We assessed the independent sampling and the chain performance using Effective Sample Size (ESS) and Auto-Correlation time by analyzing the log file in TRACER38. We used all the sampled trees used to draw the Maximum Clade Credibility (MCC) tree in the software TreeAnotater. We carried out all the analyses in CIPRES Science Gateway29 that provide access to most of the tools used. The time tree was plotted using FigTree v1.4.326.
Nucleotide selection analysis
We checked the evolutionary signatures on the VP1 genomic region of the novel lineages and topotypes identified in this study using Datamonkey 2.0 platform39,40. We used the Fixed Effective Likelihood (FEL) to test the codon-specific selective pressure41. We implemented a method known as the Fast, Unconstrained Bayesian Approximation for Inferring Selection (FUBAR)42 to compare the FEL result. We performed five MCMC chains for two million generations. We kept the burn-in to 20% while sampling 100 trees from each chain cycle. The selective pressure predictions can be tested in both ML and Bayesian frameworks thus the robustness of our results can be increased. We executed the adaptive Branch Site Random Effective Likelihood model (aBSREL)43,44 to check the clade-specific evolution and episodic diversification of newly identified topotypes and lineages.
Amino acid residue selection analysis
The Directional Evolution of Protein Sequences (DEPS) analysis was performed for the deduced amino acid sequences of the viral isolates in the topotypes CEY-I vs. ISA-II and WCSA-II vs. EA and WA in Datamonkey platform40. Initially, two sequence alignments were prepared for the novel topotypes CEY-I and WCSA-II and their sister lineages. The automatic model selection was conducted before the DEPS analysis, as described in Kosakovsky Pond et al.45. The General Discrete Model was used to calculate the site-to-site rate variation.
Results
Phylogenetic analysis
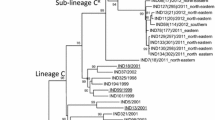
The topotypes and lineages were resolved at major nodes of the distance tree (Fig. 1). The best fit model with highest AIC value (0.733) for our dataset was GTR + I + G. The substitution rates for the model were calculated as AC = 1.112; AG = 7.827; AT = 1.236; CG = 0.506; CT = 10.392; and GT = 1.0. The proportion of invariance was 0.3183, and the gamma shape parameter was 0.85. Both BIC and DT criteria also gave the similar model in the model selection. We used the parameters of the best-selected model for Bayesian tree construction. The MCMC chains converged maximally after 500,000 cycles and first 10% of the trees sampled were discarded as the burn-in. The ESS for all the priors were above 200 indicating higher robustness in our analysis. It is important to run MCMC chains until ESS exceeds 200 thus the poor mixing of the samples can be avoided. The clade structure and the branching pattern of the MCC tree generated was similar to the best ML tree deduced from the ML bootstrap analysis. Thus, the major clades were well supported by both bootstrap and posterior probability values (Fig. S1). The MCC tree exhibits the evolution of FMD serotype O with divergence times (Fig. 2).

The midpoint rooted distance tree built using UPGMA algorithm. The X-axis represents the nucleotide divergence (ND) calculated using uncorrected pair-wise distances. The colored lines except for the black ones within the tree indicate the known topotypes as depicted in the legend. The collapsed branches represent already known topotypes and lineages. The expanded branches show novel lineages and novel topotypes along with their operational taxonomic units (OTUs). A→G indicate newly formed clades in the tree. The newly assigned names are given below the clades. A and B: Newly named topotypes CEY-1 and WCSA-1, respectively; BI, BII, C, E, F and G: Newly named lineages; BI: Srl-77; BII: Syr-62; C: Tur-69; D: the clade containing three newly named lineages (E: May-78, F: Bur-77 and G: Tai-87) within the topotype SEA.

The midpoint rooted MCC tree showing the divergence times of the topotypes and lineages in serotype O. The X-axis represents the time scale. The scale bar indicates the rate of substitutions per site per year. The colored lines except for the black ones within the tree indicate the known topotypes as depicted in the legend. The shaded boxes indicate the novel topotypes and the novel lineages. The exact midpoint rooted MCC tree showing the divergence times with tip labels, node support (Posterior Probabilities and bootstrap values) and 95% Highest Posterior Density node bars is given as Fig. S1.
Evolutionary signatures in VP1 region
The total predicted AA alignment including newly named lineages and topotypes, and FMDV serotype O VP1 reference sequence had 46 Parsimony Informative (PI) sites out of 214 positions considered. There was seven PI amino acids (AA) in G-H loop and two in C-terminus region. To infer the evolutionary aspects of newly identified clades, we primarily used ML-based methods given in Datamonkey platform. The evolution of positive (adaptive) or negative (purifying) selection was measured based on the synonymous (dS) and non-synonymous (dN) substitution rates. The FEL analysis calculates the site (codon) specific dS and dN rates in a given alignment based on likelihood ratios. The FUBAR, however, estimates the dS and dN ratios based on the posterior probabilities (>90% pp are given as positively selected codons). The FEL and FUBAR analyses inferred that all the codon positions of the studied viral isolates are under the PURIFYING selection. We have provided the codon-specific rates, likelihood ratios, p values, and pp values in the Table S2. According to the aBSREL analysis, the phylogeny did not support any episodic diversification along each clade.
Topotype and lineage characterization
The UPGMA tree separated the 55 isolates studied into four major clades that were not reported before (Fig. 1). The distance tree built using UPGMA algorithm had topotype separation at a nucleotide similarity level of 80%–85%.
We identified the separation of topotypes and lineages at major nodes in the midpoint rooted distance tree (Fig. 1). The Clade A consists of FMDV VP1 sequences of the isolates collected in Sri Lanka during 1962–1982. The Clade A separated in the UPGMA tree with an ND of 17.76% indicating the existence of a novel topotype. We named this novel topotype as ‘CEYLON-1 (CEY-1)’ due to its apparent geographical restriction within Sri Lanka. CEY-1 cladded sister to the topotype, ISA-2. We identified two unique AA substitutions within CEY-1 at the positions of 140 and 186 (T140H and T186V) from which T140H is located within the G-H loop and T186V is located within the B-C loop of the VP1 protein. CEY-1 was distantly related to the extinct Indonesian topotypes ISA-1 and ISA-2. The topotypes, CEY-1, ISA-1, and ISA-2, shared the ‘insertion’ of an AA in the B-C loop of VP1 where CEY-1 contained E/Q insertion and ISA-1 and ISA-2 contained either A/T insertion. According to the divergent time analysis, the topotype CEY-1 had diverged from ISA-1 in the year 1936 [95% highest posterior density (HPD) = 66.0308–85.676] (Fig. 2). We estimated the topotype specific substitution rate of CEY-1 as 8.1 × 10−3 substitutions per site per year.
The Clade B nested monophyletic to the African topotypes, EA-3 and EA-1 (Fig. 1). The FMDV sequences isolated in Eurasian and Middle East countries represent the Clade B (Table S1). The clade B exceeds the ND cut-off of 15%, thus can be treated as a novel topotype and we named it as ‘WEST, CENTRAL AND SOUTH ASIA 1’ (WCSA-1). The MCC time tree showed that the Time for The Most Recent Common Ancestor (TMRCA) was 70 years ago in 1955 (95% HPD = 59.288–70.333) (Fig. 2). The comparison of the AA sequences of VP1 protein revealed a unique substitution of A by either P, S or T at the 210th position located within the C-terminus. We estimated the topotype specific substitution rate of WCSA-1 as 8.8 × 10−3 substitutions per site per year.
The topotype WCSA-1 split at 11.44% of ND into two sister clades, BI and BII indicating the existence of two lineages (Fig. 1). The Clade BI solely comprised of Sri Lankan isolates and the first isolate of B1 was sampled in 1977. Thus we identified Clade BI as a novel lineage and named as ‘Srl-77’. This novel lineage is distinctive by having unique AA substitution of K by either E or V (K43E/V) at the βB sheet and E by either P or T (E47P/T) at B-C loop. The Srl-77 lineage had a prominent AA replacement of T by A (T172A) located within the βI sheet. We estimated the topotype specific substitution rate of Srl-77 as 4.8 × 10−3 substitutions per site per year.
The Clade BII, representing the other lineage sister to Srl-77 (Fig. 1), shared multiple geographical localities ranging from Central Asia, South Asia, and the Middle East. We named BII as ‘Syr-62’. The divergence time for the split between Srl-77 and Syr-62 was estimated to be at 63 years ago in 1955 (95% HPD = 54.632–63.454) (Fig. 2). Syr-62 contained two unique mutations which are, substitution of T by either V or D at 172nd position (T172V/D) located within the βI loop and substitution of D by T (D139T) found within the G-H loop. We estimated the topotype specific substitution rate of Syr-62 as 5.9 × 10−3 substitutions per site per year.
We identified a novel clade, labeled C, within the ME-SA topotype (Fig. 1). The Clade C separated from rest of the group with an ND of 11.32%. Thus we treated this clade also as a novel lineage. Even though the existence of this lineage is found in multiple regions, we named this lineage as ‘Tur-69’ as this was first detected in Turkey in 1969. Four viral strains including one Sri Lankan isolate represent Tur-69. Among these four isolates, except the one collected from Turkey (O1/Manisa/TUR/69; AY593823), substitution of I by A at 57th position (I57A) located within the C-D loop was unique to Tur-69. The deduced AA sequence of Tur-69 further revealed that replacement of A by V (A4V) located within the N-terminus was unique for all the individuals of ME-SA including Tur-69. This lineage has diverged from rest of the ME-SA group around 1965 (95% HPD = 44.404–53.340) (Fig. 2). We estimated the topotype specific substitution rate of Tur-69 as 4.7 × 10−3 substitutions per site per year.
The Clade D formed a sister clade with Mya-98 of SEA topotypes separating each other at an ND of 12.22% (Fig. 1). The UPGMA tree divided the Clade D into three sub-clades that exceed 5% ND. The Clades E and F separated at 9.04% of ND. The Clade E consisted of isolates collected from different countries (mostly from Southeast Asia and two isolates from Sri Lanka). Thus we named Clade E as ‘May-78’ a novel lineage under the topotype, SEA. Within the SEA topotype, a unique substitution of A by T (A13T) at N-terminus was prominent in May-78. We estimated the topotype specific substitution rate of May-78 as 4.5 × 10−3 substitutions per site per year.
The Clade F consisted of the viral isolates collected from Burma and Laos. The first isolate of Clade F was reported in Burma (now named Myanmar) in 1977. We identified Clade F as a novel lineage and assigned the name ‘Bur-77’. This lineage had two fixed AA substitutions of T by either L or P (T142L/P) and N by either A or T (N143A/T) located within the G-H loop. Bur-77 shared T142L/P and N143A/T with Cam-94 in the deduced VP1 capsid protein chain. Within the Clade D, May-78 and Bur-77 separated from the Clade G at an ND of 9.64%. The Clade G exclusively comprised of sequences obtained from the isolates collected in Malaysia, Thailand, and Laos in the years between 1987 and 2002. Since the first isolate was collected from Laos in 1987, we named the Clade G as ‘Tai-87’. The deduced AA sequence of VP1 region revealed prominent AA substitution of Q by H located within the N-terminus (Q28H) which was shared with the lineage, Mya-98. We identified a unique AA substitution of E by A present in Tai-87, situated within the E-F loop (E102A). Moreover, some individuals of Tai-87 showed a unique AA substitution of K by Q (K41Q) located within the αZ helix.
We used the clustering pattern of the distance tree to identify the topotypes and lineages based on the ND (Fig. 1) as described previously1,46,47. However, the MCC and ML trees had different topology within the topotype SEA. A well supported clade (bs = 94 and PP = 96%) was formed including May-78 individuals (Fig. S1). All the individuals (O/MYA/1/92, O/MYA/5/92, O/MYA/5/93, O/MYA/5/2002, O/LAO/1/87, O/LAO/1/88, O/TAI/189/87) of Tai-87 lineage in the UPGMA tree cladded sister to Mya-98 in both ML and MCC trees (bs = 98, PP = 98) (Fig. S1). The TMRCA among Mya-98, Tai-87, and May-78 was estimated as the year 1967 (95% HDP = 40.534–48.376) (Fig. 2). Moreover, Cam-9448 also formed a sister clade with the all the individuals of Bur-77 lineage (O/LAO/1/82, O/LAO/5/82, O/BUR/11/78, O/BUR/2/77, O/BUR/8/78, O/BUR/2/78) in both ML and MCC trees. Tai-87 estimated to be originated in 1965 (95% HPD = 43.1129–51.539) in the calibrated time tree (Fig. 2). The newly named two topotypes and six lineages along with the estimated year of origin and evolutionary parameters are summarized in Table 1.
Directional selection of amino acid residues
The DEPS analyses revealed that there was one directionally evolving residue when CEY-1 is compared with ISA-2 (P = 0.0013). There were eight directionally evolving residues when the topotype WCSA-1 is compared with EA and WA topotypes (0.0 ≤ P ≤ 0.0008) (Table 2). The two sites undergoing directional selection between CEY-1 and ISA-2 appeared as convergent evolution or repeated substitution. There were 19 sites undergoing directional selection between WCSA-1 vs. EA and WA. Only one site out of 19 was undergoing frequency-dependent selection. Whereas 14 sites were undergoing the selection kind of convergent evolution or repeated substitution. The remaining four sites were undergoing convergent evolution / repeated substitution or frequency-dependent selection (Table 3).
Discussion
In the present study, the FMDV isolates collected during 1962–1992 nested into four major distinct clades in both UPGMA and MCC trees (Figs 1 and 2). We used a data set containing all the described topotypes of serotype O to identify the phylogenetic positions of the FMDV strains assessed in the present study (Table S1). We used 80–85% NI level for topotype differentiation1,2,49. Since the newly identifying lineages could be replaced or extinct, we used 85%-95% NI to distinguish the lineages. A summary of newly named lineages and topotypes are given in Table 1.
The introduction of novel topotypes and lineages are essential when it comes to identification of the viral type of an FMD epidemic. The molecular characterization of FMDV is vital in taking the counter measures to control a disease spreading situation. The typing of the old viral strains allows us to understand and predict the viral evolution which could be extended to present context. The emergence of topotype and lineages to their geographic origin may help to understand the evolutionary process as well as the history of the disease distribution. Although all the topotypes and lineages we introduce in this study are extinct and no longer detected, it solidifies the platform of molecular dating in the context of sampling of ‘old topotypes/lineages.’ Thus, we believe it is necessary to report and characterize the old topotypes and lineages to the scientific community.
The topotype ISA-1 includes FMDV isolates collected in 1962, 1974 and 1983 (ISA/1/62, ISA/8/83, ISA/9/74), while the two sequences of topotype ISA-2 were from isolates collected during 1972 and 1974 (JAV/5/72, ISA/1/74). Although the common ancestry of ISA-1 and ISA-2 was ambiguous in previous studies4, these topotypes were thought to be distantly related since they shared only one insertion of a single AA between residues 45 and 46 of the B-C loop. Remarkably, the newly identified topotype CEY-1 had similar insertion and cladded with ISA-2 with higher node supports (bs = 63, PP = 92). The present analysis confirmed that FMD was introduced into Sri Lanka before 1950. CEY-1 had also started to evolve before 1950. According to our MCC tree (Fig. 2), the topotypes ISA-1, ISA-2 and CEY-1 shared monophyly with EURO-SA (bs = 97 and PP = 97), and TMRCA is around 1931 (95% HPD = 72.868–89.331). We believe that ISA-1 viruses may have originated from the introduction of FMD into Indonesia from Holland during the end of the Dutch colony there around 1930.
In Sri Lanka, a massive epizootic FMD outbreak was reported in 1987 where 85,641 cases were reported. Although the FMD researchers identified the responsible serotype of 1987 outbreak, they did not know the exact topotype or the origin of the causative strain. In the present study, we revealed that the causative viral of the FMD outbreak in 1987 was lineage Srl-77 belonged to the newly named topotype WCSA-1. The sister lineage (Syr-62) of Srl-77 shares the isolates in multiple geographic locations. A clade within Syr-62 comprised of three isolates namely O/1492/Russia/USSR/88, O/SYR/18/62 and O/Rey/IRN/66 which shared the most common recent ancestor with a strain collected from Tajikistan (O/738/Tajikistan/USSR/74). Since the Syr-62 lineage contains sequences of the oldest samples collected, we believe WCSA-1 topotype may have a Central Asian origin where it could have spread through the Middle East. The WCSA-1 topotype shares a monophyletic relationship with the clade containing topotypes EA-1, EA-2, EA-3, EA-4, and WA. They all share an AA substitution of A by either S, P or L in N-terminus (at 4th position). The previous understanding was the origin of WA topotype might have evolved from ME-SA11. Moreover, many studies on EA topotypes did not reveal the possible origin7,50. The recent WRLFMD reports51,52,53 shows that the WA topotype also clustered inside EA topotypes, which is somewhat congruent with the analysis reported in the present study. We believe that the WCSA-1 topotype may be the missing link between African topotypes and ME-SA. We reveal that the African topotypes were originated 54 years before (95% HPD = 46.236–62.432). The African topotypes share a common ancestor with WCSA-1 before 70 years (95% HPD = 58.288–70.233).
The Tur-69 lineage in ME-SA is the oldest lineage present in MCC tree generated in the present study. This Tur-69 has a Mediterranean origin. From there, Tur-69 could have moved to Asia. In 1991, an epidemic was reported in Sri Lanka. The FMDV isolates collected in 1991 from Sri Lanka, O/SRL/1/91, have not been assigned to any lineage to date. We identified the ancestral lineages to all the existing O/SEA lineages. Our time analysis calculated the oldest TMRCA for SEA topotype to be around 1968 (95% HPD = 40.534–48.376). The lineage Cam-94 within SEA could have evolved from Bur-77 according to our analysis. Similarly, the monophyletic relationship between Mya-98 and Tai-87 implies that Mya-98 may have been evolved from Tai-87 lineage. We believe that both Bur-77 and Tai-87 could have been replaced with Cam-94 and Mya-98, respectively, and these types of lineage turnovers are a common phenomenon in FMDV50. Although SEA topotype was known to be endemic to Southeast Asian countries until the end of 199054, we identified spread of O/SEA/May-78 towards South Asia as the FMDV isolates, O/SRL/2/84 and O/SRL/3/84), collected from Sri Lanka cladded under May-78. However, the FMD cases caused by May-78 in Sri Lanka may have gone unnoticed due to the existence of Srl-77 outbreak in 1987.
All the newly identified lineages had rates of diversification closer to or lower than the rates of the known epizootic outbreak. The closer and lower diversifications could be due to smaller sampling size in short period leading to a slight elevation in the rates of evolutions of the topotypes and lineages. However, the selection analysis of the present study exhibits all the lineages are under purifying selection thus we believe that new FMDV strains would have replaced all the topotypes and lineages described in the present study or reached evolutionary static states. The dominant convergent evolution or repeated substitution of the residue-sites between the novel topotypes explained in comparison to the sister groups implies that the development of specific immunity in host organisms may have shaped the evolution of FMDV topotypes.
Conclusion
We identified and named two novel topotypes, CEY-1 and WCSA-1, and six novel lineages, Syr-62, Srl-77, Tur-69, May-78, Tai-87 and Bur-77 by characterizing the VP1 genomic region of the FMDV isolates collected during 1962–1990. We also described the possible origin events and the dynamics of the topotypes and lineages based on the phylogenetic relationships and deduced protein sequences (Table 1). We found that the FMDV outbreaks in Sri Lanka had different geographic origins. We believe that introduction of genetically diverse viral strains of distinct topotypes may have caused the epizootic outbreaks in Sri Lanka. Moreover, in this study, we reconstructed the phylogeny of the serotype O of FMDV. Thus the present study will help FMD scientific community to understand the disease epidemiology, origin, and viral diversification patterns further.
Data Availability
The VP1 sequences generated in this study (n = 56) were submitted to GenBank under MK390887-MK390940, KY091288, and AJ294918. The MCC tree with HPD values and expanded OUT names are given in Supplementary Fig. S1. The data generated during the nucleotide selection analysis is provided in the Supplementary Table S1. The data generated in FUBA and FEL analyses are given in Supplementary Table S2. The deduced amino acid sequences are depicted in the Supplementary Table S3.
References
Samuel, A. R. & Knowles, N. J. Foot-and-mouth disease type O viruses exhibit genetically and geographically distinct evolutionary lineages (topotypes). J Gen Virol. 82, 609–21, https://doi.org/10.1099/0022-1317-82-3-609 (2001).
Knowles, N. J., Samuel, A. R., Davies, P. R., Midgley, R. J. & Valarcher, J. F. Pandemic strain of foot-and-mouth disease virus serotype O. Emerg Infect Dis. 11, 1887, https://doi.org/10.1016/j.meegid.2008.11.005 (2005).
Condy, J. B., Herniman, K. A. & Hedger, R. S. Foot-and-mouth disease in wildlife in Rhodesia and other African territories: a serological survey. J Comp Pathol. 79, 27–31, https://doi.org/10.1016/0021-9975(69)90023-1 (1969).
Brito, B. P., Rodriguez, L. L., Hammond, J. M., Pinto, J. & Perez, A. M. Review of the global distribution of Foot‐and‐Mouth Disease Virus from 2007 to 2014. Transbound Emerg Dis. 64, 316–332, https://doi.org/10.1111/tbed.12373 (2017a).
Jamal, S. M. & Belsham, G. J. Foot-and-mouth disease: past, present and future. Vet Res. 44, 116, https://doi.org/10.1186/1297-9716-44-116 (2013).
Knowles, N. J. & Samuel, A. R. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 91, 65–80, https://doi.org/10.1016/S0168-1702(02)00260-5 (2003).
Ayelet, G. et al. Genetic characterization of foot-and-mouth disease viruses, Ethiopia, 1981–2007. Emerg Infect Dis. 15, 1409, https://doi.org/10.3201/eid1509.090091 (2009).
Knowles, N. J., Wadsworth, J., Bachanek-Bankowska, K. & King, D. P. VP1 sequencing protocol for foot and mouth disease virus molecular epidemiology. Rev Sci Tech. 35, 741–755, https://doi.org/10.20506/rst.35.3.2565 (2016).
Subramaniam, S. et al. Emergence of a novel lineage genetically divergent from the predominant Ind2001 lineage of serotype O foot-and-mouth disease virus in India. Infect Genet Evol. 18, 1–7, https://doi.org/10.1016/j.meegid.2013.04.027 (2013).
Abdul-Hamid, N. F. et al. Phylogeography of foot-and-mouth disease virus types O and A in Malaysia and surrounding countries. Infect Genet Evol. 11, 320–328, https://doi.org/10.1016/j.meegid.2010.11.003 (2011).
Samuel, A. R., Knowles, N. J. & Mackay, D. K. Genetic analysis of type O viruses responsible for epidemics of foot-and-mouth disease in North Africa. Epidemiol Infect. 122, 529–538, https://doi.org/10.1017/S0950268899002265 (1999).
Weddell, G. N. et al. Sequence variation in the gene for the immunogenic capsid protein VP1 of foot-and-mouth disease virus type A. Proc Natl Acad Sci, USA 82, 2618–2622, https://doi.org/10.1073/pnas.82.9.2618 (1985).
Jinding, C., Mingqiu, Z., Hui, K. H. & Leung, F. C. Molecular characterization of foot-and-mouth disease virus in Hong Kong during 2001–2002. Virus Genes. 32, 139–143, https://doi.org/10.1007/s11262-005-6869-1 (2006).
Martinez, M. A. et al. Evolution of the capsid protein genes of foot-and-mouth disease virus: antigenic variation without accumulation of amino acid substitutions over six decades. J Virol. 66, 3557–3565 (1992).
Kasambula, L. et al. Serotype identification and VP1 coding sequence analysis of foot-and-mouth disease viruses from outbreaks in Eastern and Northern Uganda in 2008/9. Transbound Emerg Dis. 59, 323–330, https://doi.org/10.1111/j.1865-1682.2011.01276.x (2012).
Mateu, M. G., Camarero, J. A., Giralt, E., Andreu, D. & Domingo, E. Direct evaluation of the immunodominance of a major antigenic site of foot-and-mouth disease virus in a natural host. Virology. 206, 298–306, https://doi.org/10.1016/S0042-6822(95)80045-X (1995).
Kitson, J. D., McCahon, D. A. & Belsham, G. J. Sequence analysis of monoclonal antibody resistant mutants of type O foot and mouth disease virus: evidence for the involvement of the three surface exposed capsid proteins in four antigenic sites. Virology. 179, 26–34, https://doi.org/10.1016/0042-6822(90)90269-W (1990).
Mason, P. W., Rieder, E. & Baxt, B. RGD sequence of foot-and-mouth disease virus is essential for infecting cells via the natural receptor but can be bypassed by an antibody-dependent enhancement pathway. Proc Natl Acad Sci, USA 91, 1932–1936, https://doi.org/10.1073/pnas.91.5.1932 (1994).
Wijewardana, B. D. R. & Fernando, W. W. H. S. The position of foot-and-mouth disease in Sri Lanka during 1977-1981. Rev sci. tech. Off. Int.Epiz. 2, 171–176 (1983).
Hettiarachchi, R., Kodituwakku, M. & Hammond, J. Characterization of foot and mouth disease to strengthen disease control in Sri Lanka. SLVJ. 56&57, 21–24 (2009).
Abeyratne, S. A. et al. The phylogenetic analysis of VP1 genomic region in foot-and-mouth disease virus serotype O isolates in Sri Lanka reveals the existence of’Srl-97’, a newly named endemic lineage. PloS one. 13, e0194077 (2018).
Mao, R. et al. Establishment and evaluation of a stable bovine thyroid cell line for investigating on of Foot-and-mouth disease virus. Front Microbiol. 9, 2149, https://doi.org/10.3389/fmicb.2018.02149 (2018).
Chapman, W. G. & Ramshaw, I. A. Growth of the IB-RS-2 pig kidney cell line in suspension culture and its susceptibility to foot-and-mouth disease virus. Appl Environ Microbiol. 22, 1–5 (1971).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680, https://doi.org/10.1093/nar/22.22.4673 (1994).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Rambaut, A. FigTree, a graphical viewer of phylogenetic trees. http://tree.bio.ed.ac.uk/software/figtree (2014).
Akaike, H. A new look at the statistical model identification. IEEE Trans Automat Contr. 19, 716–23, https://doi.org/10.1007/978-1-4612-1694-0_16 (1974).
Posada, D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 25, 1253–1256, https://doi.org/10.1093/molbev/msn083 (2008).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Computing Environments Workshop (GCE), 2010. 1–8. http://www.ieeexplore.ieee.org/abstractdocument/5676129 (2010).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 57, 758–771, https://doi.org/10.1080/10635150802429642 (2008).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22, 2688–2690, https://doi.org/10.1093/bioinformatics/btl446 (2006).
Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on Mathematics in the Life Sciences. 17, 57–86 (1986).
Drummond, A. J., Ho, S. Y., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88, https://doi.org/10.1371/journal.pbio.0040088 (2006).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 7, 214, https://doi.org/10.1186/1471-2148-7-214 (2007).
Kendall, D. G. On the generalized “birth-and-death” process. The Annals of Mathematical Statistics. 19, 1–15 (1948).
Stadler., T., Kühnert, D., Bonhoeffer, S. & Drummond, A. J. Birth–death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc Natl Acad Sci USA 110, 228–233, https://doi.org/10.1073/pnas.1207965110 (2013).
Habiela, M. et al. Laboratory animal models to study foot-and-mouth disease: a review with emphasis on natural and vaccine-induced immunity. J Gen Virol. 95, 2329–2345, https://doi.org/10.1099/vir.0.068270-0 (2014).
Rambaut, A. Tracer. 4. http://beast.bio.ed.ac.uk/Tracer (2007).
Pond, S. L. & Frost, S. D. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 21, 2531–2533, https://doi.org/10.1093/bioinformatics/bti320 (2005a).
Delport, W., Poon, A. F., Frost, S. D. & Kosakovsky Pond, S. L. Datamonkey 2010; a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 26, 2455–2457, https://doi.org/10.1093/bioinformatics/btq429 (2010).
Kosakovsky Pond, S. L. & Frost, S. D. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 22, 1208–1222, https://doi.org/10.1093/molbev/msi105 (2005b).
Murrell, B. et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol. 30, 1196–1205, https://doi.org/10.1093/molbev/mst030 (2013).
Smith, M. D. et al. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol. 32, 1342–1353, https://doi.org/10.1093/molbev/msv022 (2015).
Kosakovsky Pond, S. L. et al. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 28, 3033–3043, https://doi.org/10.1093/molbev/msr125 (2011).
Kosakovsky Pond, S. L., Poon, A. F., Leigh Brown, A. J. & Frost, S. D. A maximum likelihood method for detecting directional evolution in protein sequences and its application to influenza A virus. Mol Biol Evol. 25, 1809–1824, https://doi.org/10.1093/molbev/msn123 (2008).
Tosh, C., Sanyal, A., Hemadri, D. & Venkataramanan, R. Phylogenetic analysis of serotype A foot-and-mouth disease virus isolated in India between 1977 and 2000. Arch Virol. 147, 493–513, https://doi.org/10.1007/s007050200002 (2002).
Ularamu, H. G. et al. Characterization of foot-and-mouth disease viruses collected in Nigeria between 2007 and 2014: evidence for epidemiological links between West and East Africa. Transbound Emerg Dis. 64, 1867–1876, https://doi.org/10.1111/tbed.12584 (2017).
Nguyen, T. et al. Heterogeneity and genetic variations of serotypes O and Asia 1 foot-and-mouth disease viruses isolated in Vietnam. Vet Microbiol. 145, 220–229, https://doi.org/10.1016/j.vetmic.2010.04.005 (2010).
Klein, J. Understanding the molecular epidemiology of foot-and-mouth-disease virus. Infect Genet Evol. 9, 153–161, https://doi.org/10.1016/j.meegid.2008.11.005 (2009).
Brito, B. P. et al. Dynamics of widespread foot-and-mouth disease virus serotypes A, O and Asia-1 in southern Asia: a Bayesian phylogenetic perspective. Transbound Emerg Dis. 17, 1–15, https://doi.org/10.1111/tbed.12791 (2017b).
WRLFMD. Report on FMDV O in Libiya in 2011. Pirbright: World Reference Laboratory for Foot-and-Mouth Disease (2011).
WRLFMD. Report on FMDV O in Sudan in 2009–2011. Pirbright: World Reference Laboratory for Foot-and-Mouth Disease (2012).
WRLFMD. Report on FMDV O in Egypt in 2015–2016. Pirbright: World Reference Laboratory for Foot-and-Mouth Disease (2016).
Brito, B. et al. A traditional evolutionary history of foot-and-mouth disease viruses in Southeast Asia challenged by analyses of non-structural protein coding sequences. Sci Rep. 8, https://doi.org/10.1038/s41598-018-24870-6 (2018).
Acknowledgements
We would like to thank the staff of the Veterinary Research Institute, Dept. of Animal Production and Health, Gannoruwa, Peradeniya, Sri Lanka, who submitted samples to the WRLFMD during between 1971 and 1997. The National Research Council, Sri Lanka Target Oriented Research Grant (Grant No.: NRC/TO/14-10). The sequencing was supported by the Department for Environment, Food and Rural Affairs (Defra) of the United Kindgom (Grant No. SE2943). The work of the WRLFMD was also supported by funding provided from the European Union (via a contract from EuFMD, Rome). The views expressed herein can in no way be taken to reflect the official opinion of the European Union. The Pirbright Institute receives grant-aided support from the Biotechnology and Biological Sciences Research Council of the United Kingdom (projects BB/E/I/00007035, BB/E/I/00007036 and BBS/E/I/00007037).
Author information
Authors and Affiliations
Contributions
L.T.R., U.K.W., H.S.J., N.K., J.W. and S.S.S. conceptualization of the research project. L.T.R., U.K.W., H.S.J. and S.S.S. wrote the original draft the manuscript. L.T.R. carried out the statistical data analysis. N.K., J.W., V.M., J.A., C.W. and S.S.S. reviewed and further edited the manuscript. S.S.S. and N.K. supervised the project.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ranaweera, L.T., Wijesundara, U.K., Jayarathne, H.SM. et al. Characterization of the FMDV-serotype-O isolates collected during 1962 and 1997 discloses new topotypes, CEY-1 and WCSA-1, and six new lineages. Sci Rep 9, 14526 (2019). https://doi.org/10.1038/s41598-019-51120-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51120-0
- Springer Nature Limited
This article is cited by
-
A review of foot-and-mouth disease in Ethiopia: epidemiological aspects, economic implications, and control strategies
Virology Journal (2023)
-
Phylogenetic and evolutionary analysis of foot-and-mouth disease virus A/ASIA/Sea-97 lineage
Virus Genes (2021)
-
Early origin and global colonisation of foot-and-mouth disease virus
Scientific Reports (2020)