
Abstract
Primary Ciliary Dyskinesia (PCD) is underdiagnosed in Brazil. We enrolled patients from an adult service of Bronchiectasis over a two-year period in a cross-sectional study. The inclusion criteria were laterality disorders (LD), cough with recurrent infections and the exclusion of other causes of bronchiectasis. Patients underwent at least two of the following tests: nasal nitric oxide, ciliary movement and analysis of ciliary immunofluorescence, and genetic tests (31 PCD genes + CFTR gene). The clinical characterization included the PICADAR and bronchiectasis scores, pulmonary function, chronic Pseudomonas aeruginosa (cPA) colonization, exhaled breath condensate (EBC) and mucus rheology (MR). Forty-nine of the 500 patients were diagnosed with definite (42/49), probable (5/49), and clinical (2/49) PCD. Twenty-four patients (24/47) presented bi-allelic pathogenic variants in a total of 31 screened PCD genes. A PICADAR score > 5 was found in 37/49 patients, consanguinity in 27/49, LD in 28/49, and eight PCD sibling groups. FACED diagnosed 23/49 patients with moderate or severe bronchiectasis; FEV1 ≤ 50% in 25/49 patients, eight patients had undergone lung transplantation, four had been lobectomized and cPA+ was determined in 20/49. The EBC and MR were altered in all patients. This adult PCD population was characterized by consanguinity, severe lung impairment, genetic variability, altered EBC and MR.
Similar content being viewed by others

Introduction
Motile ciliopathies are characterized by their generation of abnormal fluid flow or movement within fluids, which compromises mucus clearance and results in chronic airway disease1. Amongst motile ciliopathies, Primary Ciliary Dyskinesia (PCD, OMIM: #244400) is a genetically heterogeneous recessive disorder that results in neonatal respiratory distress, chronic oto-sino-pulmonary diseases, male infertility and organ laterality defects in ~50% of cases2. The global incidence of PCD is estimated at one case per 10,000 to 20,000 births. An international registry including 18 countries has recently gathered data on 3,013 patients diagnosed with PCD3.
According to the PCD foundation consensus4, the diagnosis of PCD includes two major clinical criteria and at least one of the following altered tests: a low nasal nitric oxide (nNO) production rate on two occasions, diagnostic ciliary ultrastructure with Transmission Electron Microscopy (TEM), bi-allelic gene mutations in one PCD-associated gene5 and wave abnormalities on high-speed video microscopy analysis (HVMA)4. In addition, some services have used High-Resolution Immunofluorescence Microscopy (IFM) to confirm ultrastructure data6.
Considering the classification of the international PCD register, PCD patients can be separated into three groups: definitive diagnosis of PCD, established by identifying hallmark TEM findings and/or biallelic PCD mutation, probable PCD (patients with abnormal video movement and/or low nNO), and clinical PCD (negative or ambiguous tests but a strong clinical characteristic)3. However, the genetic tests for variants that determine alterations of ultrastructure and transport proteins for the diagnosis of PCD are not universally available7.
Despite the extensive number of patients who have been diagnosed with PCD in Europe8 and the United States9, in many countries, PCD diagnosis is still not performed, mainly due to the lack of resources. Moreover, few studies address the clinical characterization of PCDs in adults10,11, especially in terms of disease severity and the relation to severe pulmonary commitment12. In Brazil, there are no reference centres for PCD screening, diagnosis and management. Therefore, the prevalence of PCD is unknown in this country, and there are few data that characterize PCD patients13.
In this study, we aimed to identify, diagnose and fully characterize a group of adult PCD patients who were followed up at a Bronchiectasis Outpatient Service at a large tertiary care complex in São Paulo, Brazil.
Results
Demographics and clinical characteristics
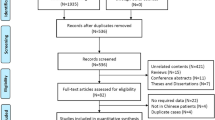
Of the 500 patients registered at the Bronchiectasis Outpatient Service over a two-year period, 55 fulfilled the eligibility criteria for PCD assessment, and 49 were diagnosed with PCD. The six patients excluded presented normal nNO and ultrastructure and no laterality disorders. The characteristics of these patients are shown in the Supplementary File (Table S1).
The mean ± SD age of the patients was 41.6 ± 12.9 y, ranging from 21 to 77 y (28 M:21 F). The mean ± SD BMI was 23.8 ± 3.6 kg/m2 (ranging from 16.8 to 32.8 kg/m2). Furthermore, 45/49 (92%) of the patients were Caucasians and four patients were Afro descendants. Consanguinity was present in 27/49 (55%) of the patients, and there were eight sibling groups in this population (n = 18). The place of birth of the patients was divided between the southeast and northeast regions of the country, but consanguinity was more frequent in patients from the northeast part of the country (31%). Relatives with similar respiratory commitment were described in 28/49 (57%) patients.
The FACED scores ranged from mild in 20/49 patients (41%) to moderate in 14/49 patients (29%) to severe in 7/49 patients (14%). Chronic P. aeruginosa colonization (cPA) was found in 20/49 (41%) patients. The mean ± SD FEV1 in PCD patients was 49.2 ± 19.8 L/s, and 25 patients had FEV1 < 50% predicted. Eight patients (16%) had previously undergone a lung transplant, and 4/49 (8%) had been lobectomized. A single patient (2%) was oxygen-dependent (for individual data, see Table 1).
Table 2 presents the signs and symptoms related to these PCD patients.
Diagnostic assessment
The PICADAR scores ranged from three to 14 points, with 37/49 (76%) patients with scores higher than five. Laterality disorders were present in 28/49 (58%) patients: 23/49 (47%) had situs inversus, 3/49 (6%) had dextrocardia, 1/49 (2%) had congenital heart disease, and 1/49 (2%) had polysplenia (Table 3).
Data on nNO production rate measurements in control patients (127.3 ± 47.6 nL/min) and in non-PCD bronchiectatic patients (88.2 ± 27.4 nL/min) are shown in the Supplementary File (Table S1). In the entire PCD population, the mean ± SD nNO was 17.2 ± 15.6 nL/min (Table 3).
Thirty-five PCD patients and all controls underwent CBP evaluation. All the PCD patients presented altered exams, except for two (just one test: movement altered - Table 3).
Non-PCD bronchiectatic patients presented normal ultrastructure evaluations. Among the PCD cases, the brushing nose technique to collect cilia cells was initially successful in 46/49 cases (93.8%) with repetition of a second brush in three patients. In PCD patients, the results were as follows: dynein arm defects or dynein deficiency [DD: 20/49 (41%)], microtubular disorganisation + inner dynein arm (MTD + IDA) [14/49 (29%)], absence of central pair (ACP) plus or not transposition (ACP + T) [6/49 (12%)], and normal ultrastructure (NU) [7/49 (14%)] (Table 3, Fig. 1). Cilia IFM was performed on 28 patients, and the results were altered in 19/28 patients (68%) (Table 3, Fig. 1).

Examples of transmission electron microscopy (TEM) and immunofluorescence microscopy (IMF) results. (a) TEM cross-section of a control respiratory cilium, showing the typical “9 + 2” arrangement with nine outer-microtubule doublets and a central pair of microtubules (left panel) and a drawing of the respiratory motile cilium (right panel). (b) ODA arm defect shown by TEM (left panel) and IMF with antibodies directed against DNAH5 (green) and RSPH4A (red). In control cilia (middle panel), both proteins colocalize along the ciliary axonemes (yellow). Cilia with an ODA defect show absence of DNAH5 from the ciliary axonemes (right panel). (c) TEM section of a cilium with absent of central pair (9 + 0) and transposition defect (8 + 1) (left panel). IMF: antibodies directed against DNAH11 (green) and RSPH9 (red) (middle panel). Cilia with radial spoke defect show absence of RSPH9 (right panel). (d) Microtubular disorganisation + Inner Dynein Arm defect by TEM (left panel) and IMF microscopy using an antibody directed against CCDC39 (red) and DNAH5 (green). In the control cells (middle panel), CCDC39 (red) colocalizes with DNAH5 (green) along the ciliary axonemes (yellow). By contrast, cells from a PCD individual with tubular disorganisation CCDC39 was completely absent from the ciliary axonemes, indicating a CCDC39 defect (right panel). The nuclei were stained with Hoechst 3342 (blue).
All PCD patients except two underwent genetic screening. Bi-allelic PCD pathogenic variants in autosomal recessive pattern inheritance were found in the following order: CCDC40 [8/49 (16%) patients], DNAH5 [4/49 (8%) patients], RSPH1 and DYX1C1 [3/49 (6%) patients], DNAAF3 [2/49 (4.1%)], and 1/49 (2%) for the following genes: CCDC39, DNAI2, DNAH11, RPGR and CCDC151. An overview and considerations regarding the genetic screening in the use of pathogenic variants are shown in brief in Table 3, and the complete overview is shown in the Supplementary File (Table S2). The genetic variants with proven pathogenicity screened in patients with phenotypes compatible with PCD are described in the Supplementary File (Table S3), and the variants with uncertain significance are summarized in the Supplementary File (Table S4). Related genotypes screened in patients with phenotypes compatible with primary ciliary dyskinesia are described in the Supplementary File (Table S5).
According to the first results of the iPCD cohort3, this patient group was categorized as follows: definite PCD diagnosis in 42/49 (86%) patients, probable PCD in 5/49 (10%) patients, and clinical PCD in 2/49 (4%) patients. Table 3 shows the characteristics of each group. The nNO was 13.3 ± 9.4 nL/min in the definite PCD group; 22.2 ± 11.5 nL/min in the probable PCD group; and 63.1 ± 21.2 nL/min in the clinical PCD group. PCD diagnostic test results are summarized in Fig. 2.

Results of positive PCD diagnostic tests.
Assessment of severity
Twenty-five patients (51%) had severe lung impairment (FEV1 < 50%), with a FACED score of 3.4 ± 1.3 (range of one to six). At PCD diagnosis, eight patients had previously had lung transplants, four had been submitted to a lobectomy and one was oxygen-dependent. The more frequent ultrastructural abnormalities in these patients were as follows: DD in 11 patients, MTD + IDA in seven patients, ACP in three patients and NU in three patients.
Inflammation and mucus rheology
EBC pH was collected from 35 patients (35/49, 71.4%), and the EBC pH of patients with PCD was 6.60 ± 0.33, which was less than that of a historical control group of healthy volunteers (7.7 ± 0.49)14. The mucus contact angle (53.2 ± 16.4°) was increased in relation to normal values (37 ± 2°) in 35/36 (97.2%) patients. Cough transportability (20.7 ± 5.8 mm) was below reference values (34 ± 9 mm) in 27/36 (75%) patients15. Viscosity values were 22.4 ± 7.7 cP, whereas plastic viscosity values were 10.9 ± 7.7 cP (no reference values available) (Table 4).
Discussion
In this study, we report the diagnostic assessment and the clinical, functional and genetic characteristics of an adult PCD population of 49 individuals in São Paulo, Brazil. Fifty-one percent of the patients presented severe functional impairment in adult life, corroborating the idea that PCD is not a mild disease12. This is the first characterization of a group of adult PCD patients in this country.
The newly diagnosed PCD patients in this study represented ten percent of the patients in the Bronchiectasis Outpatient Service of the largest tertiary care hospital in Brazil, which is in keeping with the frequency of other studies16, showing that PCD is not as rare as previously thought in Brazil. This population had a mean age of 41.6 ± 12.9 years, and 92% were Caucasians with relatively high rates (55%) of consanguinity, which remains frequent in this country. Situs inversus (50%) was the more frequent laterality disorder, in accordance with previous reports17.
The diagnosis of PCD remains challenging since none of the available tests can be used as a stand-alone test18. Referral centres differ in the combination of five tests used to assess diagnosis: nNO, HSVM, TEM, IFM and genetic tests18. We were able to perform all of these tests on 53% of the patients, and ultrastructure and/or genetic screening were performed on all patients. Few studies have evaluated the five tests in the same patient group19,20. Furthermore, differences between the North American21 and European diagnostic consensus22 increase the complexity of the diagnostic approach. In our setting, we consider the North American consensus more feasible since it requires fewer replicates of examinations. IFM contributed to diagnosis in 68% of the patients, suggesting that in medical settings where genetic screening is not affordable, this technique could represent a viable alternative. Some patients did not present positive IFM results because of inadequate samples due excess mucus or few cilia6. Therefore, we cannot exclude the possibility that the positivity of this technique could have been higher if we had repeated the exams.
Recent data indicate that PCD affects lung function in early life23. At the time of PCD diagnosis, 51% of our patients presented severe lung involvement. Eight (16%) patients had undergone lung transplantation at diagnosis, 4/49 (8%) had been lobectomized and 1/49 (2%) was oxygen dependent, confirming disease severity. In addition, a high prevalence of chronic cPa was present in this population, higher than previously reported10,24. Although dynein cilia defect was the most frequent abnormality found in the severe patients, the MTD + IDA defect was present in the younger (≤46 y) patients. These findings suggest that the MTD + IDA defect is associated with a more rapid decline in lung function20. There are few available data on the effect of early PCD diagnosis on later life lung function23,25. Nevertheless, it is highly likely that the lack of early PCD diagnosis and the lack of long-term, adequate and intensive treatment contributed to the disease severity in these patients.
Ultrastructure cilia defect indicated the diagnosis in 81.6% of the patients, and the genetic panel tests identified 49% (24/49) of the patients with the PCD bi-allelic gene. Therefore, genetic tests can only not be used to exclude the diagnosis of PCD26. Moreover, we found many variants of uncertain significance that could be associated with our genetically mixed population.
We observed phenotype-genotype inconsistencies in three patients (Table 3- Legend). So far, there has been no clear relationship between ultrastructure, genotypes, and respiratory phenotypes, mainly due to the clinical and genetic heterogeneity of PCD, and some inconsistencies are difficult to explain27. Interestingly, one male patient that had cilia ultrastructure compatible with MTD + IDA was hemizygous to a RPGR mutation linked to the X-chromosome. Such a mutation causes retinitis pigmentosa and is rarely associated with respiratory cilia defect28,29.
The PCD patients, like those with other chronic inflammatory airway diseases, presented lower EBC pH levels. It is possible that pH and other exhaled compounds could be a non-invasive tool to evaluate PCD treatment30,31. Our results indicate that reduced mucociliary transport, chronic inflammation and repeated infections in the respiratory tract produce a thicker mucus in PCD, as shown by the higher contact angle, reduced cough transportability and/or higher viscosity.
This study has several limitations. We acknowledge that our study population is small, and any conclusions should be drawn with care. However, this study represents an initial effort to adequately diagnose and fully characterize these patients in Brazil. It is possible that we have included more severe patients with bronchiectasis that were treated in a tertiary care centre. However, the prevalence of bronchiectasis in adult PCD patients seems to be very high10. The PICADAR scores were highly variable, with some patients presenting low scores, which could be explained by memory bias. We used an nNO handheld device for screening, which is less accurate21. However, in countries with limited resources, such as ours, the recommended chemiluminescence nNO analyser is generally not affordable. There is no standardization of equipment, samples, processing or analysis of cilia movement, and subtle abnormalities in CBP can be difficult to differentiate from secondary dyskinesia. In this study, CBP and nNO were used as accessory tools to strengthen the positive PCD diagnosis18.
In conclusion, we diagnosed and described the clinical condition of 49 adult PCD patients who were monitored at a Bronchiectasis service. This population was characterized by high consanguinity levels and severe pulmonary commitment. Genetically, a wide variability of pathogenic variants in genes related to PCD and variants of uncertain significance were found, which is likely to be a reflection of the genetically mixed population of Brazil. PCD is considered an orphan disease, as it has neither the prevalence of asthma nor the lethality of cystic fibrosis. We hope to use this series of patients to contribute to PCD awareness in our country and demonstrate the need for earlier diagnosis.
Methods
This cross-sectional study included 55 adults with suspicion of PCD selected from 500 patients monitored at the Bronchiectasis Outpatient Service, Pulmonology Division, São Paulo University Medical School, from 2015 to 2017. This study was approved by the Ethics Committee of the institution [CAAE: 22823414.8.0000.0068]. All subjects signed written informed consent statements.
Eligibility
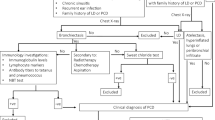
Patients characterized with idiopathic bronchiectasis after a systematic aetiology protocol evaluation were screened for this study. Our institutional protocol includes genetic and/or sweat testing for cystic fibrosis, assessment of gastroesophageal reflux disorder, immunodeficiency (HIV and immunoglobulins) tests, alpha-1 anti-trypsin serum levels, rheumatological antibodies, white blood cell counts and sputum cultures (aerobic, fungi and mycobacteria). Patients were selected to enrol in the PCD diagnosis effort if they presented at least one of the following conditions: laterality disorders or productive chronic cough associated with recurring lower respiratory infections with or without upper respiratory infections and predominance of tomographic findings (bronchiectasis and tree in bud opacities) in the lower, middle and lingula lobes. Patients were submitted to at least two of the following diagnostic tests: nNO production rate measurement, cilia movement evaluation, ciliary ultrastructure evaluation, IFM and genetic tests.
We also evaluated a group of healthy volunteers for nNO measurements and cilia movement to validate our findings. Individuals were excluded from the control group if they had experienced respiratory symptoms in the previous month and/or had a history of smoking.
Clinical characterization
We collected the following information: demographic variables including age, sex, self-reported race, place of birth, parental consanguinity, symptoms in relatives, body mass index (BMI), and pulmonary function tests. We obtained data on the presence of chronic Pseudomonas aeruginosa colonization (two or more isolates of the same organism at least three months apart in one year), spermiogram, and previous surgery interventions and/or oxygen dependence. Patients were further characterized by PICADAR32 and FACED scores33. In a subset of patients, exhaled breath condensate and mucus rheology were performed.
Diagnostic assessment
Nasal nitric oxide production rate (nNO)
The nNO production rate measurements were made using a NIOX MINO (AEROCRINE AB®, Solna, Sweden) device, according to the manufacturer’s instructions34, in patients who had fasted for at least eight hours and were free from acute respiratory disease34,35,36. See the Supplementary File for methodological details. Control groups and patients with no PCD and details regarding the protocol were registered.
Cilia beat frequency and pattern (CBF and CBP) and ciliary ultrastructure
Cilia were collected to study movement and ultrastructure procedures. The detailed method is described in the Supplementary File13.
Evaluation of CBF and CBP
Several strips of ciliated epithelium movement were recorded for each patient. The recorded cilia cell movement videos were studied a second time, and the CBF and CBP were classified as follows: recognisability of regular forward and recovery strokes (normal), static cilia, almost static cilia with minimal residual movement, stiff beating due to a reduced bending capacity/amplitude, and abnormal circular beating37,38. Cilia movement with no agreement with any previous description but without an effective stroke were considered altered. Only strips of ciliated epithelium without damaged epithelium and no isolated cilia cells were evaluated. CBF was evaluated according to previous studies13, and the final results of 10 measurements were recorded. In our study, we assumed CBP to be more important than CBF for evaluating and defining movement39. Therefore, if the CBP was altered, we assumed the final result of the movement to be altered.
Analyses of ciliary Ultrastructure by TEM
The collected material was immersed in 2% glutaraldehyde and processed according to standardized norms40 using cross-section thicknesses of 50 nm. Quantitative and qualitative analyses were conducted. At least 100 cross-sections of cilia were evaluated. High-quality cross-sections were assessed for the presence of dynein arms. Details of the TEM evaluation are described in the Supplementary File41,42,43.
High-Resolution Immunofluorescence Microscopy (IFM)
High-resolution IFM was performed at the University Hospital of Muenster, Germany, following the laboratory protocols. The following antibodies were used: anti-DNAH5, anti-RSPH4A, anti-RSPH9, anti-CCDC39, anti-GAS8 and anti-DNAH116. High-resolution immunofluorescence images were taken using a Zeiss Apotome Axiovert 200 (processed with AxioVision 4.8) or Zeiss LSM880 (processed with ZEN2 software) (see the Supplementary File).
Genetics analysis
Detailed methodologies used for the genetic analysis are described in the Supplementary File. DNA extraction was performed using the FlexiGene DNA Kit extraction kit (Qiagen®, Valencia, CA, 91355, USA). After DNA extraction, the sample was quantified in Qubit 2.0 (Life Technologies®, São Paulo/SP, Brazil) and then submitted to panel sequencing.
Pulmonary function test (PFT)
Spirometry (Koko Legend, Inspire Health Inc., Longmont, USA) was performed according to the recommendations of the American Thoracic Society and the European Respiratory Society. Data were interpreted based on the methods proposed by Pereira and collaborators who examined the Brazilian population44. The forced vital capacity (FVC) and FEV1 were considered PFT parameters.
PCD diagnosis and severity assessment
The possibility of having PCD was considered for patients who had altered results in at least two of the following tests: nNO production rate measurement, evaluation of ciliary movement (CBP), ultrastructure analyses of the cilia (TEM), IFM and genetic tests related to PCD gene sequencing. Patients with confirmed ciliary ultrastructural defect and/or with bi-allelic causing-PCD gene variants were definitively diagnosed with PCD3,4,39. Probable PCD was assumed for patients with only one abnormal test, such as altered movement (CBP) and/or low nNO production rate compared to the control the group, but all typical clinical symptoms were present. Patients with negative or ambiguous tests but strong clinical characteristics were defined as a clinical PCD diagnosis3.
We considered patients to have severe disease when the forced expiratory volume in one second (FEV1) was < 50% of that predicted.
pH in exhaled breath condensate (EBC)
The patients fasted for at least eight hours before EBC collection. For EBC collection and analysis, see the Supplementary File. Immediately after EBC collection, the EBC pH was analysed, and the results were compared with historical normal values (7.7 ± 0.49)14.
Mucus rheology
Patients were asked to cough three times, and the sputum samples were stored at −80 °C for further analyses of contact angle, cough transportability and (plastic) viscosity. For the methodological details, see the Supplementary File15.
Statements
Approval
Written approval was obtained from the patients for re search purposes, and from the institutional ethical committee (Comissão de Ética para Análise de Projetos de Pesquisa – CAPPesq).
Accordance
The methods were used in accordance with the relevant guidelines and regulations.
Informed Consent
An informed consent was obtained from all participants.
Data Availability
All the data generated or analysed during this study are included in this published article, and its Supplementary Information Files.
References
Mitchison, H. M. & Valente, E. M. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol [Internet]. John Wiley & Sons, Ltd; [cited 2018 Jan 11]; 241(2), 294–309. Available from: https://doi.org/10.1002/path.4843 (2017 Jan 1).
Knowles, M. R., Daniels, L. A., Davis, S. D., Zariwala, M. A. & Leigh, M. W. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med [Internet]. [cited 2017 Feb 26]; 188(8), 913–22. Available from: https://doi.org/10.1164/rccm.201301-0059CI (2013 Oct 15).
Goutaki, M. et al. The international primary ciliary dyskinesia cohort (iPCD Cohort): methods and first results. Eur Respir J [Internet]. [cited 2017 Feb 26]; 49(1), 1601181. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28052956 (2017 Jan).
Shapiro, A. J. et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr Pulmonol [Internet]. [cited 2017 Feb 26]; 51(2), 115–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26418604 (2016 Feb).
Werner, C. et al. An international registry for primary ciliary dyskinesia. Eur Respir J [Internet]. [cited 2017 Feb 26]; 47(3), 849–59. Available from: https://doi.org/10.1183/13993003.00776-2015 (2016 Mar).
Werner, C., Onnebrink, J. G. & Omran, H. Diagnosis and management of primary ciliary dyskinesia. Cilia [Internet]. [cited 2017 Feb 26]; 4(1), 2. Available from: http://www.ciliajournal.com/content/4/1/2 (2015).
Shoemark, A. et al. Accuracy of Immunofluorescence in the Diagnosis of Primary Ciliary Dyskinesia. Am J Respir Crit Care Med [Internet]. [cited 2017 Mar 4];rccm.201607-1351OC. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28199173 (2017 Feb 15).
Kuehni, C. et al. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur Respir J.; 36(6), 1248–58 (2010 Jul 6).
Knowles, M. R. & Leigh, M. W. Primary Ciliary Dyskinesia Diagnosis. Is Color Better Than Black and White? Am J Respir Crit Care Med [Internet]. [cited 2017 Aug 18]; 196(1), 9–10, Available from: https://doi.org/10.1164/rccm.201702-0426ED (2017 Jul 1).
Noone, P. G. et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med [Internet]. [cited 2017 Feb 26]; 169(4), 459–67. Available from: https://doi.org/10.1164/rccm.200303-365OC (2004 Feb 15).
Contarini, M. et al. Why, when and how to investigate primary ciliary dyskinesia in adult patients with bronchiectasis. Multidiscip Respir Med [Internet]. BioMed Central; [cited 2019 May 4]; 13(Suppl 1), 26. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30151188 (2018).
Saglani, S. Lung function in primary ciliary dyskinesia: breaking the myth that this is a mild disease. Eur Respir J [Internet]. [cited 2018 Sep 26]; 52(2), 1801365. Available from: https://doi.org/10.1183/13993003.01365-2018 (2018 Aug 23).
Olm, M. A. K. et al. Primary ciliary dyskinesia: evaluation using cilia beat frequency assessment via spectral analysis of digital microscopy images. J Appl Physiol [Internet]. [cited 2017 Feb 26]; 111(1), 295–302. Available from: https://doi.org/10.1152/japplphysiol.00629.2010 (2011 Jul 1).
Vaughan, J. et al. Exhaled breath condensate pH is a robust and reproducible assay of airway acidity. Eur Respir J [Internet]. [cited 2018 Jul 4]; 22(6), 889–94. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14680074 (2003 Dec).
Goto, D. M. et al. Furosemide impairs nasal mucociliary clearance in humans. Respir Physiol Neurobiol [Internet]. [cited 2018 Jul 4]; 170(3), 246–52. Available from: http://linkinghub.elsevier.com/retrieve/pii/S156990481000042X (2010 Mar 31).
Shoemark, A., Ozerovitch, L. & Wilson, R. Aetiology in adult patients with bronchiectasis. Respir Med [Internet]. [cited 2018 Oct 12]; 101(6), 1163–70. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0954611106005968 (2007 Jun).
Goutaki, M. et al. Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J [Internet]. [cited 2017 Aug 18]; 48(4), 1081–95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27492829 (2016 Oct).
Rumman, N., et al. Diagnosis of primary ciliary dyskinesia: potential options for resource-limited countries. Eur Respir Rev [Internet]. [cited 2017 Mar 4]; 26(143), 160058. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28096286 (2017 Jan 17).
Abitbul, R. et al. Primary ciliary dyskinesia in Israel: Prevalence, clinical features, current diagnosis and management practices. Respir Med [Internet]. [cited 2018 Oct 14]; 119:41–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27692146 (2016 Oct).
Davis, S. D. et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med [Internet]. American Thoracic Society; [cited 2017 Nov 21]; 191(3), 316–24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25493340 (2015 Feb 1).
Shapiro, A, J. et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med [Internet]. [cited 2018 Aug 31]; 197(12), e24–39. Available from: https://doi.org/10.1164/rccm.201805-0819ST (2018 Jun 15).
Dalrymple, R. A. & Kenia, P. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia: a guideline review. Arch Dis Child - Educ Pract Ed [Internet]. [cited 2018 Aug 31];edpract-2017-312902. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30076157 (2018 Aug 3).
Halbeisen, F. S. et al. Lung function in patients with primary ciliary dyskinesia: an iPCD Cohort study. Eur Respir J [Internet]. [cited 2018 Oct 12]; 52(2), 1801040. Available from: https://doi.org/10.1183/13993003.01040-2018 (2018 Aug).
Maglione, M. et al. Multicenter analysis of body mass index, lung function, and sputum microbiology in primary ciliary dyskinesia. Pediatr Pulmonol [Internet]. [cited 2018 Jul 9]; 49(12), 1243–50. Available from: https://doi.org/10.1002/ppul.22984 (2014 Dec).
Kuehni, C. E. & Lucas, J. S. Diagnosis of primary ciliary dyskinesia: summary of the ERS Task Force report. Breathe [Internet]. [cited 2018 Oct 12]; 13(3), 166–78. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28894478 (2017 Sep).
Haarman, E. G. & Schmidts, M. Accuracy of diagnostic testing in primary ciliary dyskinesia: are we there yet? Eur Respir J [Internet]. [cited 2017 Feb 26]; 47(3), 699–701. Available from: https://doi.org/10.1183/13993003.01914-2015 (2016 Mar 29).
Horani, A., Ferkol, T. W., Dutcher, S. K. & Brody, S. L. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev [Internet]. NIH Public Access; [cited 2019 May 4]; 18:18–24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26476603 (2016 Mar).
Moore, A. et al. RPGR is mutated in patients with a coplex X linked phenotype combining primary ciliary dyskinesia. J Med Genet. 43_ _:326–33 (2006).
Bukowy-Bieryłło, Z. et al. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr Pulmonol [Internet]. [cited 2019 May 4]; 48(4), 352–63. Available from: https://doi.org/10.1002/ppul.22632 (2013 Apr).
Davis, M. D. & Hunt, J. Exhaled Breath Condensate pH Assays. Immunol Allergy Clin North Am [Internet]. [cited 2018 Oct 14]; 32(3), 377–86. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22877616 (2012 Aug).
Bikov, A. & Horvath, I. Methodological Issues and Possible Clinical Implications for Exhaled Breath Condensate pH in Asthma. Curr Top Med Chem [Internet]. [cited 2018 Oct 14]; 16(14), 1550–60. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26420368 (2016).
Behan, L. et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J [Internet]. European Respiratory Society; [cited 2017 Aug 18]; 47(4), 1103–12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26917608 (2016 Apr).
Athanazio, R. et al. Latin America validation of FACED score in patients with bronchiectasis: an analysis of six cohorts. BMC Pulm Med [Internet]. BioMed Central; [cited 2018 Mar 11]; 17(1), 73. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28446170 (2017 Apr 26).
Marthin, J. K. & Nielsen, K. G. Hand-held tidal breathing nasal nitric oxide measurement–a promising targeted case-finding tool for the diagnosis of primary ciliary dyskinesia. Watz H, editor. PLoS One [Internet]. [cited 2017 Feb 26]; 8(2), e57262. Available from: https://doi.org/10.1371/journal.pone.0057262 (2013 Feb 20).
Harris, A. et al. Validation of portable nitric oxide analyzer for screening in primary ciliary dyskinesias. BMC Pulm Med. 14(18), 1–8 (2014).
Montella, S. et al. Measurement of nasal nitric oxide by hand-held and stationary devices. Eur J Clin Invest [Internet]. [cited 2017 Aug 18]; 41(10), 1063–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21413977 (2011 Oct).
Chilvers, M. A., Rutman, A. & O’Callaghan, C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol [Internet]. [cited 2017 Feb 26]; 112(3), 518–24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/13679810 (2003 Sep).
Raidt, J. et al. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur Respir J. Sep 3 44(6), 1579–88 (2014).
Lucas, J. S. et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J [Internet]. European Respiratory Society; [cited 2017 Nov 21]; 49(1), 1601090. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27836958 (2017 Jan 1).
Rutland, J., Dewar, A., Cox, T. & Cole, P. Nasal brushing for the study of ciliary ultrastructure. J Clin Pathol.; 35_ _(3), 357–9 (1982).
Shoemark, A., Dixon, M., Corrin, B. & Dewar, A. Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J Clin Pathol. 65(3), 267–71 (2012 Mar).
Carlén, B, & Stenram, U. Primary ciliary dyskinesia: a review. Ultrastruct Pathol.; 29(3–4), 217–20 (2005 May).
Roomans, G. M., Ivanovs, A., Shebani, E. B. & Johannesson M. Transmission electron microscopy in the diagnosis of primary ciliary dyskinesia. Ups J Med Sci [Internet]. [cited 2017 Feb 26]; 111(1), 155–68. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16553254 (2006).
de Pereira, C. A., Duarte, A. A. O., Gimenez, A. & Soares, M. R. Comparison between reference values for FVC, FEV1, and FEV1/FVC ratio in White adults in Brazil and those suggested by the Global Lung Function Initiative. J Bras Pneumol [Internet]. [cited 2018 Aug 3]; 40(4), 397–402. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25210962 (2012).
Acknowledgements
The authors would like to thank the PCD patients for their cooperation. The authors would like to thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP: #2014/18049-9, #2015/12183-8 and #2015/12858-5) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico for supporting this research (CNPq: #471078/2014-0 and # 3305428/2014).
Author information
Authors and Affiliations
Contributions
M.A.K.O. and F.A.L.M. contributed equally to this paper. They were responsible for the acquisition, analysis, diagnoses, interpretation of data, and wrote the manuscript. R.A.A. was responsible for the design of the work and selection of patients, and critically read the article. S.Z.R. and R.S. selected the patients and drafted the work. N.K.N. and M.M. conducted the mucus studies. N.T.L. and H.O. performed the IFM studies. C.S.B. drafted the genetic sequencing studies. P.H.N.S., J.D.R., M.H.J. participated in the draft, concept and analysis of data. T.M. contributed with the draft of the work, analysed and interpreted data, prepared the manuscript, and critically read the article. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Olm, M.A.K., Marson, F.A.L., Athanazio, R.A. et al. Severe pulmonary disease in an adult primary ciliary dyskinesia population in Brazil. Sci Rep 9, 8693 (2019). https://doi.org/10.1038/s41598-019-45017-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45017-1
- Springer Nature Limited
This article is cited by
-
Characterization of pathogenic genetic variants in Russian patients with primary ciliary dyskinesia using gene panel sequencing and transcript analysis
Orphanet Journal of Rare Diseases (2024)