
Abstract
Of the ten human-restricted Neisseria species two, Neisseria meningitidis, and Neisseria gonorrhoeae, cause invasive disease: the other eight are carried asymptomatically in the pharynx, possibly modulating meningococcal and gonococcal infections. Consequently, characterizing their diversity is important for understanding the microbiome in health and disease. Whole genome sequences from 181 Neisseria isolates were examined, including those of three well-defined species (N. meningitidis; N. gonorrhoeae; and Neisseria polysaccharea) and genomes of isolates unassigned to any species (Nspp). Sequence analysis of ribosomal genes, and a set of core (cgMLST) genes were used to infer phylogenetic relationships. Average Nucleotide Identity (ANI) and phenotypic data were used to define species clusters, and morphological and metabolic differences among them. Phylogenetic analyses identified two polyphyletic clusters (N. polysaccharea and Nspp.), while, cgMLST data grouped Nspp isolates into nine clusters and identified at least three N. polysaccharea clusters. ANI results classified Nspp into seven putative species, and also indicated at least three putative N. polysaccharea species. Electron microscopy identified morphological differences among these species. This genomic approach provided a consistent methodology for species characterization using distinct phylogenetic clusters. Seven putative novel Neisseria species were identified, confirming the importance of genomic studies in the characterization of the genus Neisseria.
Similar content being viewed by others

Introduction
The genus Neisseria includes a number of species that colonize the epithelium of animals1. Human-restricted species are Gram-negative, oxidase and catalase positive bacteria with two different morphologies: coccoid (Neisseria cinerea, Neisseria gonorrhoeae, Neisseria polysaccharea, Neisseria lactamica, Neisseria meningitidis, Neisseria mucosa, Neisseria oralis and Neisseria subflava); and rods (Neisseria elongata and Neisseria bacilliformis). All are asymptomatic commensal inhabitants of the mucosa, but two are also potentially pathogenic species: (i) N. meningitidis, which colonizes the oropharynx, sometimes causing meningitis and/or septicaemia; and (ii) N. gonorrhoeae, which typically inhabits the genitourinary tract, causing local and occasionally systemic gonococcal disease2. The remaining eight species are generally harmless inhabitants of the oro- and/or nasopharynx.
Conventional Neisseria phenotypic taxonomy reliably identifies the three species most studied at the time of writing: N. meningitidis; N. gonorrhoeae; and N. lactamica. This approach has, however, presented difficulties in defining and identifying other species3, which has led to the application of molecular approaches such as DNA-DNA hybridization (DDH)3 and phylogenetic analyses of nucleotide sequence data of the 16S ribosomal gene4. Analysis with single loci, such as 16S rRNA genes, poorly differentiate Neisseria species, due to distortions resulting from low resolution and horizontal genetic transfer (HGT)5. Phylogenies based on multi-locus sequence analysis (MLSA)4, such as (i) the seven loci used in Multi Locus Sequence Typing (MLST)5,6, (ii) the 53 ribosomal genes of ribosomal MLST (rMLST)7, and (iii) the 246 loci of the Neisseria genus core genome MLST (cgMLST)3, have greatly improved Neisseria species classification. These approaches led to the reclassification of some Neisseria species, with the consolidation of N. subflava biovar sublfava, perflava and flava, and Neisseria flavescens into a single species named Neisseria subflava. Isolates previously classified as Neisseria sicca were identified as variants of N. mucosa3 and “Neisseria mucosa var. heidelbergensis”, was shown to be genetically identical to Neisseria oralis8. A single-locus sequence species identification assay, which uses a fragment of the ribosomal gene rplF, can identify different Neisseria species reliably, but as this is a single locus test, it is not on its own an appropriate tool for the definition of novel species9. Measuring the Average Nucleotide Identity (ANI) between genomes pairs has also facilitated bacterial taxonomic research and been proposed as an alternative to DDH10,11, which is highly specialized, labor intensive and difficult to standardize and compare among laboratories12.
Non-pathogenic Neisseria (NPN) are rarely isolated and characterized, mostly because they seldom cause invasive disease, except in immunocompromised individuals1; however, their possible role in modulating nasopharyngeal carriage of N. meningitidis has increased interest in them. Several studies have indicated that nasopharyngeal colonization with N. lactamica can be protective against N. meningitidis infection13,14,15 and that N. lactamica has potential to be used in the design an outer-membrane vesicle (OMV) meningococcal vaccines16. Neisseria are known to be abundant in the human microbiome17, reinforcing the importance of understanding the diversity of this genus. This has been exemplified by the findings that: (i) in both high-income countries and in the African meningitis belt, the incidence of carriage of N. meningitidis increases with age as that of N. lactamica declines13,18,19; (ii) the risk factors for carrying N. meningitidis in the African Meningitis Belt (AMB) are inversely related to those of carrying NPNs20; and (iii) the microbiota has a major influence on other diseases, including pneumonia21 and cystic fibrosis22,23.
N. polysaccharea has been described as polyphyletic, potentially containing more than one species. In particular, isolate 15883, identified by Berger in 1985 in Germany24, has been proposed as a new species3,25. The increase in the number of non-N. meningitidis carriage isolates collected in previously under-sampled and geographically distinct regions20, combined with the concomitant decrease in the cost of whole genome sequencing (WGS), have allowed an increasing number of NPN isolates to be sequenced, although numbers remain low compared to N. meningitidis and N. gonorrhoeae.
Based on the phylogeny of partial rplF nucleotide sequences, isolates genetically close to isolate 15883, were identified in MenAfriCar carriage studies, suggesting that this putative novel species, which had not been found since Berger, could be present in the African continent20. Similar non-defined isolates were identified in other studies conducted in and outside of the African meningitis belt25,26,27,28 and in the UKMenCar4 carriage study29. Here, phylogenetic relationships of members of the human-restricted Neisseria species were examined using WGS data to characterize their genetic and phenotypic diversity and clarify the identity of isolates belonging to the Neisseria genus but currently of unknown species.
Results
Hierarchical gene-by-gene analysis identifies polyphyletic groups
WGS data of the 181 isolates included in this study (Supplemental Table 1) permitted an hierarchical approach to the analysis of bacterial isolate relationships, with increasing numbers of loci providing higher resolution (Fig. 1). A maximum-likelihood phylogeny generated from f_rplF sequence data identified four clusters including monophyletic N. meningitidis and N. gonorrhoeae clusters, and more diverse N. polysaccharea and Nspp isolates (Fig. 1A). Eighteen f_rplF alleles were identified: two among N. gonorrhoeae (f_rplF alleles 5 and 7); five among N. meningitidis (f_rplF 1, 2, 3, 4 and 8); six among N. polysaccharea (f_rplF alleles 9, 39, 44, 63, 90 and 120); and six among Nspp isolates (f_rplF alleles 16, 62, 69, 70, 79 and 146).

Hierarchical gene by gene analysis. Maximum-likelihood trees generated from the aligned sequences of f_rplF (A); the concatenated sequences of 51 rMLST loci (B) and the concatenated sequences of the 1114 complete cgMLST loci out of the 1441 (C). The orange stars correspond to the isolates selected in the first ANI analysis (Table 2) and the purple stars to those added in the ANI analysis done only among the N. polysaccharea clusters. The purple circles group Np isolates from specific geographical location. Countries of isolation of Nspp clusters are indicated. Np: N. polysaccharea and Nspp: Neisseria spp.
A phylogeny reconstructed from the sequences of 51 ribosomal loci identified distinct clusters among the Nspp isolates, each separated by long branches. This also confirmed the polyphyletic relationships of the N. polysaccharea isolates (Fig. 1B). A total of 115 rMLST profiles were identified: 9 among N. gonorrhoeae isolates; 18 among N. meningitidis isolates, with an additional isolate missing two loci and for which an rMLST profile could not be defined; 39 among N. polysaccharea isolates; and 49 among Nspp isolates. A network analysis using rMLST, which included additional genomes from the remaining human-restricted Neisseria3, showed that these Nspp isolates were distinct from other known species (Supplemental Fig. 1).
Nine distinct clusters were observed in a maximum-likelihood phylogeny derived from the 1114 cgMLST loci with complete sequences for all isolates; the cgMLST was defined for this study as explained in the “genomic diversity” heading of the method section. These were provisionally designated Nspp 1 to Nspp 9. At least two clusters could be differentiated among the N. polysaccharea isolates, designated N. polysaccharea 1 and N. polysaccharea 2 (Fig. 1C). Phylogeographical clustering was observed in both Nspp and N. polysaccharea groups as follows: (i) all the isolates from Nspp 1 (N = 36) and Nspp 8 (N = 1) were collected in Mali; (ii) Nspp 2 (N = 20), Nspp 4 (N = 14) and Nspp 5 (N = 21) were collected in Malawi; (iii) Nspp 3 (N = 9) came from Chad; (iv) Nspp 7 (N = 2) from The Gambia; and (v) Nspp 9 (N = 1) from Germany. The only cluster with isolates from different locations was Nspp 6 collected in Malawi (N = 2) and in the UK (N = 1). Similarly, all N. polysaccharea 2 isolates were collected in The Gambia (N = 6) and Mali (N = 7), two countries of the African meningitis belt, whereas N. polysaccharea 1 included isolates both from Europe and Africa. Even within the N. polysaccharea 1 cluster, these remained polyphyletic with clusters separated by deep branches. The ten isolates from Malawi (N = 10) formed a divergent branch and the remaining three African isolates, from Mali (N = 1) and The Gambia (N = 2), clustered more closely together compared to European isolates (Fig. 1C).
Average nucleotide identity identified seven putative novel Neisseria species
The cgMLST ANI calculated for the 13 representative genomes of the different clusters varied from 93.1% (between N. gonorrhoeae and Nspp 6 and N. gonorrhoeae and Nspp 7) to 96.6% (between Nspp 1 and Nspp 2). Only three pairwise comparisons yielded values >95%, these were between: (i) Nspp 1 and Nspp 2; (ii) Nspp 1 and Nspp 9; and (iii) Nspp 2 and Nspp 9. Using a threshold of 95% ANI, these findings indicate that these should be considered as distinct bacterial species. All the remaining ANI values were lower than 95% (Fig. 2A). Similar results were obtained using whole genome ANI (Supplemental Fig. 2A).

Heatmap of pairwise comparison of the 1114 core genes with complete sequences among the different clusters. Two-way Average Nucleotide Identity using concatenated cgMLST loci sequences among all the clusters (A) and only among the N. polysaccharea (B); ANI measured in percentage are presented with a colour gradient, yellow for values <94 and red for values >95%, which is the threshold above which genomes are considered to be from the species. Pairwise allelic comparison of the cgMLST loci (C), Number of loci with at least one allele shared are displayed with a colour gradient, pink for values <50 and purple for values >500. Nm: N. meningitidis; Ng: N. gonorrhoeae; Np: N. polysaccharea and Nspp: Neisseria spp.
Using the 95% threshold, the ANI data were used to classify the Nspp isolates into seven distinct species groups, each of which assigned a proposed species name:
-
1.
57 isolates including clusters Nspp 1, Nspp 2 and Nspp 9; the name Neisseria bergeri sp. nov. is proposed for these organisms, after the late Ulrich Berger who isolated the first strain (type strain: 15883; pubMLST ID: 5193) in Germany in 198424.
-
2.
The Nspp 8 cluster comprised a single isolate collected in Mali in 2010; the name “Neisseria maigaei” is suggested, after the late Aziz Maiga who played an important role in the isolate characterization of the MenAfriCar study in Mali.
-
3.
The name Neisseria uirgultaei sp. nov. is proposed for the nine isolates from cluster Nspp 3 (type strain: 12_13955_XS2_1; pubMLST ID: 43129), after Sir Brian Greenwood, the director of the MenAfriCar project (greenwood is uirgulta in Latin).
-
4.
As only two isolates have been identified in Nspp 7, the name “Neisseria basseii” is suggested, after the town of Basse in The Gambia, where the isolates were collected.
-
5.
The name Neisseria blantyrii is proposed for the 14 Nspp 4 isolates (type strain: M-041; pubMLST ID: 46402), after the city of Blantyre in Malawi where the isolates were collected.
-
6.
Neiseria viridiae sp. nov. is proposed for the 21 Nspp 5 isolates (type strain: M-226; pubMLST ID: 46439), after Jenny MacLennan (nee Green) who collected and characterized all the isolates from Malawi and The Gambia included in this study (green is viridi in Latin).
-
7.
Finally, the name “Neisseria benedictiae” is suggested for the three Nspp 6 isolates, after Julia Bennett (bennet is benedictus in Latin) who pioneered the genomic work of N. bergeri isolates and the speciation of Neisseria with cgMLST approaches (Fig. 3).
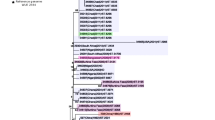
Figure 3 
Maximum-likelihood tree of the putative novel Neisseria species defined following 95% cgMLST ANI.

The pairwise allelic comparison of the 1114 core loci with complete sequences between each defined cluster identified that, despite sharing the same loci, most clusters had distinct alleles, with only small numbers of shared allele between clusters. Nspp 1 and Nspp 2, however, had the largest number of loci (N = 693), sharing at least one allele, and similarly for Nspp 1 and Nspp 9 (N = 145) and Nspp 2 and Nspp 9 (N = 134) (Fig. 2C), consistent with the ANI analysis suggesting their grouping into a single species.
Neisseria polysaccharea is polyphyletic
The two N. polysaccharea clusters, designated N. polysaccharea 1 and N. polysaccharea 2, had an ANI value of 94.6%, confirming that they can be considered as two distinct species (Fig. 2A). The pairwise allelic comparison based on the two clusters found that N. polysaccharea 1 also had large number of loci sharing at least one allele with various clusters (N. polysaccharea 2; Nspp 1, Nspp 2, Nspp 4, Nspp 5 and Nspp 6), suggesting that the cluster was not homogeneous and probably included more than one species. Three additional genomes of the N. polysaccharea 1 cluster were selected to attempt to characterize this further (Fig. 1C). 95% cgMLST and whole genome ANI (Fig. 2B and Supplemental Fig. 2B) comparison among the genomes of these 5 isolates identified another distinct cluster, N. polysaccharea 3 (Fig. 3).
Phenotypic analysis of clusters
Macroscopic analysis of bacterial colony morphology established that colonies from all isolates tested were grey, exhibited γ-haemolysis, and formed a round shape on blood agar (BAP). The putative novel species exhibited edges with a regular shape and smooth texture; however, Neisseria blantyrii (Nspp 4) visually appeared to be moister. The size range of the colonies varied from 0.1 mm to 1 mm; N. meningitidis colony size range extended to 2 mm (Table 1).
Scanning electron microscopy (SEM) of representative isolates from each proposed species revealed that they were all coccoid bacteria, with a mix of completely formed diplococci and incompletely formed diplococci, which were characterized by a “heart-shaped” morphology (Fig. 4). Variation over the extracellular structures observed around the cell membrane was identified: for example, isolates from the three sub-clusters of N. bergeri (Nspp 1, 2, and 9) exhibited filaments covering their extracellular membranes, “pili-like” structures could also be seen connecting cells of “N. maigaei” (Nspp 8), “N. blantyrii“(Nspp 4) cells were covered in an intense filamentous network, “N. viridiae” (Nspp 5) and “N. benedictiae” (Nspp 6) exhibited visible “bulbs” on the surface of their extracellular membranes. Conversely, “Neisseria uirgultaei” (Nspp 3) possessed a smooth surface with no visible extracellular elements and “Neisseria basseii” (Nspp 7) showed few visible filaments and bulbs (Fig. 4).

Scanning Electron Microscopy images of the different clusters identified by genomic analysis. A-N. meningitidis, B-N. polysaccharea cluster 2, C-N. polysaccharea cluster 3, D-Nspp 1 (N. bergeri), E-Nspp 2 (N. bergeri), F-Nspp 3 (N. uirgultaei), G-Nspp 4 (N. blantyrii), H-Nspp 5 (N. viridiae), I-Nspp 6 (N. benedictiae), J-Nspp 7 (N. bassei), K- Nspp 8 (N. maigaei) and L-Nspp 9 (N. bergeri). The scale bar represents 0.25 µm.
Biochemical data obtained using DiatabsTM system identified the putative novel species as GGA, ONPG, and Tributyrin negative, Leucine and Proline Aminopeptidase positive. They were also positive for the maltose degradation and negative for lactose degradation. Results were inconsistent for glucose and sucrose when repeated: N. bergeri subcluster 1 (Nb1; Nspp 9) and 2 (Nb2; Nspp 2), “N. uirgultaei” (Nspp 3), “N. basseii” (Nspp 7) and “N. viridiae” (Nspp 5) being mostly positive for glucose after at least 3 repeats, and N. bergeri subcluster 3 (Nb3; Nspp 1), “N. maigaei” (Nspp 8), “N. blantyrii” (Nspp 4) and “N. benedictiae” (Nspp 6) mostly negative. Similarly, “N. viridiae” (Nspp 5) and “N. benedictiae” (Nspp 6) were mostly positive for sucrose while all the other species were negative. GGA, proline aminopeptidase and lactose/ONPG tests were useful for distinguishing new species from N. meningitidis and N. lactamica; however, none of these allowed a distinction from N. polysaccharea (Table 2). Results obtained from the API®-NH tests showed that all the isolates were positive for the degradation of glucose, fructose, maltose, and saccharose, which contradicted the pattern observed for glucose with the DiatabsTM for some isolates. Similarly, to the DiatabsTM assay, the β-galactosidase, proline aminopeptidase and gamma-glutamyl transferase (GGT) reactions were useful to distinguish putative novel species from N. lactamica and N. meningitidis but no biochemical test could distinguish them from N. polysaccharea (Table 3).
Discussion
The concept and definition of species in prokaryotes continues to be subject to debate, as do the best methods for microbial taxonomy30,31. Many bacterial species have been defined solely on phenotypic characteristics4. For example, N. meningitidis and N. gonorrhoeae, were defined by their very different clinical manifestations, although they are very closely related and do not meet the 70% DDH criterion necessary to separate them into distinct species taxonomically32. The integration of ecological and genetic data has greatly improved classification. With the advent of high-throughput nucleotide sequencing studies, multi locus sequence analysis (MLSA) became an approach of choice33. The analysis of the seven MLST loci has proven to be sufficient to distinguish N. meningitidis, N. gonorrhoeae, and N. lactamica34, for example, which single-locus 16S RNA sequence analysis has failed to achieve3. Although housekeeping genes have predominantly been used, there have been no specific guidelines on the loci or the number of loci to be chosen for MLSA. The increased availability of whole or nearly complete genome sequence data allows species characterization at a higher resolution, as demonstrated through the use of the Neisseria genus cgMLST to identify multiple Neisseria species3.
The phylogenomic approach35 adopted here combines rMLST, MLSA, and ANI scores, using nucleotide sequence data of core genes, supplemented with a quantitative measure of genetic relatedness (Fig. 2A,B). This allows phylogenetic relationships to be inferred among the collection of isolates under study (Fig. 1C). The 95% cgMLST ANI cut-off identified seven distinct species (Fig. 3); however, three clusters identified by phylogenomic analyses were indistinguishable by ANI and were therefore grouped into a single species named N. bergeri sp. nov. These three clusters can be described as subspecies (Nb Subspecies 1, 2, and 3). Three putative novel Neisseria species were assigned names, as clusters contained more than three representative isolates: N. uirgultaei sp. nov., N. blantyrii sp. nov., and N. viridiae sp. nov. The remaining clusters had fewer than three isolates and therefore will not be formally named; however, the species names (“N. maigaei”, “N. basseii” and “N. benedictiae”) are proposed for future reference. Identical ANI results were obtained using the full draft WGS (Supplemental Fig. 2A), confirming that using genes core to these human-restricted Neisseria species provided optimal genomic resolution with exclusion of intergenic regions and accessory genes not affecting results. Congruence between ANI results and cgMLST allelic comparison (Fig. 2A,C), suggests that allelic diversity plays an important role in the speciation process, alongside gene presence/absence. The low number of shared alleles confirms the infrequency of cross-species recombination as previously observed in a study comparing N. meningitidis, N. lactamica, and N. gonorrhoeae36. Analyses identified distinct clusters among the N. polysaccharea isolates included in this study: two phylogenetic clusters, N. polysaccharea 2 and N. polysaccharea 3, could be reclassified as distinct species based on the 95% cgMLST and ANI (Fig. 2B and Supplemental Fig. 2B). However, a subset (N. polysaccharea 1) remained polyphyletic.
A major limitation when undertaking taxonomic studies is ensuring the natural diversity of the bacterial population has been appropriately sampled. Studies are often geographically limited, leading to an underestimation of diversity. The different nasopharyngeal carriage studies undertaken in the UK29,37, countries of the African Meningitis Belt19,26,38, and in Malawi (outside the belt)28 have led to the collection of a large number of Neisseria isolates; however, the NPNs were not always characterized at the genomic level. Our results indicate a phylogeographic clustering of genotypes, with isolates from the same country clustering together (Fig. 1C). Even though most recent carriage studies, done in the UK and in African countries, used the same laboratory methodology, most of the previously non-speciated isolates were recovered in Africa with only one Nspp found in the UK, suggesting a potentially greater diversity of Neisseria species in African countries than in the UK. However, differences in the age groups targeted, the whole population (AMB and Malawi) as opposed to 15–19 years old (UK), may have played a role in the differences observed.
WGS analysis of additional carried Neisseria isolates from various geographical regions will improve the taxonomy of this genus. Isolates previously characterized as “N. bergeri”, based on nucleotide sequences of the rplF locus20, included additional putative novel Neisseria species, such as those identified in this study. Genome sequences obtained from the Malian collection of the MenAfriCar project found that most of Nspp genomes were classified as N. bergeri sp. nov. using the 95% cgMLST ANI analysis presented here; however, one was found to form a distinct putative species, “N. maigaei”. Although the sequencing of the f_rplF locus provides species identity for previously defined species, our study confirms that it does not provide sufficient resolution for the characterization of new species. Each of the clusters identified had distinct f_rplF alleles except N. benedictiae (Nspp 6) and N. basseii (Nspp 7) which shared allele 69, consistent with recent HGT or shared ancestry.
All the putative novel species and the N. polysaccharea isolates were indistinguishable using standard biochemical tests, despite being clearly distinct by 95% cgMLST ANI. However, additional tests including biochemical analyses, pH, or chemotaxonomy (analysis of chemical composition of the cell) combined with a larger numbers of isolates, including additional type strains, may identify further differences39. Phenotypic tests are labor intensive, and the interpretation of the results can be subjective. The DiatabsTM tests were, overall, more straightforward than API®-NH tests, which have a strict McFarland Standard concentration. Sugar degradation by the DiatabsTM were accurate for the N. meningitidis and N. lactamica controls but were elsewhere unstable with repeats giving different results. On the other hand, the degradation of sugars assessed by the API®-NH were highly reproducible, but failed to give the expected results for N. meningitidis24. Coccoid cells, some of which exhibiting distinct extracellular membrane structures, were visible following SEM analyses (Fig. 4). After 20 hours of incubation, most of the putative novel species had “Heart-shaped” cells (Fig. 4). These cells were still visible after 48 h of incubation (data no shown) but additional time points may establish whether these forms were intermediate shapes of coccoid cells or remained distinctive. Overall, phenotypic tests were inconclusive.
The genomic analyses presented here, allowed putative novel species predominantly found in African countries to be identified. Determining the broader implications of these findings will require more sampling, genomic characterization and analyses of the interactions of these different species in the nasopharynx. This information will determine any role that they play in modulating carriage and invasion by N. meningitidis. Future microbiome and immunological studies will be crucial to shed further light on the dynamics of carriage, but genomic characterizations of the isolates will be necessary to adequately assess the diversity. Another novel, non-human-restricted, Neisseria species has been recently identified in the US, indicating the potential for other unsampled species40.
Results from this study suggest that Neisseria diversity may be greater in countries outside the AMB, for example Malawi, than within the belt. This could have important implications regarding the impact of those species in modulating the carriage and invasiveness of N. meningitidis, as the disease epidemiology is known to be very different between these two regions of Africa; however, it is likely that analysis of more genome sequences of the Neisseria isolates from the AMB, such as those from the other countries sampled alongside Mali and Chad during the MenAfriCar project20, will improve our understanding of the diversity seen within the AMB and allow more adequate hypotheses to be formulated. The analytical methods developed here provide a framework for future speciation endeavors and can easily be repeated to incorporate additional genomes when available. It is now evident that the diversity of human restricted Neisseria had been underestimated and more carriage studies using genomic methods should be encouraged to define a more accurate picture of the genus.
Materials and Methods
Isolates and genomes
Isolate collection
A total of 107 Neisseria isolates unclassified to species level (Nspp) were identified in the pubMLST Neisseria database; some of these had been recorded as “Neisseria bergeri”, following preliminary analysis20,25,28. The majority of the Nspp were collected in carriage studies performed in Africa and included: (i) 57 isolates collected from Ndirande, Blantyre, Malawi, from27,28,41; (ii) 37 isolates collected in Mali and 9 from Chad20,38; and (iii) 2 isolates obtained in Basse, Gambia26. Two additional isolates were collected in Europe: the first in Germany in24; and the second in the United Kingdom29.
DNA preparation, Genome sequencing and assembly
All the African Nspp isolates were sequenced de novo using the Illumina MiSeq platform. Isolates were cultured on blood agar plates (BAP) and incubated overnight for 16–24 hours at 37 °C, in 5% CO2. For 96 of the isolates, genomic DNA was extracted using the Wizard genomic DNA Purification kit (Promega) following the manufacturer’s instructions; for the remaining nine isolates from Chad, DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen), as previously described42. DNA samples were sequenced by pair-end sequencing on an Illumina HiSeq, with read lengths of 100 (N = 55), 125 (N = 46) and 150 (N = 4) base pairs at the Wellcome Trust Sanger Institute, using previously described methods43.
Resultant short reads were assembled using the Velvet genome assembly program (v1.2.08)44: all odd-numbered kmer lengths between 21–99, 21–124 and 21–149 were sampled for read lengths of 100, 125, and 150 base pairs respectively, using the VelvetOptimiser software (v2.2.4)45 to automatically establish the optimal assembly parameters for Velvet (default optimization functions used). The assembled contigs were deposited in the Neisseria PubMLST database46 which uses the Bacterial Isolate Genome Sequence Database (BIGSdb) software47 and to the European Nucleotide Archive (Accession numbers in Table 1). Draft genomes were automatically curated at all loci defined in the database using the BLAST algorithm48 and a sequence similarity threshold of >98%, allowing rapid annotation of known alleles and sequences which were very similar to defined loci43. Manual verification was then performed for variable loci, such as those containing internal stop codons, frame shifts or those with sequence similarities lower than 98%. Incomplete gene sequences at the beginning or end of a contig were identified as such.
Additional genomes
In addition to the 105 African Nspp sequenced de novo, and the two European strains previously sequenced and deposited into the http://pubMLST.org/neisseria database, 74 other genomes deposited in the database were chosen as described below, to complete the genome collection analyzed in this study. These represented three known human-associated Neisseria species and included all the genomes available at the time of analysis for N. polysaccharea (N = 45), all but one finished genomes of N. gonorrhoeae (N = 9) and representative genomes of the most common clonal complexes of N. meningitidis (N = 20)43, with a preference for finished genomes when available. Additional information and meta-data for each of the 181 genomes are available in Supplemental Table 1.
Genomic diversity
Phylogenetic analysis
A hierarchical gene-by-gene approach to Whole Genome Sequence (WGS) analysis was applied to all 181 genomes, including the analysis of: (i) f_rplf, a fragment of the ribosomal gene rplF9, (ii) 51 of the 53 rMLST loci (BACT0060 (rpmE) and BACT0065 (rpmI) were excluded as they are known to be paralogous in Neisseria species); and (iii) a newly defined cgMLST scheme (Human-restricted Neisseria cgMLST v1.0), including 1441 loci present in at least 95% of isolates in this collection. All the sequences were aligned using MAFFT in pubMLST46,49.
Two methods where used in parallel to generate the list of core loci using: (i) the Genome Comparator tool of PubMLST was used to compare all genomes, using the N. meningitidis finished genome FAM18 (Accession number: AM421808) as a reference with parameters set at a minimum identity of 70%, a minimum alignment length of 50%, a BLASTN word size of 20 base pair and a core threshold of 95% (paralogous loci were excluded from this analysis and variable loci aligned and concatenated into a single sequence for each genome); (ii) Proteinortho, assembled draft genomes were downloaded from PubMLST for each isolate and used as input files for the annotation program Prokka, using default settings with the option “–addgenes” and “–compliant”50. The protein sequence outputs of Prokka were used as input files for Proteinortho, with default settings, to detect orthologous group of genes51.
Both methods generated a list of core loci present in 95% of the genomes included in the analysis; 1421 loci were identified by Genome comparator and 1366 by Proteinortho. An inclusive list of 1441 loci including any that were considered core to 95% of isolates by both methods was generated and used in the phylogenetic analysis. Within these 1441 loci, 1114 had complete sequences in all the genomes, whereas the remaining 327 fell at the beginning or the end of a contig in at least one genome and were classified as incomplete genes and excluded from the phylogenetic analysis (Supplemental Table 2).
Phylogenetic analyses were performed for f_rplF, rMLST, and cgMSLT loci using a finished genome of Neisseria lactamica (pubMLST ID: 8851/isolate ID: 020-06) as an out-group. The f_rplF and rMLST phylogenies were generated in MEGA6 using the Maximum likelihood method with the following options: General Time Reversible model52, 100 bootstrap replications, Gamma distributed rate with Invariant sites and 5 discrete Gamma Categories. The cgMLST phylogeny was generated in FastTree53 using the General Time Reversible model (“-gtr”) and all other defaults options. Tree annotations were performed in MEGA654.
Another rMLST phylogeny was reconstructed with the Neighbor Joining model in MEGA6 using the 181 genomes previously described and all 40 additional human-restricted Neisseria species included in the paper published by Bennett et al., which included the available type strains genomes3.
Pairwise allelic variations at the core genes, between each identified clusters, were assessed using the allele-overlap script comparing alleles of each gene between two defined sets of genomes55.
Statistical methods
Average nucleotide identity
The Average Nucleotide Identity (ANI) was calculated by comparing concatenated sequences of cgMLST loci from representative isolates of each phylogenetic cluster of interest: 13 isolates for the analysis of the whole phylogeny and 5 for the further characterization of the N. polysaccharea cluster. An online ANI tool, “ the ANI calculator”56,57 was used to calculate a two-way ANI between each pair of selected isolates using the method described previously11. Pairs of isolates with ANI values higher than 95% were considered as indistinguishable at the species level as suggested by the program. The analysis was also computed using the whole genomes sequences of the isolates.
Phenotypic analyses
Phenotypic characteristics of the clusters identified were evaluated using representative isolates from cluster Nspp1-9, Np1-2, and three positive controls: (i) N. meningitidis (pubMLST ID: 35956/isolate ID: Z1318)58; (ii) N. polysaccharea (pubMLST ID: 19095/isolate ID: CCUG 18031)59; and (iii) N. lactamica (pubMLST ID: 8851/isolate ID: 020-06)59 (Supplemental Table 1). Each isolate was cultured on Columbia Horse Blood Agar plates (Oxoid) and incubated at 37 °C for 16 to 24 hours, in an atmosphere containing 5% CO2. A single isolated colony was then sub-cultured under the same conditions for 20 hours before carrying out the tests described below.
Morphological analysis
Macroscopic assessment. Cell morphology was characterized, and the color, texture, shape and colony contour were visually assessed after 20 h of incubation. The size of individual colonies was measured using a graduated ruler.
Scanning Electron microscopy (SEM). Sub-cultured isolates were prepared for SEM after 20 hours of incubation, following adaptation of a protocol previously described60. Briefly, a piece of agar with bacterial cells attached was excised and fixed with a 2.5% glutaraldehyde solution in 0.1 M Phosphate buffer and incubated at room temperature for 1 hour. Specimens were then rinsed in 0.1 M Phosphate buffer and post-fixed with 1% osmium tetroxide for 1 hour. Specimens were then rinsed in Mili-Q water and dehydrated using an ethanol series: 50% for 10 min, 70% overnight (stored at 4 °C), 90% for 10 min, 95% for 10 min, 100% for 20 min and in pure ethanol three times for 20 min each. Ethanol was removed, and the samples were dried by incubating specimens in a 50% Hexamethyldisilazane (HMDS) and 50% ethanol solution for 15 min, followed by incubation in pure HMDS for an additional 15 min. Once HMDS was removed, samples were left in a fume hood overnight to allow HMDS fumes to evaporate. Samples were mounted on SEM stubs with a conductive carbon adhesive, then coated with 10–15 nm of gold using a Quorum Technologies Q150R ES sputter coater and were imaged with a Zeiss Sigma 300 field emission gun SEM at an accelerating voltage of 2 kV.
Sugar degradation and other biochemical tests
Oxidase tests were conducted with the oxidase test disks (Sigma-Aldrich) following the manufacturer’s instructions. Catalase tests were undertaken by putting the bacteria in contact with a drop of hydrogen peroxide; the formation of bubbles was considered a positive reaction. Haemolysis was assessed for each sample by visual inspection of the cells on BAP.
DiatabsTM (Rosco Diagnostica) were used according to the manufacturer’s instructions to test for sugar degradation of glucose, maltose, sucrose, and lactose, and other selected microbial enzymatic properties including: the ability to enzymatically hydrolyse aminopeptidase substrates like gamma glutamyl aminopeptidase (GGA), leucine aminopeptidase and proline aminopeptidase; the hydrolysis activity of the enzyme β-galactosidase on the orthonitrophényl-β-galactoside (ONPG) substrate; and the enzymatic hydrolysis of tributyrin into butyric acid and glycerol often used for the differentiation of Moraxella catarrhalis from Neisseria species. The results were assessed following the instructions of the test (Supplemental Table 3).
The API-NH® kit (Biomerieux) for Neisseria characterisation was also tested in parallel, following the manufacturer’s instructions (Supplemental Table 4).
Accession number(s)
The Accession numbers for the de novo sequenced isolates are provided in Table 1. Accession numbers or reference are provided for the other genomes. All genomes will be made available on the pubMSLT database upon publication of the manuscript.
References
Liu, G., Tang, C. M. & Exley, R. M. Non-pathogenic Neisseria: members of an abundant, multi-habitat, diverse genus. Microbiology. https://doi.org/10.1099/mic.0.000086 (2015).
Unemo, M. & Dillon, J. A. Review and international recommendation of methods for typing neisseria gonorrhoeae isolates and their implications for improved knowledge of gonococcal epidemiology, treatment, and biology. Clin Microbiol Rev 24, 447–58 (2011).
Bennett, J. S. et al. A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology 158, 1570–80. (2012).
Gevers, D. et al. Opinion: Re-evaluating prokaryotic species. Nat Rev Microbiol 3, 733–9 (2005).
Maiden, M. C. et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA 95, 3140–5 (1998).
Bennett, J. S. et al. Species status of Neisseria gonorrhoeae: evolutionary and epidemiological inferences from multilocus sequence typing. BMC Biol 5, 35 (2007).
Jolley, K. A. et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158, 1005–15 (2012).
Bennett, J. S., Jolley, K. A. & Maiden, M. C. Genome sequence analyses show that Neisseria oralis is the same species as ‘Neisseria mucosa var. heidelbergensis’. Int J Syst Evol Microbiol 63, 3920–6 (2013).
Bennett, J. S., Watkins, E. R., Jolley, K. A., Harrison, O. B. & Maiden, M. C. Identifying Neisseria species by use of the 50S ribosomal protein L6 (rplF) gene. J Clin Microbiol 52, 1375–81 (2014).
Konstantinidis, K. T. & Tiedje, J. M. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 102, 2567–72 (2005).
Goris, J. et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57, 81–91 (2007).
Rossello-Mora, R. Updating prokaryotic taxonomy. J Bacteriol 187, 6255–7 (2005).
Gold, R., Goldschneider, I., Lepow, M. L., Draper, T. F. & Randolph, M. Carriage of Neisseria meningitidis and Neisseria lactamica in infants and children. J Infect Dis 137, 112–21 (1978).
Evans, C. M. et al. Nasopharyngeal colonization by Neisseria lactamica and induction of protective immunity against Neisseria meningitidis. Clin Infect Dis 52, 70–7 (2011).
Deasy, A. M. et al. Nasal Inoculation of the Commensal Neisseria lactamica Inhibits Carriage of Neisseria meningitidis by Young Adults: A Controlled Human Infection Study. Clin Infect Dis 60, 1512–20 (2015).
Gorringe, A. R. et al. Phase I safety and immunogenicity study of a candidate meningococcal disease vaccine based on Neisseria lactamica outer membrane vesicles. Clin Vaccine Immunol 16, 1113–20 (2009).
Keijser, B. J. et al. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res 87, 1016–20 (2008).
Cartwright, K. A., Stuart, J. M., Jones, D. M. & Noah, N. D. The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect 99, 591–601 (1987).
Kristiansen, P. A. et al. Carriage of Neisseria lactamica in 1- to 29-year-old people in Burkina Faso: epidemiology and molecular characterization. J Clin Microbiol 50, 4020–7 (2012).
Diallo, K. et al. Pharyngeal carriage of Neisseria species in the African meningitis belt. J Infect 72, 667–77 (2016).
de Steenhuijsen Piters, W. A. et al. Dysbiosis of upper respiratory tract microbiota in elderly pneumonia patients. ISME J 10, 97–108 (2016).
Filkins, L. M. et al. Prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J Bacteriol 194, 4709–17 (2012).
de Koff, E. M., Groot, K. M. & Bogaert, D. Development of the respiratory tract microbiota in cystic fibrosis. Curr Opin Pulm Med 22, 623–628 (2016).
Berger, U. First isolation of Neisseria polysacchareae species nova in the Federal Republic of Germany. Eur J Clin Microbiol 4, 431–3 (1985).
Bennett, J. S. et al. Ribosomal MLST analysis reveals a distinct species of Neisseria, previously identified as Neisseria polysaccharea that is closely related to Neisseria meningitidis, p 138. In (ed).
MacLennan, J. M. et al. Carriage of serogroup W-135, ET-37 meningococci in The Gambia: implications for immunisation policy? Lancet 356, 1078 (2000).
Mandala, W. L. et al. Lymphocyte subsets in healthy Malawians: implications for immunologic assessment of HIV infection in Africa. J Allergy Clin Immunol 125, 203–8 (2010).
MacLennan, J. M. et al. High carriage rates of commensal Neisseria species in Malawi including a previously un-named species, p 335. In (ed).
MacLennan, J. M. & Maiden, M. C. J. Group UMC. UKMENCAR4: A meningococcal carrige study in 21,000 teenagers to understand changing meningococcal epidemiology and evaluate National vaccination policy, p 47. In (ed).
Staley, J. T. Biodiversity: are microbial species threatened? Curr Opin Biotechnol 8, 340–5 (1997).
Cohan, F. M. What are bacterial species? Annu Rev Microbiol 56, 457–87 (2002).
Tonjum, T. 2001. Genus I. Neisseria. In Bergey’s Manual of Systemic Bacteriology Springer.
Naser, S. M. et al. Application of multilocus sequence analysis (MLSA) for rapid identification of Enterococcus species based on rpoA and pheS genes. Microbiology 151, 2141–50 (2005).
Hanage, W. P., Fraser, C. & Spratt, B. G. Fuzzy species among recombinogenic bacteria. BMC Biol 3, 6 (2005).
Staley, J. T. The bacterial species dilemma and the genomic-phylogenetic species concept. Philos Trans R Soc Lond B Biol Sci 361, 1899–909 (2006).
Bennett, J. S. et al. Independent evolution of the core and accessory gene sets in the genus Neisseria: insights gained from the genome of Neisseria lactamica isolate 020-06. BMC Genomics 11, 652 (2010).
Maiden, M. C. & Stuart, J. M. Group UKMC. 2002. Carriage of serogroup C meningococci 1 year after meningococcal C conjugate polysaccharide vaccination. Lancet 359, 1829–31 (2002).
MenAfriCar, C. Meningococcal carriage in the African meningitis belt. Trop Med Int Health 18, 968–78 (2013).
Tindall, B. J. et al. Notes on the characterization of prokaryote strains for taxonomic purposes. Int J Syst Evol Microbiol 60, 249–66 (2010).
Wroblewski, D. et al. Neisseria dumasiana sp. nov. from human sputum and a dog’s mouth. Int J Syst Evol Microbiol 67, 4304–4310 (2017).
MacLennan, C. A. et al. Dysregulated humoral immunity to nontyphoidal Salmonella in HIV-infected African adults. Science 328, 508–12 (2010).
Hill, D. M. et al. Genomic epidemiology of age-associated meningococcal lineages in national surveillance: an observational cohort study. Lancet Infect Dis, https://doi.org/10.1016/S1473-3099(15)00267-4. (2015).
Bratcher, H. B., Corton, C., Jolley, K. A., Parkhill, J. & Maiden, M. C. A gene-by-gene population genomics platform: de novo assembly, annotation and genealogical analysis of 108 representative Neisseria meningitidis genomes. BMC Genomics 15, 1138 (2014).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–9 (2008).
Gladman, S. & Seemann, T. VelvetOptimiser, on Victorian Bioinformatics Consortium, http://bioinformatics.net.au/software.velvetoptimiser.shtml. Accessed (2012).
Jolley, K. PubMLST, Neisseria sequence Typing, http://pubmlst.org/neisseria/. Accessed (2010).
Jolley, K. A. & Maiden, M. C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–10 (1990).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–66 (2002).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–9 (2014).
Lechner, M. et al. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12, 124 (2011).
Nei, M. & Kumar, S. Molecular evolution and phylogenetics. Oxford University Press, Oxford; New York (2000).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26, 1641–50 (2009).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–9 (2013).
Hauck, S. allele-overlap, vv1.10, https://github.com/sohauck/big-batch/tree/master/allele-overlap (2016).
Rodriguez, R. L. & Konstantinidis, K. Bypassing Cultivation To Identify Bacterial Species. Microbe Magazine, https://doi.org/10.1128/microbe.9.111.1 (2014).
Rodriguez, R. L. Average Nucleotide Identity calculator, http://enve-omics.ce.gatech.edu/ani/.
Wang, J. F. et al. Clonal and antigenic analysis of serogroup A Neisseria meningitidis with particular reference to epidemiological features of epidemic meningitis in the People’s Republic of China. Infect Immun 60, 5267–82 (1992).
Bennett, J. S. et al. Genetic diversity and carriage dynamics of Neisseria lactamica in infants. Infect Immun 73, 2424–32 (2005).
Bozzola, J. J. Conventional specimen preparation techniques for scanning electron microscopy of biological specimens. Methods Mol Biol 369, 449–66 (2007).
Acknowledgements
Kanny Diallo was supported by a Wellcome Trust Training Fellowship in Public Health and Tropical Medicine (grant number: 103957/Z/14/Z). Martin Maiden was supported by the Wellcome Trust (grant number: 087622/Z/08/Z). The funding sources had no role in the study design, collection, analysis and interpretation of the data, in the writing of the report or in the decision to submit the paper for publication. This publication made use of the Neisseria Multi Locus Sequence Typing website (https://pubmlst.org/neisseria/) developed by Keith Jolley and sited at the University of Oxford (Jolley & Maiden 2010, BMC Bioinformatics, 11:595). The development of this site has been funded by the Wellcome Trust and European Union. Electron microscopy work was performed at the Dunn School EM Facility. We are grateful to Prof Mohamed Taha and Dr Eva Hong from Institut Pasteur, Dr Mustapha Mustapha, from the Infectious Diseases Epidemiology Research Unit at the University of Pittsburgh, Dr Adam Retchless from the Center for Disease Control and Cecilia Fazio from the Istituto Superiore di Sanità (ISS) Rome, Italy, for allowing us to include some of their unpublished genomes into our analysis.
Author information
Authors and Affiliations
Contributions
K.D., J.M. and O.B.H. participated to the design of the study, performed the laboratory work, did the results analyses and wrote the first draft of the paper; K.D., J.M., C.M., S.O.S., D.M.D., C.T., C.A.M., J.P. and R.B. participated in the collection and characterisation of the isolates; EJ participated in the electron microscopy work; J.M., B.M.G. and M.C.J.M. provided guidance on the design and data analysis of the study. All authors made significant input in the writing process of this manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Diallo, K., MacLennan, J., Harrison, O.B. et al. Genomic characterization of novel Neisseria species. Sci Rep 9, 13742 (2019). https://doi.org/10.1038/s41598-019-50203-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50203-2
- Springer Nature Limited
This article is cited by
-
Distributed genotyping and clustering of Neisseria strains reveal continual emergence of epidemic meningococcus over a century
Nature Communications (2023)
-
Genomic sequence of the non-pathogen Neisseria sp. strain MA1-1 with antibiotic resistance and virulence factors isolated from a head and neck cancer patient
Archives of Microbiology (2022)
-
Comparative genomics of a novel clade shed light on the evolution of the genus Erysipelothrix and characterise an emerging species
Scientific Reports (2021)