
Abstract
Targeting chokepoint enzymes in metabolic pathways has led to new drugs for cancers, autoimmune disorders and infectious diseases. This is also a cornerstone approach for discovery and development of anthelmintics against nematode and flatworm parasites. Here, we performed omics-driven knowledge-based identification of chokepoint enzymes as anthelmintic targets. We prioritized 10 of 186 phylogenetically conserved chokepoint enzymes and undertook a target class repurposing approach to test and identify new small molecules with broad spectrum anthelmintic activity. First, we identified and tested 94 commercially available compounds using an in vitro phenotypic assay, and discovered 11 hits that inhibited nematode motility. Based on these findings, we performed chemogenomic screening and tested 32 additional compounds, identifying 6 more active hits. Overall, 6 intestinal (single-species), 5 potential pan-intestinal (whipworm and hookworm) and 6 pan-Phylum Nematoda (intestinal and filarial species) small molecule inhibitors were identified, including multiple azoles, Tadalafil and Torin-1. The active hit compounds targeted three different target classes in humans, which are involved in various pathways, including carbohydrate, amino acid and nucleotide metabolism. Last, using representative inhibitors from each target class, we demonstrated in vivo efficacy characterized by negative effects on parasite fecundity in hamsters infected with hookworms.
Similar content being viewed by others

Introduction
Metabolic potential is a critical determinant for a pathogen’s development and growth, infectivity and maintenance1,2,3,4,5. Consequently, the enzymes comprising the metabolic network of a pathogen are potential targets for drug discovery. In fact, multiple drugs in current clinical and agricultural use as anti-infective agents target these enzymes2,3,5. However, this approach has not been suitable for the discovery of anthelmintics until recently, due to insufficient knowledge regarding the metabolism of parasitic nematodes - partly due to large metabolic diversity within the phylum, especially amongst those inhabiting distinct trophic niches. Moreover, until recently, there has also been a lack of a comprehensive knowledgebase of their genomes and biochemistry. This newly generated genomics information4,6 significantly facilitates and expedites the development of drugs targeting specific parasites, and also enables comparative studies to infer shared similarities in diverse parasites that could be exploited to design anthelmintics with a broad-spectrum potential. Such anthelmintics are especially desirable because concomitant infections with different parasites are predominant in endemic countries7,8,9.
In an earlier study to identify novel broadly effective anthelmintics1, we followed an omics-driven target and drug prioritization approach to find potential metabolic enzyme drug targets using data from 10 Nematoda genomes, including four parasitic nematodes (a whipworm, a filarial worm, and two plant parasitic species). The premise of that work was that certain enzymes in the metabolic network form nodes that are especially vulnerable and hence are attractive candidates for drug-targeting. Specifically, targeting the enzymes that either uniquely produce or uniquely consume a substrate, called ‘chokepoint enzymes’10 is expected to harm the parasite by changing the concentration of the substrate to either too low or too high, potentially leading to morbidity or mortality. If carefully selected, employing insight into the biology and metabolic requirements of both the host and the pathogen, such targets have the potential to selectively harm the parasites without unacceptable side effects for the host. Based on the target and hit prioritization, seven compounds were selected and assayed in vitro in three highly divergent nematodes - C. elegans, a hookworm and a filarial species. Perhexiline, a putative inhibitor of carnitinepalmitoyl transferase (CPT) was effective against all three species. This work formed the basis of further exploration of putative CPT inhibitors repurposed for their anthelmintic potential11.
One limitation of the previous studies was the small number of nematode genomes available at the time, especially for parasitic worms. Moreover, even among the 10 genomes that were used, five belonged to the free-living genus Caenorhabditis. Fortunately the total number of nematode genomes and the diversity represented in those species has substantially increased in recent years, particularly with the publication of a large number of helminth genomes and their comparative analyses by International Helminth Genomes Consortium (IHGC12). Among the multiple analyses reported by IHGC was a comparison of the metabolic potential of nematodes, requiring us to develop a comprehensive method for enzyme annotation, and generate pathway reconstructions13,14,15, resulting in novel insights and confirmation of previous results that can be important for drug discovery and for understanding parasite biology and host-parasite interactions. The genomic data now available to us provide an opportunity to expand upon previous studies using a more representative set of parasitic worms for prioritizing chokepoint enzymes as drug targets and advancing the chemogenomic analysis, and phenotypic and in vivo assays.
Our primary objective was to identify drugs effective against whipworm and hookworm, which are among the most important human parasites. Moreover, we also aimed at covering ever broader phylogenetic and physiological range. Therefore, in this study, we strategically selected a set of 17 representative parasitic nematode species, spanning 4 of the 5 nematode phyla, including both intestinal and filarial worms, and used systems biology and evolutionary principles to reconstruct metabolic networks for these diverse parasites11,13,15. We were able to carry out more accurate gene- and single nucleotide-level comparative studies leveraging the recent advancements in genomic resources and metabolic databases. For instance, comparing a potential target gene among a diverse group of parasites and with the host gene sequence, critical sequence variations may be identified which are specific to the parasites, or are highly conserved among nematodes yet sufficiently divergent from the host16. Such omics-driven knowledge-based target prioritization approach17,18 followed by chemogenomic screening using large-scale drug databases (e.g. ChEMBL19) facilitated the identification of drug-like compounds with broad-spectrum control potential. We selected representative intestinal parasites at the extremes of the phylogeny20 (whipworm Trichuris muris from clade I of Nematoda and hookworm Ancylostoma duodenale from clade V) along with a phylogenetically distant lymphatic parasite (filarial Brugia pahangi from clade III) to conduct phenotypic screening of adult developmental stages of these worms to validate our predictions. The approach enabled the identification of inhibitors of key chokepoint enzymes that shared potentially pan-intestinal and pan-phylum efficacy. We also pursued iterative phenotypic screening guided by structure-activity relationships (SAR) to expand our list of lead compound candidates. The in vivo effect of representative compounds was also tested.
Results and Discussion
Identifying and prioritizing the metabolic enzyme chokepoints in parasitic nematodes
The overall analysis process is outlined in Fig. 1 (for more details see Supplementary Fig. S1). A total of 669 unique ECs (Enzyme Commission IDs) were identified across 17 nematode species. Analysis of individually reconstructed metabolic networks led to identification of 389 ECs that were chokepoints in at least one species (Supplementary Table S1). Among these, 186 were taxonomically conserved20 across at least Trichuris muris (Clade I), Ancylostoma ceylanicum (Clade V) (the two species used for screening – “target species”) and at least one other Clade III or Clade IVa species (to ensure broad nematode conservation).

Flowchart outlining the overall analysis pipeline. ww = ”whipworm”.
These 186 chokepoint ECs were ranked based on a weighted scoring method (Supplementary Fig. S1; Methods) which included phylogenetic conservation, number of metabolic pathways they are involved in, RNAi phenotype of the C. elegans ortholog, previous identification in literature, and RNAseq based expression levels in especially relevant developmental stage(s). Every nematode chokepoint from the two target species - intestinal nematodes representing the phylogenetic extremes of the phylum Nematoda (clade I, the whipworm T. muris and clade V, the hookworm A. ceylanicum) - was linked to ‘drug-like’ compounds in the ChEMBL19 database based on similarity to ChEMBL targets, resulting in 448 and 606 unique ChEMBL targets corresponding to 664 A. ceylanicum proteins and 756 T. muris proteins, respectively. Using pChEMBL19 values a total of 188,454 target:compound pairs were identified (pChEMBL score ≥5, corresponding to 10 μM IC50 etc.). Of these, 83,134 pairs included both a gene with an EC assignment and a compound with a “Quantitative Estimate of Druglikeness” (weighted QED) score recorded in the database. Intersection of these with the 186 selected nematode chokepoint ECs resulted in 22,498 conserved chokepoint ChEMBL target:compound pairs (82 targets/64 ECs paired to 12,395 unique compounds). The final ChEMBL target:compound scores were calculated, prioritizing target:chokepoint pairs that have high drug likeness and high target affinity. Multiplying the final nematode chokepoint EC score and the final ChEMBL target:compound score (Supplementary Fig. S1C) resulted in the final prioritization score. Thereafter, the 22,498 scored compound:target pairs (64 ECs) were reduced to 11,869 pairs (50 ECs) with a PDB21 (Protein Data Bank) structural match to the target. In the final step, the top 3 target:compound pairs per EC were selected (142 total target:compound pairs, 128 compounds; Supplementary Fig. S1D, Supplementary Tables S2, S3), and these were ranked according to the final prioritization score.
Out of the top 50 ECs, 10 ECs were prioritized based on associated compound availability and guided by literature review of the EC functions and potential for biological efficacy (Table 1). These ECs were categorized into five classes. The first class of target ECs was dehydrogenases, and included (i) L-lactate dehydrogenase (LDH; 1.1.1.27), previously suggested to be a potential anthelmintic target in Clonorchis sinensis22 and Taenia asiatica23. A wide variety of existing effective anthelmintics including Praziquantel, Levamisole, and Mebendazole have been found to result in LDH inhibition in trematode family Paramphistomidae24; (ii) Malate dehydrogenase (MDH; 1.1.1.37), inhibited by several major benzimidazole anthelmintics including Albendazole, although it is not their primary target25; (iii) Aldehyde dehydrogenase (NAD+) (ALDH; 1.2.1.3), previously explored as a potential target due to its impact on alcohol tolerance and cancer chemotherapy resistance. ALDH may be related to broad based drug resistance, and its inhibition might facilitate efficacy of other drugs26 and (iv) Dihydroorotate dehydrogenase (quinone) (DHODH; 1.3.5.2), studied as a target for protozoan (malaria and trypanosome) parasites27,28,29,30. The second class of EC targets was kinases, and included (i) Hexokinase (HK; 2.7.1.1), a possible target of phenothiazine, which had been widely used in the mid-20th century as an anthelmintic31; (ii) Pantothenate kinase (PanK/CoA; 2.7.1.33), which is involved in CoA biosynthesis and possibly available at high levels in cells32 and (iii) 1-phosphatidylinositol 4-kinase (PI4K; 2.7.1.67), with several inhibitors under studies against malaria33 and Cryptosporidium34. The other classes were represented by one EC each, and included Fructose-bisphosphatase (FBPase; 3.1.3.11), a key enzyme in gluconeogenesis that has not been identified as a target in helminths; 3′,5′-cyclic-nucleotide phosphodiesterase (PDE; 3.1.4.17), inhibitors for which have been suggested to be good potential drugs for targeting parasitic nematodes due to their ability to disrupt the C. elegans life cycle and nematode-specific active binding sites35; and Leucyl aminopeptidase (LAP-1/cathepsin III; 3.4.11.1), a potential drug target in Leishmania32, known to be an excretory/secretory (ES) product and a modulator of host immune system36, and previously studied as a potential vaccine candidate for the liver fluke, Fasciola hepatica37,38.
First generation compound screening with parasitic nematodes in a phenotypic motility assay
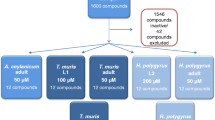
To test the knowledge-based compound predictions for the 10 prioritized ECs (15 ChEMBL targets; 2,167 compound:target pairs; 1,581 drugs), 94 compounds were purchased and assayed in adult hookworm. Eleven (Table 2) had deleterious effects in hookworm (Motility Index, MI ≤0.75 for severe phenotype, 0.76 to 1.8 for moderate phenotype or had noticeable phenotype; 1.81 to 3, no phenotype) and were putatively targeting six of the 10 enzymes (success rate of 60% based on the target numbers), representing 3 different enzyme classes (2 dehydrogenases, 3 kinases, and a phosphodiesterase; Table 2). The 11 compounds causing severe or moderate phenotype in hookworm were also assayed in whipworm and seven were hits (i.e. had moderate to severe motility inhibition), hence classified as potential pan-intestinal compounds. Four of the seven compounds were potential pan-intestinal hits resulting in severe phenotypes (MI < 1.3) in both species, and two (33 and 69) had severe phenotype only in whipworm. One compound (9) showed moderate/noticeable phenotype in both species. Based on our analysis (Supplementary Fig. S1) the active compounds were linked to drug-like compounds shown to target MDH, PDE and PI4K/PI3K/mTOR in other species. The eight compounds (out of the 11 hits) associated with these enzymes (Table 2) were screened in non-intestinal nematode, adult stage of the filarial nematode Brugia pahangi, to examine their pan-phylum potential. Four (15, 47, 58, 59) were effective at day 2, and 3 additional (20, 30, 33) at day 6 in B. pahangi, hence showing pan-Nematoda potential (motility inhibition of at least 80% at day 2 or day 5 was used as a cut-off for activity).
Identification of second generation compounds with pan-phylum potential and screening with parasitic nematodes in a phenotypic motility assay
Based on the results from the 94 first generation compounds we identified three effective target classes - MDH, PDE and PI4K/PI3K/mTOR - and expanded the compound list by structural similarity and pharmacophore searches, resulting in identification and screening of 26 second generation compounds (Supplementary Table S4). This compound expansion and screening in the intestinal and filarial species led to the identification of four (102, 105, 108, 113) further active potential pan-intestinal molecules, of which two (102 and 105) have pan-Phylum molecules (Fig. 2, Supplementary Table S4) in this series. The putative target-associated compounds that were not active in this assay, were not necessarily structurally related to the first-generation inhibitors.

Structural similarity of the 17 active compounds, identified in the primary, secondary and tertiary screen of compounds for the identified targets. The clustering was based on (1 − Tanimoto similarity measure) as distance metric, calculated using ChemmineR105 package, and agglomerated using “complete linkage” method.
Overall the screening of the first and second generation compounds resulted in identification of twelve active compounds that putatively target three homologous therapeutic targets - MDH, PI3K/mTOR and PDEs (Table 3) in at least one parasitic species.
Screening of known azoles
Four of our active first-generation compounds were prioritized based on their known activities against MDH39,40 although they have numerous discrete canonical targets (Table 3). Gossypol is a polyphenol known to target lactate dehydrogenase41,42 and BCL2 family proteins involved in apoptosis and APE-1, which participates in DNA damage repair43,44,45. The other three were azoles (Miconazole, Econazole and Sulconazole), which are imidazole derivatives known to be antifungal agents. They inhibit P-gp and multiple CYP proteins. One of the CYP proteins (14 alpha-demethylase) is thought to be the main target that leads to antifungal activity by inhibiting synthesis of ergosterol, an important steroid necessary for permeability of the fungal cellular membrane46,47. These azoles also display other activities, including inhibition of certain ion channels and receptors48,49. We included Butaconazole, also an azole, among our second-generation compounds based on the successful motility inhibition shown by these three azoles. Like the other three azoles, Butaconazole is also predicted to inhibit 14 alpha-demethylase48. In our in vitro assays, Sulconazole had IC50s of 11.8 µM against the whipworm T. muris and 4.6 µM against the filarial worm B. pahangi.
Screening of known PI3K, PI4K lipid kinases and mTOR inhibitors
Our discovery that the mixed mTOR/PI3K/PI4K inhibitor Torin1 (15) showed efficacy in inhibiting worm motility in both hookworms and whipworms prompted us to test other known and more selective inhibitors of each of these kinases. The high prioritization score of 15 was based on its high potency as an inhibitor of mTOR with an IC50 of 2 nM and 10 nM in the protein complexes mTORC1 and mTORC2, respectively50. In the same study, it exhibited 1000-fold selectivity of mTOR over PI3K in mammalian cells. In our assays, the IC50 for 15 was 13.3 µM in the whipworm T. muris and 3.9 µM in the filarial worm B. pahangi. Intriguingly, the structurally related inhibitor Torin-2 (23) did not exhibit similar activity, nor did the other PI4K kinase inhibitors (95, 106, 109, 116, and 120) (Supplementary Table S4). Therefore, we identified and evaluated additional mTOR inhibitors and found that Omipalisib51 (105) and WYE-35452 (113) showed equivalent or better activity against both these nematodes (Fig. 2). Interestingly, 105 showed better activity than 113, a more selective inhibitor of mTOR. Because the structurally related inhibitor Torin-2 did not exhibit activity and Omipalisib did, we screened more selective PI3K inhibitors and additional mTOR inhibitors to help better indicate the mechanism of action (Supplementary Fig. S3). The three additional drugs were: 1. CC-223 (124): a potent, selective, and orally bioavailable mTOR inhibitor with IC50 of 16 nM, >200-fold selectivity over the related PI3K-α53, 2. MLN1117 (Serabelisib; 125): selective p110α (a PI3K) inhibitor with an IC50 of 15 nM54, and 3. XL388 (126): a highly potent ATP-competitive mTOR inhibitor with an IC50 of 9.9 nM55 which simultaneously inhibits both mTORC1 and mTORC2. None of the three showed significant activity. Dual inhibitors such as PF-04979064 exist56 and testing them can provide further clarification.
Screening of known PDE inhibitors
We identified a weak PDE1/PDE5 inhibitor (69) (IC50 1.9 µM/0.7 µM respectively57) in the worm assays and, subsequently, a more potent inhibitor Tadalafil58 (108) among the second-generation compounds. The additional seven PDE inhibitors tested (Supplementary Table S4) are from several chemical series, which encompass varied PDE family selectivity profiles. Intriguingly, Tadalafil was the most active PDE inhibitor, while other classes including Sildenafil (110) and Vardenafil (89) did not show activity against the representative hookworm or whipworm (Supplementary Table S4). The compounds we tested so far are largely selective for PDE5, PDE1 and PDE6 although our lead Tadalafil also shows significant activity for PDE11 (IC50 15 nM)59, for which a direct ortholog is not found in nematodes even though PDE5 is the most closely related nematode enzyme to mammalian PDE11s (Supplementary Fig. S4). Tadalafil is a PDE5 inhibitor with IC50 of 9.4 nM and was at least 1000 times more selective for PDE5 than most of the other families of PDEs, with the exception of PDE1158. Subsequent screening of BC-11-38 (123), a selective PDE11 inhibitor60, showed no activity against whipworm (Supplementary Table S4). To further explore the Tadalafil scaffold, and to test the hypothesis that we are hitting PDE (and that worm PDE is similar to human), Tadalafil and N-cyclopropyl Tadalafil were docked into A. ceylanicum PDE5 homology model (Tadalafil docking shown in Fig. 3a). Acknowledging that uptake may be differentiated, we chose two compounds N-Desmethyl Tadalafil (121) and N-Desmethyl N-cyclopentyl Tadalafil (122) with varying ClogP (1.864 and 3.7944, respectively) for screening in whipworm. Both compounds showed activity (Supplementary Table S4). We note that most of the residues important for interactions with Tadalafil are conserved among Nematoda, but are divergent from the hosts (Fig. 3b). Additional studies are needed to determine the target, in light of the difference in PDE geneset among species (e.g. lack of PDE5 and two additional PDE2 in whipworm; Supplementary Fig. S4) and the PDE expression profiles in adult worms (Fig. 3c; Supplementary Table S5). Both 69 and 108 were not active against the filarial nematode B. pahangi. Taxonomically restricted residues in PDE5 exist among intestinal and filarial nematodes (Fig. 3c) and further focused studies are needed to determine their role in the observed differential activity.

Characterizing PDE as a potential target (a) Tadalafil docked to human (magenta) and A. ceylanicum (cyan) PDE5 homology model, showing the interacting residues. (b) Sequence alignment of PDE5 homologs from 6 worms (Ac = A. ceylanicum, Na = N. americanus, Ce = C. elegans, Bm = B. malayi, Ov = O. volvulus, Wb = W. bancrofti) and 3 paralogs each from the hosts human (Hs) and mouse (Mm). No homolog was identified in T. muris. The region shown here is selected to illustrate that most of the interacting residues from panel A are conserved among nematodes and divergent from host species (indicated with red asterisks). Residues at or near the active site that are different between filarial and non-filarial worms are highlighted with a black asterisk. The residue numbers are based on A. ceylanicum PDE5. The residues matching the aligned residue in A. ceylanicum are colored, with the color depicting the residue type. (c) A maximum-likelihood phylogenetic tree based on sequence similarity of PDEs present in A. ceylanicum, T. muris and C. elegans, and their expression levels in adult stages (FPKM – fragments per kilobase per million mapped reads - plotted as bars in the outer track). PDE family clusters are indicated using shaded ellipses. The sequences were aligned using MAFFT106. The phylogenetic tree is estimated using PhyML 3.0107 and the node support values are calculated using “aLRT SH-like” option. All node support values are >=0.79.
Putative nematode targets are involved in diverse metabolic pathways
The compounds that caused the most severe phenotypes putatively target three nematode enzymes that are involved in six metabolic pathways. PIK (EC2.7.1.67) and PDE (EC3.1.4.17) are involved in inositol phosphate metabolism and purine metabolism, respectively and MDH (EC1.1.1.37) is involved in four other pathways (described below).
PIK is a transferase that transfers phosphorus-containing groups (phosphotransferases with an alcohol group as acceptor) which is involved in carbohydrate metabolism (inositol phosphate metabolism). A previous study has identified another enzyme in the inositol phosphate metabolism pathway, inositol polyphosphate multikinase (IPMK, EC2.7.1.151), as a druggable and lethal target in kinetoplastid parasites61, showing vulnerability of this pathway in other parasites.
PDEs are hydrolases that cleave ester bonds (phosphoric-diester hydrolases) and are involved in purine nucleotide metabolism. Despite the widespread notion that nematodes cannot synthesize purines62, complete or near-complete purine synthesis pathways were recently found in most members of clades I, IIIb (Ascaridomorpha) and V12. This critical function highlights purine metabolism as a promising drug target, as evidenced here by the efficacy of targeting PDE; notably, inhibitors of PDE have been suggested as promising potential drugs against parasitic nematodes given their ability to disrupt the C. elegans life cycle and existence of nematode-specific active binding sites35. Ten other high priority chokepoints are part of the purine metabolism pathway (Supplementary Table S6). For example, previously we have prioritized pyruvate kinase (EC 2.7.1.40), an enzyme upstream of PDE, as a top promising conserved chokepoint drug target,1 and due to its critical function and diversification in the active site compared to the human host it has also been identified as a promising drug target for malaria63 and Staphylococcus aureus64. Pyruvate kinase is also a chokepoint in pyruvate metabolism pathway.
MDH is an oxidoreductase that acts on the CH-OH group of donors with NAD+ as acceptor. It is involved in three carbohydrate metabolism pathways (pyruvate, glyoxylate/dicarboxylate, and TCA cycle) and also in amino acid metabolism (cysteine and methionine). It is essential to energy production through both aerobic and anerobic pathways, making it essential for parasite’s survival even in very diverse niches. Some anthelmintics appear to target MDH activity65 and it was originally thought to be the possible target of benzimidazole anthelmintics, with Mebendazole shown to inhibit MDH activity in vitro66. Multiple enzymes from carbohydrate metbolism and glycolytic pathways, including MDH, have been studied for their potential as anthelmintic targets67,68,69. There are 7 other enzymes among our top 50 chokepoints list (Supplementary Table S2) that are also part of pathways that involve MDH (4 in pyruvate metabolism and 4 in cysteine/methionine metabolism; one enzyme - EC 1.1.1.27 - is involved in both these pathways). Many of these have been suggested and explored as potential anti-parasitic, antifungal or anthelmintic drugs63,70,71,72,73,74,75,76,77,78,79,80.
In vivo efficacy of lead compounds
To ascertain whether the newly discovered classes of pan-nematode chokepoint inhibitors possessed therapeutic utility in parasitic nematodes, Syrian hamsters (Mesocricetus auratus) infected with A. ceylanicum were treated with representative drugs from each of the three target classes at 100 mg/kg per os, based on previously reported effective dosage of currently used major anthelmintics81. Three (Butaconazole, Tadalafil and Torin-1) of the four tested compounds that were active in vitro had significant and noteworthy impact on eggs per gram of feces indicating treatment affected parasite fecundity, although the treatment had no marked effect on worm burden (Fig. 4a,b). Rolipram, a drug that was not active in vitro, did not have an impact on eggs per gram or on worm burden (Fig. 4c,d).

Butaconazole, Tadalafil, Torin-1 reduced fecal egg count but not total worm load and rolipram did not reduce either fecal egg count or total worm load. (a) Treatment with each of the three drugs significantly reduced fecal egg count in Syrian hamsters infected with the hookworm A. ceylanicum compared with untreated control animals. (b) The fecal egg count reduction was not accompanied by a reduction of worm load, which was not statistically significant between control and treated animals. No reduction in fecal egg count (c) or worm burden (d) in Syrian hamsters infected with the hookworm A. ceylanicum when treated with rolipram compared to control animals.
In conclusion, omics-driven knowledge-based identification of drug targets is a powerful approach to identify targets essential for organisms survival. Undertaking this approach we were able to prioritize 10 of the 186 identified conserved chokepoint enzymes and undertake a target class repurposing approach to test and identify drugs with broad spectrum anthelmintic activity. First, we tested 94 compounds using an in vitro phenotypic assay, and identified 11 actives. Based on the findings and SAR, we tested 32 additional compounds, identifying 6 more actives. Among the 17 actives 5 are potential pan-intestinal and 6 are potential pan-Nematoda drugs. The active compounds target three different target classes. Using representative drugs from each target class, we demonstrated efficacy in hamsters infected with hookworm, as characterized by negative effects on parasite fecundity. While the leading scaffolds require optimization, the study identified target classes and essential pathways for parasitic nematode survival.
Materials and Methods
Enzyme annotation, nematode chokepoints identification and taxonomic classification
High confidence annotation of nematode metabolic enzymes was performed as described12. In short, the multi-step process assigns Enzyme Commission (EC) numbers to sequences in proteomes of interest. Multiple EC-annotation approaches that were employed included KAAS82, PRIAM83, DETECTv284 and BRENDA85. For species with good quality draft genomes (including the representative intestinal species, T. muris and A. ceylanicum), the pathway hole-filling algorithm of the Pathway Tools package86 was used to identify additional metabolic enzymes.
Identification and prioritization of nematode chokepoint targets and drug
For nematode chokepoint EC prioritization we first quantified pan-Nematoda chokepoint conservation by performing chokepoint analysis on 17 species spanning the phylum (Supplementary Fig. S1 and Supplementary Table S1): Ancylostoma ceylanicum87, Ancylostoma duodenale12, Ascaris lumbricoides12, Ascaris suum88, Brugia malayi89, Dirofilaria immitis90, Heligmosomoides bakeri12, Loa loa91, Necator americanus92, Nippostrongylus brasiliensis12, Onchocerca volvulus87, Strongyloides ratti93, Strongyloides stercoralis93, Trichuris muris94, Trichuris suis12, Trichuris trichiura94, and Wuchereria bancrofti12. Second, chokepoint ECs were assigned a score with a maximum value of 6, based on five different criteria: (i) ECs with higher levels of pan-Nematoda conservation received a higher score, calculated using (2× [Number of species/17]), with a maximum value of 2; (ii) Chokepoints annotated to multiple KEGG95 pathways received a higher score, calculated using (number of pathways/4), to a maximum value of 1; (iii) If the C. elegans ortholog has a “lethal” or “sterile” phenotype annotated by WormBase96, it received 1 point; (iv) The chokepoints among the top 25% targets scored in the “Comparative genomics of the major parasitic worms” study (International Helminth Genomes Consortium12) received a score calculated by (score/137), maximum value 1. This score was based on multiple factors, including lack of human homolog, presence of a C. elegans or Drosophila melanogaster homolog with a deleterious phenotype, expression in key life cycle stages, widespread conservation among helminths, lack of paralogs, presence of a related structure in PDB, etc. The top quartile among all the scored proteins were given a positive score by us, as they represented potentially good anthelmintic targets; and (v) RNA-Seq-based gene expression levels were used to prioritize ECs based on data from A. ceylanicum (0.5 x the percentile of the EC’s adult-stage expression compared to all other genes) and T. muris (0.5 x the percentile of the EC’s [adult stage/free-living larval stages] fold change value compared to all other genes), with a maximum value of 1. The distribution of all of the contributing scores and the final scores are shown in Supplementary Fig. S2.
For the ChEMBL target:drug pair prioritization, ChEMBL19 targets were annotated among the two target species (minimum 40% identity across 75% of the protein’s length, P ≤ 10−10) and subsequently ChEMBL was used to match drugs, identifying target:drug pairs with a pChEMBL score ≥5. The drugs that had a “Quantitative Estimate of Druglikeness” (weighted QED) score available and were associated with one of the prioritized 186 nematode metabolic chokepoints were identified. The final ChEMBL target:drug scores were calculated by multiplying (i) the pChEMBL drug:target affinity scores, scaled between 0.5 and 1, and (ii) the weighted QED scores, scaled between 0 and 1. Final prioritization score was calculated by multiplying the final nematode chokepoint EC score and the final ChEMBL target:drug score (Supplementary Fig. S1). Last, significant sequence similarity to either a human or a mouse protein with a structure in PDB (sequence match with an E value < 10−10) was required for a drug:target pair to be included for further consideration.
For each of the 50 target ECs with available PDB structure, the nematode sequence was aligned with that of the PDB structure using MOE (Chemical Computing Group, Inc.). A two-step visual inspection of the ligand binding site ranked targets based on (i) identification of a ‘druggable’ binding site97, with compound bound where possible, and (ii) protein sequence differences between the nematode and host sequences in the region of the binding site (this step was preceeded by manual improvement of the nematode gene models guided by transcriptional evidence). This process resulted in 10 priority targets.
Identification of small molecule inhibitors
First generation compounds: For the 10 prioritized EC targets, we identified all reported active compounds in ChEMBL. These were assessed for drug-likeness; PAINS98 were eliminated. Of these, 94 compounds were available for purchase. Both hookworm and whipworm assays were carried out for an initial batch of 30 compounds. Based on the results of these, it was decided to employ a stepwise approach for the remaining first generation compounds i.e. hookworm, then whipworm, then Brugia, contingent on activity in the previous step(s). Second generation compounds: Based on the results from 94 first generation compounds, five proteins were reasoned as the potential biologically relevant targets for our active hits and thus more readily commercially available compounds known to target each of these were included (n = 26). These were five PI4K/PI3K inhibitors, nine mTOR inhibitors, five PDE inhibitors, two potential malate dehydrogenase inhibitors, and five Econazole analogs. The mTOR inhibitors were included based on CHEMBL1256459 (Torin1) - a potent inhibitor of mTORC1/2. Third generation compounds: Based on the results from the 26 second generation compound, additional compounds were identified and screened (n = 6). These included three PDE inhibitors, two mTOR and one PI3K (p110α) inhibitor.
Compound screening with adult stage of intestinal nematode Ancylostoma ceylanicum
In vitro assays were carried out as described81. Briefly, young adult A. ceylanicum were harvested from the small intestine of infected hamsters on day 11 post-inoculation (p.i.) and washed three times using prewarmed medium (RPMI1640, 100 U penicillin, 100 μg/ml streptomycin, 10 μg/ml amphotericin). Three worms were manually picked into each well of the 96-well, 4 wells/test drug, containing 99 μl of medium. 1 μl of 3 mM (100% DMSO) drugs were transferred into the corresponding well giving a final concentration of 30 µM (1% DMSO) and 100 µl final volume. Assay plates were incubated for 48 hours at 37 °C and 5% CO2. Drug activity was determined every 24 hours using the standard motility index. Motility index of 3 were given to vigorous worms, 2 for motile worms, 1 for motile after stimulation by touching and 0 for dead worms. Pyrantel pamoate 30 µM was used as positive control prepared exactly as the test drugs, while 1% DMSO was used as negative control.
Compound screening with adult stage of intestinal nematode Trichuris muris
In vitro assays were carried out as described81. Briefly, T. muris adult whipworms were harvested from the cecum and large intestines of infected STAT6 −/− mice. Three worms were manually picked into each well of a 24-well plate, 2 wells/ test drug, containing 990 µl. 10 µl of 3 mM (100% DMSO) drugs were transferred into the corresponding well giving a final concentration of 30 µM (1% DMSO) and a final volume of 1 ml of medium. Test plates were incubated for 48 hours at 37 °C and 5% CO2. Drug activity was assayed using the standard motility index as per hookworm adults. Levamisole 30 µM was used as positive control prepared exactly as the test drugs, while 1% DMSO was used as negative control. IC50 values at 48 hours were established using a non-linear regression with Prism 7 (Graphpad, La Jolla, CA). Only IC50s displaying R2 values ≥ 0.7 are reported.
Compound screening with adult stage of the filarial nematode Brugia pahangi
Adult female B. pahangi worms were obtained from Dr. Brenda Beerntsen, University of Missouri, Columbia, MO. Individual females were placed in each well of a 24-well plate in culture medium (RPMI-1640 with 25 mM HEPES, 2.0 g/L NaHCO3, 5% heat inactivated FBS, and 1X Antibiotic/Antimycotic solution.
Initial screening of the compounds was performed at 10 μM for Miconazole, Econazole, and Sulconazole. Gossypol, Torin2, PP121, and Torin1 were initially screened at 30 μM and 10 μM. Four worms were used as replicates at each concentration in 500 uL of media. Worms treated only with 1% DMSO served as the negative control. The cultures were maintained at 37 °C, 5% CO2 incubator for seven days - the duration of the assay.
The Worminator, a visual imaging software, was used to determine the effect of each treatment on worm motility99. Movements of individual worms were calculated by determining the number of pixels displaced per second per well. Worm movements were averaged over the time of recording (60 seconds) and mean movement units (MMUs) were determined for individual worms. Percent inhibition of motility was calculated by dividing the MMUs of the treated worms by the average MMUs of the 1% DMSO treated control worms, subtracting the value from 1.0, flooring the values to zero and multiplying by 100%. Videos of the worms during the assay were recorded on days 0, 1, 2, 3, and 6 of the incubation. Compounds showing ≥ 75% inhibition of motility at either concentration on day 3 of the assay were investigated further to determine IC50s. Worms were treated in the same fashion as above and motility was measured each day for three days using the Worminator. IC50 values on day 3 (Torin 1 and Sulconazole) were established using a non-linear regression curve fit with Prism 7 (Graphpad, La Jolla, CA). All IC50s with R2 values ≥ 0.7 are reported.
In vivo A. ceylanicum assay
Assays were conducted essentially as previously81,100. Briefly, 5 weeks old golden Syrian hamsters were infected with 120 third-stage larvae of A. ceylanicum. Infected rodents were grouped based on the fecal egg count on day 18 p.i. Each group had five hamsters. Drugs were delivered in water suspension after completely dissolved in 100% DMSO giving a final dose of 100 mg/kg and 0.1% DMSO volume to body weight. Control group was dosed only with water and 0.1% DMSO. Fecal samples were collected 21 days p.i. for fecal egg counts. On day 22 p.i., the hamsters were euthanized, and the small intestine was opened longitudinally. Adult parasites within the intestine were counted under a dissecting microscope.
A 100 mg/kg dosing for in vivo experiments was chosen based on similar studies from other groups as maximum single dosing for anthelmintic compounds81,101,102,103. For comparison, with current clinically approved anthelmintics high efficacy is seen with doses at 5–10 mg/kg103,104.
Ethics statement
All animal experiments were carried out under protocols approved by University of Massachusetts Medical School (UMMS; A-2483 and A-2484) Institutional Animal Care and Use Committees (IACUC). All housing and care of laboratory animals conformed to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals in Research (see 18-F22) and all requirements and all regulations issued by the United States Department of Agriculture (USDA), including regulations implementing the Animal Welfare Act (P.L. 89–544) as amended (see 18-F23). Euthanasia was accomplished by CO2 asphyxiation, followed by bilateral pneumothorax.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Taylor, C. M. et al. Discovery of anthelmintic drug targets and drugs using chokepoints in nematode metabolic pathways. PLoS Pathog 9, 1 (2013).
Bayly, A. M. & Macreadie, I. G. Folic acid antagonism of sulfa drug treatments. Trends Parasitol 18, 49–50 (2002).
Timmins, G. S. & Deretic, V. Mechanisms of action of isoniazid. Mol Microbiol 62, 1220–1227, https://doi.org/10.1111/j.1365-2958.2006.05467.x (2006).
Bolt, B. J. et al. Using WormBase ParaSite: An Integrated Platform for Exploring Helminth Genomic Data. Methods Mol Biol 1757, 471–491, https://doi.org/10.1007/978-1-4939-7737-6_15 (2018).
Umeda, T. et al. Molecular basis of fosmidomycin’s action on the human malaria parasite Plasmodium falciparum. Sci Rep 1, 9, https://doi.org/10.1038/srep00009 (2011).
Martin, J., Tyagi, R., Rosa, B. A. & Mitreva, M. A Multi-Omics Database for Parasitic Nematodes and Trematodes. Methods Mol Biol 1757, 371–397, https://doi.org/10.1007/978-1-4939-7737-6_13 (2018).
Raso, G. et al. Multiple parasite infections and their relationship to self-reported morbidity in a community of rural Cote d’Ivoire. Int J Epidemiol 33, 1092–1102, https://doi.org/10.1093/ije/dyh241 (2004).
Bethony, J. et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet 367, 1521–1532, https://doi.org/10.1016/S0140-6736(06)68653-4 (2006).
Vercruysse, J. et al. Assessment of the anthelmintic efficacy of albendazole in school children in seven countries where soil-transmitted helminths are endemic. PLoS Negl Trop Dis 5, e948, https://doi.org/10.1371/journal.pntd.0000948 (2011).
Yeh, I., Hanekamp, T., Tsoka, S., Karp, P. D. & Altman, R. B. Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Res 14, 917–924, https://doi.org/10.1101/gr.2050304 (2004).
Tyagi, R. et al. Small Molecule Inhibitors of Metabolic Enzymes Repurposed as a New Class of Anthelmintics. ACS. Infect Dis 4, 1130–1145, https://doi.org/10.1021/acsinfecdis.8b00090 (2018).
International Helminth Genomes, C. Comparative genomics of the major parasitic worms. Nat Genet, https://doi.org/10.1038/s41588-018-0262-1 (2018).
Tyagi, R., Rosa, B. A., Lewis, W. G. & Mitreva, M. Pan-phylum Comparison of Nematode Metabolic Potential. PLoS Negl Trop Dis 9, e0003788, https://doi.org/10.1371/journal.pntd.0003788 (2015).
Seshadri, S., Tyagi, R., Mitreva, M. & Parkinson, J. Annotating metabolic enzymes in parasitic worm proteomes. https://doi.org/10.1038/protex.2018.1048 (2018).
Tyagi, R., Seshadri, S., Parkinson, J. & Mitreva, M. Comparative analysis of metabolism in parasitic worms. Nature, Protocol Exchange, https://doi.org/10.1038/protex.2018.1047 (2018).
Wang, Q. et al. Characterization of parasite-specific indels and their proposed relevance for selective anthelminthic drug targeting. Infect Genet Evol 39, 201–211, https://doi.org/10.1016/j.meegid.2016.01.025 (2016).
Taylor, C. M. et al. Using existing drugs as leads for broad spectrum anthelmintics targeting protein kinases. PLoS Pathog 9, e1003149, https://doi.org/10.1371/journal.ppat.1003149 (2013).
Wang, Q. et al. Targeting Lysine Deacetylases (KDACs) in Parasites. PLoS Negl Trop Dis 9, e0004026, https://doi.org/10.1371/journal.pntd.0004026 (2015).
Bento, A. P. et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res 42, 7 (2014).
Blaxter, M. L. et al. A molecular evolutionary framework for the phylum Nematoda. Nature 392, 71–75, https://doi.org/10.1038/32160 (1998).
Rose, P. W. et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Research 41, D475–D482, https://doi.org/10.1093/nar/gks1200 (2013).
Yang, G. et al. Molecular cloning and characterization of a novel lactate dehydrogenase gene from Clonorchis sinensis. Parasitology Research 99, 55, https://doi.org/10.1007/s00436-005-0125-4 (2006).
Huang, J. et al. Identification, expression, characterization, and immunolocalization of lactate dehydrogenase from Taenia asiatica. Parasitol Res 104, 287–293, https://doi.org/10.1007/s00436-008-1190-2 (2009).
Veerakumari, L. & Munuswamy, N. In vitro effect of some anthelmintics on lactate dehydrogenase activity of Cotylophoron cotylophorum (Digenea: paramphistomidae). Veterinary parasitology 91, 129–140 (2000).
Sanchez-Moreno, M. et al. Malate dehydrogenase in helminth parasites. Inhibition by benzimidazoles and pyrimidine derivatives. Arzneimittel-Forschung 37, 1327–1331 (1987).
Koppaka, V. et al. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev 64, 520–539, https://doi.org/10.1124/pr.111.005538 (2012).
Pinheiro, M. P., Emery Fda, S. & Nonato, M. C. Target sites for the design of anti-trypanosomatid drugs based on the structure of dihydroorotate dehydrogenase. Curr Pharm Des 19, 2615–2627 (2013).
Vyas, V. K. & Ghate, M. Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev Med Chem 11, 1039–1055 (2011).
Phillips, M. A. & Rathod, P. K. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets 10, 226–239 (2010).
Arakaki, T. L. et al. Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target; structural, kinetic and RNAi studies. Mol Microbiol 68, 37–50, https://doi.org/10.1111/j.1365-2958.2008.06131.x (2008).
Allenby, G. M. & Collier, H. B. Enzyme inhibition by derivatives of phenothiazine. V. Inhibition of rat-brain hexokinase by phenothiazone. Can J Med Sci 30, 549–551 (1952).
Crowther, G. J. et al. Identification of attractive drug targets in neglected-disease pathogens using an in silico approach. PLoS Negl Trop Dis 4, e804, https://doi.org/10.1371/journal.pntd.0000804 (2010).
Paquet, T. et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci Transl Med 9, https://doi.org/10.1126/scitranslmed.aad9735 (2017).
Manjunatha, U. H. et al. A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 546, 376–380, https://doi.org/10.1038/nature22337 (2017).
Schuster, K. D., Cahill, K. B., Morris, K., Thomas, W. K. & Cote, R. H. PDE inhibitors as potential pesticides targeting parasitic nematodes. The FASEB Journal 27, 1052.1056 (2013).
Moreau, E. & Chauvin, A. Immunity against helminths: interactions with the host and the intercurrent infections. J Biomed Biotechnol 2010, 428593, https://doi.org/10.1155/2010/428593 (2010).
Lopez-Aban, J. et al. Progress in the development of Fasciola hepatica vaccine using recombinant fatty acid binding protein with the adjuvant adaptation system ADAD. Veterinary parasitology 145, 287–296, https://doi.org/10.1016/j.vetpar.2006.12.017 (2007).
Kueakhai, P. et al. Vaccine potential of recombinant saposin-like protein 2 against Fasciolosis gigantica in mice. Vaccine 31, 5518–5523, https://doi.org/10.1016/j.vaccine.2013.09.027 (2013).
Choi, S. R. et al. Design, synthesis, and biological evaluation of Plasmodium falciparum lactate dehydrogenase inhibitors. J Med Chem 50, 3841–3850, https://doi.org/10.1021/jm070336k (2007).
Seidler, J., McGovern, S. L., Doman, T. N. & Shoichet, B. K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem 46, 4477–4486, https://doi.org/10.1021/jm030191r (2003).
Conners, R. et al. Mapping the binding site for gossypol-like inhibitors of Plasmodium falciparum lactate dehydrogenase. Mol Biochem Parasitol 142, 137–148, https://doi.org/10.1016/j.molbiopara.2005.03.015 (2005).
Tripathi, A. K. et al. An alpha-proteobacterial type malate dehydrogenase may complement LDH function in Plasmodium falciparum. Cloning and biochemical characterization of the enzyme. Eur J Biochem 271, 3488–3502, https://doi.org/10.1111/j.1432-1033.2004.04281.x (2004).
Bauer, J. A. et al. Reversal of cisplatin resistance with a BH3 mimetic, (−)-gossypol, in head and neck cancer cells: role of wild-type p53 and Bcl-xL. Mol Cancer Ther 4, 1096–1104, https://doi.org/10.1158/1535-7163.MCT-05-0081 (2005).
Etxebarria, A., Landeta, O., Antonsson, B. & Basañez, G. Regulation of antiapoptotic MCL-1 function by gossypol: mechanistic insights from in vitro reconstituted systems. Biochem Pharmacol 76, 1563–1576, https://doi.org/10.1016/j.bcp.2008.08.003 (2008).
Qian, C. et al. Identification of a novel potential antitumor activity of gossypol as an APE1/Ref-1 inhibitor. Drug Des Devel Ther 8, 485–496, https://doi.org/10.2147/DDDT.S62963 (2014).
Marriott, M. S. Inhibition of sterol biosynthesis in Candida albicans by imidazole-containing antifungals. J Gen Microbiol 117, 253–255, https://doi.org/10.1099/00221287-117-1-253 (1980).
Reddy, K. K., Singh, S. K., Tripathi, S. K., Selvaraj, C. & Suryanarayanan, V. Shape and pharmacophore-based virtual screening to identify potential cytochrome P450 sterol 14α-demethylase inhibitors. J Recept Signal Transduct Res 33, 234–243, https://doi.org/10.3109/10799893.2013.789912 (2013).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic Acids Res 45, D945–D954, https://doi.org/10.1093/nar/gkw1074 (2017).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46, D1074–D1082, https://doi.org/10.1093/nar/gkx1037 (2018).
Liu, Q. et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem 53, 7146–7155, https://doi.org/10.1021/jm101144f (2010).
Basu, D. et al. The Dual PI3K/mToR Inhibitor Omipalisib/GSK2126458 Inhibits Clonogenic Growth in Oncogenically-transformed Cells from Neurocutaneous Melanocytosis. Cancer Genomics Proteomics 15, 239–248, https://doi.org/10.21873/cgp.20082 (2018).
Zask, A. et al. ATP-competitive inhibitors of the mammalian target of rapamycin: design and synthesis of highly potent and selective pyrazolopyrimidines. J Med Chem 52, 5013–5016, https://doi.org/10.1021/jm900851f (2009).
Mortensen, D. S. et al. Discovery of mammalian target of rapamycin (mTOR) kinase inhibitor CC-223. J Med Chem 58, 5323–5333, https://doi.org/10.1021/acs.jmedchem.5b00626 (2015).
Falasca, M. et al. Class II Phosphoinositide 3-Kinases as Novel Drug Targets. J Med Chem 60, 47–65, https://doi.org/10.1021/acs.jmedchem.6b00963 (2017).
Takeuchi, C. S. et al. Discovery of a novel class of highly potent, selective, ATP-competitive, and orally bioavailable inhibitors of the mammalian target of rapamycin (mTOR). J Med Chem 56, 2218–2234, https://doi.org/10.1021/jm3007933 (2013).
Cheng, H. et al. Discovery of the Highly Potent PI3K/mTOR Dual Inhibitor PF-04979064 through Structure-Based Drug Design. ACS Med Chem Lett 4, 91–97, https://doi.org/10.1021/ml300309h (2013).
Yamazaki, K. et al. Identification of phosphodiesterase-1 and 5 dual inhibitors by a ligand-based virtual screening optimized for lead evolution. Bioorg Med Chem Lett 16, 1371–1379, https://doi.org/10.1016/j.bmcl.2005.11.046 (2006).
Bischoff, E. Potency, selectivity, and consequences of nonselectivity of PDE inhibition. Int J Impot Res 16(Suppl 1), S11–14, https://doi.org/10.1038/sj.ijir.3901208 (2004).
Weeks, J. L., Zoraghi, R., Francis, S. H. & Corbin, J. D. N-Terminal domain of phosphodiesterase-11A4 (PDE11A4) decreases affinity of the catalytic site for substrates and tadalafil, and is involved in oligomerization. Biochemistry 46, 10353–10364, https://doi.org/10.1021/bi7009629 (2007).
Ceyhan, O., Birsoy, K. & Hoffman, C. S. Identification of biologically active PDE11-selective inhibitors using a yeast-based high-throughput screen. Chem Biol 19, 155–163, https://doi.org/10.1016/j.chembiol.2011.12.010 (2012).
Cestari, I., Haas, P., Moretti, N. S., Schenkman, S. & Stuart, K. Chemogenetic Characterization of Inositol Phosphate Metabolic Pathway Reveals Druggable Enzymes for Targeting Kinetoplastid Parasites. Cell chemical biology 23, 608–617, https://doi.org/10.1016/j.chembiol.2016.03.015 (2016).
Harder, A. In Advances in Parasitology Vol. 93 (eds Robin B. Gassoer & Georg Von Samson-Himmelstjerna) 69-94 (Academic Press, 2016).
Chan, M., Tan, D. S. & Sim, T. S. Plasmodium falciparum pyruvate kinase as a novel target for antimalarial drug-screening. Travel medicine and infectious disease 5, 125–131, https://doi.org/10.1016/j.tmaid.2006.01.015 (2007).
Zoraghi, R. et al. Identification of pyruvate kinase in methicillin-resistant Staphylococcus aureus as a novel antimicrobial drug target. Antimicrobial agents and chemotherapy 55, 2042–2053, https://doi.org/10.1128/aac.01250-10 (2011).
Lwin, T. & Probert, A. J. Effect of certain fasciolicides on malate dehydrogenase activity of Fasciola hepatica: A possible biochemical mode of action of hexachlorophene and oxyclozanide. Pesticide Science 2, 121–128 (1975).
Tejada, P., Sanchez-Moreno, M., Monteoliva, M. & Gomez-Banqueri, H. Inhibition of malate dehydrogenase enzymes by benzimidazole anthelmintics. Veterinary parasitology 24, 269–274 (1987).
Vykhrestiuk, N. P. & Khamatova, A. Cytosol malate dehydrogenase in Calicophoron ijimai trematodes and the effect of antiparasitic preparations on its activity. Parazitologiia 17, 397–402 (1983).
Tandon, V. & Das, B. In vitro testing of anthelmintic efficacy of Flemingia vestita (Fabaceae) on carbohydrate metabolism in Rallietina echinobothrida. Methods 42, 330–338, https://doi.org/10.1016/j.ymeth.2007.01.005 (2007).
Parvatham, K. & Veerakumari, L. Drug target prediction using elementary mode analysis in Ascaris lumbricoides energy metabolism. Biotechnology and Bioprocess Engineering 18, 491, https://doi.org/10.1007/s12257-012-0390-x (2013).
Rana, A. K. & Misra-Bhattacharya, S. Current drug targets for helminthic diseases. Parasitol Res 112, 1819–1831, https://doi.org/10.1007/s00436-013-3383-6 (2013).
Penna-Coutinho, J., Cortopassi, W. A., Oliveira, A. A., Franca, T. C. & Krettli, A. U. Antimalarial activity of potential inhibitors of Plasmodium falciparum lactate dehydrogenase enzyme selected by docking studies. PLoS One 6, e21237, https://doi.org/10.1371/journal.pone.0021237 (2011).
Bork, S. et al. Identification of Babesia bovis L-lactate dehydrogenase as a potential chemotherapeutical target against bovine babesiosis. Mol Biochem Parasitol 136, 165–172 (2004).
Krauth-Siegel, R. L., Bauer, H. & Schirmer, R. H. Dithiol proteins as guardians of the intracellular redox milieu in parasites: old and new drug targets in trypanosomes and malaria-causing plasmodia. Angew Chem Int Ed Engl 44, 690–715, https://doi.org/10.1002/anie.200300639 (2005).
Jamal, S. & Scaria, V. Cheminformatic models based on machine learning for pyruvate kinase inhibitors of Leishmania mexicana. BMC Bioinformatics 14, 329, https://doi.org/10.1186/1471-2105-14-329 (2013).
Suliman, H. S., Appling, D. R. & Robertus, J. D. The gene for cobalamin-independent methionine synthase is essential in Candida albicans: a potential antifungal target. Arch Biochem Biophys 467, 218–226, https://doi.org/10.1016/j.abb.2007.09.003 (2007).
Pascon, R. C., Ganous, T. M., Kingsbury, J. M., Cox, G. M. & McCusker, J. H. Cryptococcus neoformans methionine synthase: expression analysis and requirement for virulence. Microbiology 150, 3013–3023, https://doi.org/10.1099/mic.0.27235-0 (2004).
Jastrzebowska, K. & Gabriel, I. Inhibitors of amino acids biosynthesis as antifungal agents. Amino Acids 47, 227–249, https://doi.org/10.1007/s00726-014-1873-1 (2015).
Taylor, M. C., Kaur, H., Blessington, B., Kelly, J. M. & Wilkinson, S. R. Validation of spermidine synthase as a drug target in African trypanosomes. Biochem J 409, 563–569, https://doi.org/10.1042/BJ20071185 (2008).
Muller, I. B., Das Gupta, R., Luersen, K., Wrenger, C. & Walter, R. D. Assessing the polyamine metabolism of Plasmodium falciparum as chemotherapeutic target. Mol Biochem Parasitol 160, 1–7, https://doi.org/10.1016/j.molbiopara.2008.03.008 (2008).
Grover, A., Katiyar, S. P., Singh, S. K., Dubey, V. K. & Sundar, D. A leishmaniasis study: structure-based screening and molecular dynamics mechanistic analysis for discovering potent inhibitors of spermidine synthase. Biochim Biophys Acta 1824, 1476–1483, https://doi.org/10.1016/j.bbapap.2012.05.016 (2012).
Hu, Y. et al. An extensive comparison of the effect of anthelmintic classes on diverse nematodes. PLoS One 8, e70702, https://doi.org/10.1371/journal.pone.0070702 (2013).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35, W182–185, https://doi.org/10.1093/nar/gkm321 (2007).
Claudel-Renard, C., Chevalet, C., Faraut, T. & Kahn, D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res 31, 6633–6639 (2003).
Hung, S. S., Wasmuth, J., Sanford, C. & Parkinson, J. DETECT–a density estimation tool for enzyme classification and its application to Plasmodium falciparum. Bioinformatics 26, 1690–1698, https://doi.org/10.1093/bioinformatics/btq266 (2010).
Schomburg, I. et al. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res 41, D764–772, https://doi.org/10.1093/nar/gks1049 (2013).
Karp, P. D. et al. Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology. Brief Bioinform 17, 877–890, https://doi.org/10.1093/bib/bbv079 (2016).
Cotton, J. A. et al. The genome of Onchocerca volvulus, agent of river blindness. Nat Microbiol 2, 16216, https://doi.org/10.1038/nmicrobiol.2016.216 (2016).
Jex, A. R. et al. Ascaris suum draft genome. Nature 479, 529–533, https://doi.org/10.1038/nature10553 (2011).
Ghedin, E. et al. Draft Genome of the Filarial Nematode Parasite Brugia malayi. Science (New York, N.Y.) 317, 1756–1760, https://doi.org/10.1126/science.1145406 (2007).
Godel, C. et al. The genome of the heartworm, Dirofilaria immitis, reveals drug and vaccine targets. FASEB J 26, 4650–4661 (2012).
Desjardins, C. A. et al. Genomics of Loa loa, a Wolbachia-free filarial parasite of humans. Nat Genet 45, 495–500, https://doi.org/10.1038/ng.2585 (2013).
Tang, Y. T. et al. Genome of the human hookworm Necator americanus. Nature genetics 46, 261–269, https://doi.org/10.1038/ng.2875 (2014).
Hunt, V. L. et al. The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat Genet 48, 299–307, https://doi.org/10.1038/ng.3495 (2016).
Foth, B. J. et al. Whipworm genome and dual-species transcriptome analyses provide molecular insights into an intimate host-parasite interaction. Nat Genet 46, 693–700, https://doi.org/10.1038/ng.3010 (2014).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research 44, D457–D462, https://doi.org/10.1093/nar/gkv1070 (2016).
Howe, K. L. et al. WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res 44, D774–780, https://doi.org/10.1093/nar/gkv1217 (2016).
Perot, S., Sperandio, O., Miteva, M. A., Camproux, A. C. & Villoutreix, B. O. Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov Today 15, 656–667, https://doi.org/10.1016/j.drudis.2010.05.015 (2010).
Baell, J. B. & Holloway, G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53, 2719–2740, https://doi.org/10.1021/jm901137j (2010).
Marcellino, C. et al. WormAssay: a novel computer application for whole-plate motion-based screening of macroscopic parasites. PLoS Negl Trop Dis 6, e1494, https://doi.org/10.1371/journal.pntd.0001494 (2012).
Hu, Y. et al. Bacillus subtilis strain engineered for treatment of soil-transmitted helminth diseases. Appl Environ Microbiol 79, 5527–5532, https://doi.org/10.1128/AEM.01854-13 (2013).
Vermeire, J. J., Lantz, L. D. & Caffrey, C. R. Cure of hookworm infection with a cysteine protease inhibitor. PLoS Negl Trop Dis 6, e1680, https://doi.org/10.1371/journal.pntd.0001680 (2012).
Hu, Y., Georghiou, S. B., Kelleher, A. J. & Aroian, R. V. Bacillus thuringiensis Cry5B protein is highly efficacious as a single-dose therapy against an intestinal roundworm infection in mice. PLoS Negl Trop Dis 4, e614, https://doi.org/10.1371/journal.pntd.0000614 (2010).
Hu, Y. et al. Mechanistic and single-dose in vivo therapeutic studies of Cry5B anthelmintic action against hookworms. PLoS Negl Trop Dis 6, e1900, https://doi.org/10.1371/journal.pntd.0001900 (2012).
Tritten, L., Silbereisen, A. & Keiser, J. In vitro and in vivo efficacy of Monepantel (AAD 1566) against laboratory models of human intestinal nematode infections. PLoS Negl Trop Dis 5, e1457, https://doi.org/10.1371/journal.pntd.0001457 (2011).
Cao, Y., Charisi, A., Cheng, L. C., Jiang, T. & Girke, T. ChemmineR: a compound mining framework for R. Bioinformatics 24, 1733–1734, https://doi.org/10.1093/bioinformatics/btn307 (2008).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321, https://doi.org/10.1093/sysbio/syq010 (2010).
Acknowledgements
We would like to thank Joseph F. Urban Jr. for providing adult T. muris for calculating IC50 values for Sulconazole and Torin1. This work was supported by National Institute of Allergy and Infectious Diseases (NIAID) grants AI081803 to M.M. and AI056189 to R.V.A., Food Research Initiative (AFRI) Competitive Grant no. 2015-11323 from the USDA National Institute of Food and Agriculture to R.V.A and by the Bill & Melinda Gates Foundation to J.S.
Author information
Authors and Affiliations
Contributions
M.M., R.A., P.J.B. and J.W.J., conceived and designed the experiments. R.T., B.A.R., S.A.W. and J.H. performed the analysis to identify and prioritize drug targets and drugs/drug-like compounds. S.A.W. and J.H. performed the chemogenomic screening and modeling. M.E., C.A.B., J.S. and R.V.A. designed and performed the in vitro and in vivo studies, and related data analyses. M.M. and R.T. wrote the manuscript. All authors edited the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tyagi, R., Elfawal, M.A., Wildman, S.A. et al. Identification of small molecule enzyme inhibitors as broad-spectrum anthelmintics. Sci Rep 9, 9085 (2019). https://doi.org/10.1038/s41598-019-45548-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45548-7
- Springer Nature Limited
This article is cited by
-
Multivariate chemogenomic screening prioritizes new macrofilaricidal leads
Communications Biology (2023)
-
A genome-scale metabolic model of parasitic whipworm
Nature Communications (2023)
-
Identification of antiparasitic drug targets using a multi-omics workflow in the acanthocephalan model
BMC Genomics (2022)
-
A fragment-based approach identifies an allosteric pocket that impacts malate dehydrogenase activity
Communications Biology (2021)