
Abstract
Regulation of gene expression can occur via epigenetic effects as mediated by DNA methylation. The potential for epigenetic effects to be transmitted across generations, thus modulating phenotypic variation and affecting ecological and evolutionary processes, is increasingly appreciated. However, the study of variation in epigenomes and inter-generational transmission of epigenetic alterations in wild populations is at its very infancy. We studied sex- and age-related variation in DNA methylation and parent-offspring resemblance in methylation profiles in the barn swallows. We focused on a class of highly conserved ‘clock’ genes (clock, cry1, per2, per3, timeless) relevant in the timing of activities of major ecological importance. In addition, we considerably expanded previous analyses on the relationship between methylation at clock genes and breeding date, a key fitness trait in barn swallows. We found positive assortative mating for methylation at one clock locus. Methylation varied between the nestling and the adult stage, and according to sex. Individuals with relatively high methylation as nestlings also had high methylation levels when adults. Extensive parent-nestling resemblance in methylation levels was observed. Occurrence of extra-pair fertilizations allowed to disclose evidence hinting at a prevalence of paternal germline or sperm quality effects over common environment effects in generating father-offspring resemblance in methylation. Finally, we found an association between methylation at the clock poly-Q region, but not at other loci, and breeding date. We thus provided evidence for sex-dependent variation and the first account of parent-offspring resemblance in methylation in any wild vertebrate. We also showed that epigenetics may influence phenotypic plasticity of timing of life cycle events, thus having a major impact on fitness.
Similar content being viewed by others
Introduction
Environmental conditions can induce regulation of the expression and function of genes via epigenetic effects, without directly altering nucleotide sequence1,2,3. Epigenetic programming can produce differential gene expression among individuals, thus generating phenotypic differences even from the same genetic substrate2,4,5,6,7.
Epigenetic alterations can occur in response to a wide variety of extrinsic environmental stimuli ranging from nutritional conditions to social and chemical stress3,7,8. Epigenetic alterations, in turn, are heritable3,9,10,11,12 and thus have the potential to result in transmission of new phenotypes across multiple generations. The possibility that ancestors’ experiences result in epigenetic transmission across generation borders of phenotypic traits has obvious, tremendous and yet poorly explored implications for ecological and evolutionary processes2,7. Indeed, heritable epigenetic effects are known to affect phenotypic variation at major life-history traits including physiology, morphology and behavior3,9,13,14.
Epigenetic modulation of gene expression occurs through major classes of mechanisms that include DNA methylation, histone modifications, the effect of non-coding RNAs, and the activity of chaperones3,7,15,16. DNA methylation, particularly at the palindrome dinucleotide sequences 5′CpG3′ at the 5 position of cytosine, is generally regarded as the most common epigenetic mark in mammals, although information for other vertebrates is scanty. Methylation of cytosines located in gene promoters modulates gene transcription, is involved in alternative promoter usage and regulation of enhancer activity, and often causes downregulation of the gene9,15,17,18. Epigenetic effects mediated by DNA methylation may be a key mechanism that controls variation in behavioral traits13,18,19,20,21. For example, methylation at the dopamine receptor D4 (DRD4) gene is related to exploratory behavior22, while methylation at the dopamine receptor and serotonin transporter is related to novelty seeking behaviour in great tits (Parus major)21. In addition, methylation at the agouti-related peptide is significantly related to sexual coloration in the black grouse (Lyrurus tetrix)23.
DNA methylation can also be transmitted across generations, generally meaning that offspring resemble their parents in methylation levels, as shown exclusively by laboratory studies9,11,12. Such inheritance of methylation states can arise via different pathways. First, ‘true’ transgenerational inheritance occurs via the germline if methylation profile of the gametes is carried over to the zygote and later developmental stages. Rigorously demonstrating ‘true’ transgenerational inheritance of methylation profiles is extremely difficult, particularly in the wild, as it requires showing that ancestors resemble their F3 or later-generation descendants. Despite these difficulties, some studies have provided evidence for transgenerational inheritance of responses to parental experiences including diet, stressful conditions and exposure to toxins7,9, and via gamete-mediated inheritance of methylation marks. Such transmission by germline methylation has been demonstrated in studies of the effects of vinclozolin in mice on anxiety-like behaviors24,25 and also in a single study of birds under laboratory conditions26. However, it must be emphasized that while environmentally driven changes in DNA methylation has been repeatedly documented to be inherited in plants, the evidence for transgenerational environmental effects on methylation is still scanty and contentious for taxa like vertebrates27.
An alternative pathway of methylation inheritance is via ‘soma-to-soma’ interactions. These can occur when the prenatal environment in terms for example of in utero conditions or egg composition directly affect the methylome of the developing offspring7,16. In addition, RNA molecules stored in sperm and components of the seminal fluid also have the potential for transgenerational transmission of epigenetic traits9,15,28. Such soma-to-soma effects can also occur via general environmental effects whenever ancestors’ experience of environmental conditions causes a new epigenetic mark and phenotype to arise29,30 and ancestors tend to reinstate similar environmental (e.g. physical or social) conditions across subsequent generations, causing the same epigenetic mark to re-appear in descendants9. Finally, parent offspring resemblance may arise because of transgenerational genetic effects, whereby similarity in methylation levels in parents and offspring arises because of similarity in their genetic background7,31.
Inheritance of methylation may not necessarily occur symmetrically from either parental genome, as exemplified at a paradigmatic extreme by parental sex-specific genetic imprinting32. The evidence for the relative contribution of maternal versus paternal methylation states to descendant methylation patterns in humans and other vertebrates is mixed and parental sex-specific inheritance effects may vary among taxa3,33,34,35.
It is worth emphasizing that, independently of the exact mechanisms that determine resemblance in methylation states across generations, any such resemblance has extremely important ecological and evolutionary consequences because it implies that new epigenetic phenotypes can be carried over to subsequent generations independently of genetic inheritance. Yet, to the best of our knowledge, no study has been published to date on any vertebrate species in the wild where resemblance in methylation at the single-locus level between parent and offspring has been investigated.
The first main aim of this study thus was to investigate whether offspring, at their nestling stage and also once they attain sexual maturity, resemble their parents in CpG methylation at five highly conserved genes (clock, per2, per3, cry1, timeless), using a small, migratory bird, the barn swallow (Hirundo rustica) as a study system. In doing so, we also tested for any differential effect of methylation state of either parent on methylation of offspring of either sex, and on age- (nestling vs adult) and sex-specific variation in methylation21,36. Stemming from current interest on control of timing of major life cycle stages in the barn swallow37, in the present study we focused on methylation at ‘clock’ genes whose products dynamically interact to elicit rhythmic patterns of transcription, translation, biochemical and physiological processes, and behavior38,39 and are thus involved in circadian rhythmicity and photoperiodic responses (see also Supplementary Material). In addition, we tested for resemblance in methylation levels between pair members because similarity in methylation profiles at photoperiodic genes might influence mate choice or, reciprocally, synchronization in reproductive behavior might result in resemblance in methylation levels between pair members.
To our goals, we measured methylation at the focal loci in blood cells of 65 barn swallows (from 58 social pairs) when they were nestlings and when they were subsequently recruited as 1-year-old sexually mature adults, and in their parents. Because nucleated erythrocytes in barn swallows and other birds represent a large fraction of nucleated blood cells40, our methylation data essentially reflect methylation of erythrocytes. Parent-offspring resemblance in methylation can result from diverse mechanisms, which are extremely difficult to tease apart (see above), especially in the wild. When extra-pair fertilizations occur, however, two types of families are naturally established: first, families where parental fathers (i.e. the males that attend the offspring) are also the genetic fathers of the offspring (hereafter, within-pair offspring, WPO) that they attend; second, families where the parental father is the ‘social’, but not the genetic father (hereafter, ‘social father’) of one or more of the offspring (hereafter, extra-pair offspring, EPO) that they attend. Extra-pair fertilizations, which are common in the barn swallow41,42, afford an opportunity to get an insight into the contribution of different mechanisms of parent-offspring resemblance in methylation states, and we therefore subjected the 65 sampled offspring to genetic parentage tests. If genetic father-WPO resemblance in methylation is stronger than social father-EPO resemblance, a mechanism of inheritance mediated by the paternal germline or via non-genetic sperm and seminal fluid constituents can be invoked. Because the reciprocal condition whereby mothers attend non-genetically related offspring in their nest is very uncommon in barn swallows43,44,45, as in the majority of altricial birds46, no such approach could be undertaken to assess the relative contribution of maternal germline/egg effects as compared to postnatal environmental effects.
Very limited background knowledge on methylation in wild birds, however, prevented us from formulating explicit predictions on the differential father-offspring relationships according to paternity and on sex- and age-dependency of methylation levels.
In a previous study focusing on two clock loci (clock poly-Q and clock 5′-UTR) we showed that methylation at the clock poly-Q was statistically associated with spring migration and breeding date in the same barn swallow population37. The previous analysis was restricted to relatively old (two or more years) individuals and focused on just two loci. Here, we expand the analysis of the association between methylation at these two loci and breeding date, which is a major fitness trait because in barn swallows it predicts both seasonal reproductive success and offspring quality37,42,47,48, on a much larger sample of birds and we also assess whether methylation at other genes besides clock is related to breeding date. The importance of such replication studies to test for consistency of results has been repeatedly advocated49. In the analyses of breeding date in relation to methylation at clock 5′-UTR and clock poly-Q, we thus also considered the data included in Saino et al.37 (see also below).
Results
Methylation in pair members
LMMs showed that methylation of males at clock 5′-UTR significantly and positively predicted methylation of their female mate (F1,39 = 14.75, P < 0.001†, coefficient: 0.310 (0.081); Fig. 1) whereas the relationships between mates at the other loci were non-significant (Table 1S).

Relationship between methylation at clock 5′-UTR in females and in their male mates.
Methylation in relation to sex and life stage
LMMs with sex and life stage as fixed-effect factors showed a composite pattern of variation in methylation at the six loci (Table 1; Fig. 2). Methylation at clock 5′-UTR was lower in females than in males and in nestlings compared to adults (Table 1; Fig. 2). However, the sex-difference also depended on life stage, as shown by the significant two way-interaction (Table 1; Fig. 2). Specifically, there was a significant difference between nestling and adult females but not males and, in addition, there was a significant difference between nestling (but not adult) males and females (Table 1; Fig. 2). For clock poly-Q and timeless, methylation was significantly higher in nestlings than in adults and in females compared to males (Table 1; Fig. 2). Methylation at cry1, per2 and per3 did not vary according to sex or life stage (Table 1; Fig. 2).

Mean (+SE) methylation at six loci of adult and nestling male and female barn swallows from 58 families. Means for the adult life stage are computed by pooling parents with their offspring considered at the adult (1-year old) stage. S: significant difference according to sex; LS: significant difference according to life stage (adult vs nestling); S × LS: significant effect of the sex by life stage interaction. See also Table 1.
In these models we also tested the random effects of year of birth of the offspring and colony. At no locus did colony predict variation in methylation (χ2 < 0.3, df = 1, P > 0.65 for all loci). Methylation was found to significantly vary among years for clock 5′-UTR (χ2 = 25.0, df = 1, P < 0.001†), and per3 (χ2 = 13.1 df = 1, P < 0.001†), and marginally non-significantly (after FDR) so for timeless (χ 2 = 4.2, df = 1, P = 0.040), but not for the other loci (χ 2 < 2.1, df = 1, P > 0.14 in all cases).
Methylation at the nestling and adult stage
Methylation at the nestling stage significantly and positively predicted methylation at the 1-year-old adult stage for clock 5′-UTR, clock poly-Q, per2 and timeless (Table 2; Fig. 3). The relationship was significantly negative for cry1 (Table 2; Fig. 3). Finally, no relationship was observed between the nestling and the adult stage for methylation at per3 (Table 2; Fig. 3).

Relationships between methylation at the six loci of offspring at the time when they were recruited as 1-year old adults and their own methylation as nestlings.
Methylation in parents and offspring
Methylation of nestlings at clock 5′-UTR, clock poly-Q, cry1, per3 and timeless significantly increased with methylation of their genetic mother (Table 3; Fig. 4). At the adult offspring stage, however, a significant positive relationship with maternal methylation persisted only for timeless (Table 3; Fig. 4).

Relationships between offspring methylation at the nestling or 1-year-old adult stage and methylation of their mother. Statistically significant relationships are shown.
Nestling methylation differentially covaried with methylation of the genetic as compared to the social (but non-genetic) father at clock poly-Q (Table 3; Fig. 5). The effect of the paternity by paternal methylation term was either marginally non-significant (clock 5′-UTR) after FDR correction or far from significance for the other loci (Table 3). A closer inspection of within-paternity group coefficients (see Statistical analyses and SOM for a justification) showed that nestling methylation was significantly positively related to methylation of the genetic father at clock 5′-UTR and clock poly-Q, whereas the relationships for the social father were either significantly negative (clock poly-Q) or non-significant (Table 3).

Relationships between within- or extra-pair offspring methylation at the nestling stage and methylation of the genetic or the social, non-genetic father. Statistically significant relationships are shown.
At no locus did offspring methylation at the adult stage significantly, differentially covary with methylation of the genetic as compared to the social father (Table 3). Again, however, inspection of the within paternity group coefficients showed significant positive relationships between offspring and genetic father’s methylation at some loci (cry1, per2, per3) whereas no significant relationship with methylation of the social father existed at any locus (Table 3). Removal of the statistically non-significant paternity by paternal methylation interaction effect disclosed a significant positive relationship between adult offspring methylation and paternal (independent of genetic parentage) methylation at cry1, per2 and per3.
The effect of the interaction between maternal methylation and offspring sex significantly predicted offspring methylation at per2 at both offspring life stages (Table 3). Maternal methylation positively predicted methylation of daughters but not sons (Table 3). However, analyses restricted to the genetic fathers and their within-pair offspring did not disclose any significant interaction between methylation of the father and sex of the offspring on offspring methylation (F values associated to P values always >0.06; see also SOM).
Thus, there was a composite pattern of differential covariation between methylation in the offspring at either the nestling or the adult stage and methylation of the genetic versus the social father, with some hint for a stronger relationship between methylation in the offspring and in the genetic father as compared to the social father.
Methylation and breeding date
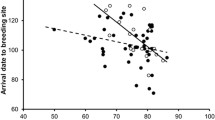
Breeding date was significantly predicted by the interaction between methylation at clock poly-Q and sex, whereas the interaction effect between methylation at clock poly-Q and age was non-significant (Table 4; Fig. 6). In females, breeding date was significantly earlier as methylation increased (Table 4; Fig. 6). In both age classes, the relationship between breeding date and methylation was significantly negative, but it was marginally non-significantly more steep for yearlings than for older individuals (Table 4; Fig. 6).

Relationship between breeding date and methylation at clock poly-Q in individuals of two age classes and according to sex. Methylation data were standardized to a mean of zero and variance of 1 within each of the two datasets (the present one and that from Saino et al.37) included in the analyses. The relationships were significantly negative within both age classes and for females but not for males.
A model excluding the non-significant interaction effects showed a marginally non-significant trend for breeding date to be earlier as methylation at clock 5′-UTR increased (Table 4).
The effect of methylation at the other loci did never attain statistical significance (P always >0.06).
Discussion
The studies of DNA methylation potentially mediating epigenetic effects in wild animal populations are at their infancy, and even basic information on individual variation in methylation levels, consistency in methylation at different life stages, and inter-generational resemblance is lacking. Yet, DNA methylation and other pathways of epigenetic alterations of gene expression can have profound effects on the formation and maintenance of phenotypic variation and thus major potential, and yet largely unexplored, consequences for ecological and evolutionary processes.
In this correlational study, we contribute to start filling some of these major gaps of knowledge by focusing on a class of highly conserved genes contributing to the circadian clock system. We first analyzed age- and sex-related variation in methylation levels at six focal genetic loci in a wild population of barn swallow, and found variation in methylation according to age and between males and females. Second, individuals were highly consistent in relative methylation levels between the nestling and the yearling life stage at most loci. Third, offspring resembled their genetic mother and their attending father in methylation particularly at the nestling stage. The occurrence of extra-pair fertilizations in the barn swallows allowed to identify some hints of stronger resemblance between the offspring and their genetic father compared to the non-genetic social father, possibly suggesting inheritance of methylation levels not only mediated by common-environment effects. In addition, methylation at clock 5′-UTR was found to positively covary between breeding mates. Finally, by largely expanding a previous dataset, we confirmed that methylation at clock poly-Q negatively predicts a major phenological fitness trait, breeding date, of females but not males and we provided novel evidence that breeding date is not predicted by methylation at the other loci. Admittedly, in the present study we did not investigate the environmental factors that govern variation in methylation levels, which was found to occur among years (clock 5′-UTR and per3) but not among colonies. The ecological causes and the ontogenetic and sex-dependent mechanisms behind variation in methylation levels thus remain to be elucidated.
Methylation in mates
We found evidence for non-random mating (i.e. assortative mating according to its definition in34) according to methylation at clock 5′-UTR but not at the other loci. To the best of our knowledge, this is the first evidence for positive assortative mating according to methylation profile in any vertebrate species. Since the clock gene is believed to be involved in photoperiodic responses, including timing of spring phenological events in birds50,51, positive assortative mating for methylation may result from an effect of methylation at this locus on timing of arrival from migration and pre-breeding activities, favoring the pairing between individuals with synchronized life cycles. An alternative interpretation is that similarity in methylation during the breeding season arises as an effect of synchrony in breeding activities, therefore being the consequence, rather than the cause, of synchronization of spring life-cycle events.
Methylation in relation to sex and life stage
We found clear evidence for variation in methylation in relation to age and sex. At clock poly-Q and timeless, nestlings had higher methylation than older individuals and females had higher methylation than males. The pattern was more complex and partly reversed for clock 5′-UTR, as methylation was lower in nestlings than in adults among females but not males, and was lower in female than male nestlings whereas no difference existed at the adult life stage.
We are aware of two cases where age-related variation in methylation in birds has been studied in the wild. Methylation at the avian glucocorticoid receptor gene did not differ between nestling and adult superb starlings (Lamprotornis superbus) whereas methylation at the agouti-related neuropeptide varied with age in the black grouse23,36. In humans, DNA methylation changes over the lifespan, but predominantly in the first year of life and throughout later childhood and adolescence, becoming more stable in the adulthood52. As a general pattern, repetitive elements are usually heavily methylated and become hypomethylated with age; gene promoters can be hypo- or hypermethylated according to their biological function and the presence of multiple methylated CpG sites in promoters causes stable silencing of genes53. At the mechanistic level, changes in the expression of enzymes regulating methylation could lead to generalized or gene-specific modulation of methylation with age54. The present age-related variation in methylation may contribute to cause the age-dependent variation in phenological traits, which is strongly expressed in barn swallows48.
Sex differences in DNA methylation have been studied in humans and other mammalian model organisms55, seldom in domestic birds56, while information on sex differences from the few studies of non-artificially selected avian models in the wild is extremely limited36. Barn swallows show variation in behaviors that may depend on circadian rhythmicity and photoperiodic responses according to sex, including for example timing of migration37,57. The sex-related differences that we observed in methylation may therefore partly account for such behavioral sex differences. In the present study, however, we did not investigate the environmental factors, if any, that could contribute to generate the observed variation of methylation levels according to age and sex, and very limited knowledge of methylation in wild vertebrates prevented us from speculating on which factors could in fact be responsible for such age- and sex-dependency.
Within-individual consistency in methylation at the nestling and adult stage
The DNA methylation landscape is dynamically patterned during development and also at later life stages58, implying that methylation can change during life, as present results suggest to be the case at four loci (see above). However, individuals that at the nestling stage had high methylation also did so at the 1-year old stage at four of the six loci that we investigated, implying that relative methylation levels tended to remain constant across individuals during the first year of life. Consistency of methylation level has been previously documented during the first life stages (e.g. from childhood to first pregnancy) in humans59. The present results thus suggest that individual consistency in behavioral patterns that is observed at phenological or other behavioral traits that may depend on circadian clocks, can partly result from consistency of individual methylation level during life47,48. Cry1 and per3 represented an exception to the positive relationship between methylation levels at different life stages, with cry1 even showing a negative relationship between life stages. However, the mechanism and the function, if any, of such negative relationship for cry1 remains obscure at present.
Methylation in parents and offspring
The results from the mother-offspring resemblance analyses clearly showed high resemblance at the nestling stage but such associations almost completely vanished as offspring reached the adult stage. Mother-offspring resemblance could arise via shared-environment effects. Additional, not necessarily mutually exclusive mechanisms that can induce resemblance in methylation are via the maternal germline, genetically-based variation in susceptibility to methylation and/or early maternal effects mediated by egg quality.
Interestingly, for per2 we found evidence for differential resemblance of maternal methylation levels with methylation of offspring of either sex. This result is consistent with observations of offspring sex-specific inheritance of methylation levels of either parent in ICR (Imprinting Control Regions)60.
Extra-pair fertilizations afforded an opportunity to indirectly test for environmental effects on father-offspring resemblance in methylation. The results, in this respect, were complex and only partly conclusive. For clock poly-Q there was a significant paternity by paternal methylation interaction effect on offspring methylation at the nestling stage, conclusively showing that resemblance with the offspring was larger for genetic that for non-genetic fathers. Hence, for clock poly-Q there was evidence for inheritance of methylation levels independent of environmental effects. Such father-offspring resemblance could arise because of transmission of paternal methylation levels via germinal cells and/or via genetic effects on susceptibility to methylation in prenatal or early postnatal life stages. Inheritance of methylation levels at clock poly-Q can have major ecological consequences because in the barn swallow methylation at clock poly-Q predicts variation at spring phenological traits37. Father- (and mother-) offspring resemblance in methylation could possibly contribute to establish parent-offspring resemblance in phenology traits.
For the other five loci, the interaction effect between paternity and paternal methylation at the nestling stage was either marginally non-significant after FDR correction (clock 5-UTR), or far from statistical significance (remaining loci), implying no statistically significant difference in the slopes of the relationships between methylation of the offspring and that of their genetic compared to non-genetic, social fathers. However, a closer inspection of the relationships within either paternity group suggested that the relationships between offspring and genetic-fathers at some loci had larger associated effect sizes (t values) than those between offspring and social fathers at the nestling or at the adult stages of the offspring. Albeit non-conclusive, these results may thus suggest father-offspring resemblance of methylation levels independent of environmental effects also at the other loci, besides clock poly-Q. In fact, the relationships between methylation of the non-genetic, social fathers and the offspring that they attended were not statistically significant. However, for clock poly-Q the relationship between methylation of the offspring at the nestling stage and that of their social father was negative, rather than null or positive as expected in the case of no or, respectively, non-null environmental effects. The interpretation of the latter result is open to speculation. Because the genetic father-offspring relationship was positive, whereas the relationship was negative for the social father, it might be speculated that mothers generating extra-pair offspring were fertilized by extra-pair males whose methylation level was negatively correlated with that of their mate. The function of such differential assortative mating34 with the social mate compared to the extra-pair mate might consist in increasing the epigenetic diversity of the offspring, with potential advantages in terms of adaptation to unpredictable ecological conditions, in line with the theory that considers the function of extra-pair fertilizations as a mean to increase phenotypic diversity of the offspring61,62,63.
Overall, the parent-offspring resemblance analyses suggested that genetic parents of either sex can differentially contribute to determine offspring methylation at individual loci and at different offspring life stages. Genetic fathers, in particular, seemed to contribute to offspring methylation at some loci of the clock genes also at the adult stage, whereas this was not the case for genetic mothers. In addition, the effect of methylation of the social (non-genetic) father on methylation of the extra-pair offspring was very weak, suggesting only minor, if any, effects of shared environment between fathers and offspring on resemblance in methylation.
Methylation and breeding date
Genetic polymorphism at clock has been shown to be associated with phenological variation in several, though not all, vertebrate species tested so far50,64,65,66, including the barn swallow51. Besides genetic polymorphism, however, methylation state may also intervene in regulating clock-dependent photoperiodic responses. In a previous study we showed that methylation at the clock poly-Q predicted timing of breeding of old female barn swallows from the same geographical area where the present study was carried out37. To assess the robustness of the previously reported relationship, here we considerably expanded the sample size of the previous study and confirmed that large methylation at clock poly-Q is associated with early breeding in females but not in males. The relationship between breeding date and methylation at clock poly-Q held for both yearling (not tested in a previous study37) and older individuals, which are known to breed on average at different times48. As previously noted37, the sign of the relationship between methylation at clock poly-Q and breeding date is consistent with the expectation. This is the case because increasing methylation levels are expected to reduce the expression of CLOCK/BMAL1 transcription factor, thus reducing the transcriptional activation of the clock genes.
In addition, the present analyses on an expanded sample disclosed a marginally non-significant (P = 0.062) negative association between breeding date, independently of sex and age, and methylation at the clock 5′-UTR locus, which was not previously detected37, suggesting that methylation at other regions of clock may also be functionally relevant to timing of breeding. However, we did not find any hint for an association between methylation at other four circadian clock genes and breeding date in both sexes, which were not tested in the previous study.
Populations of birds and other organisms have undergone rapid phenotypic changes in phenological traits during the last decades, likely in response to climate change67, which have occurred at a pace that may be difficult to explain by invoking relatively slow micro-evolutionary processes67. The observed association between methylation at clock and breeding date (present study) or migration dates37, and the observation of parent-offspring resemblance in methylation at this gene may help explaining rapid phenological change in the barn swallow and, potentially, also in several other bird species67,68. This is the case because methylation at clock poly-Q, which shows high resemblance between genetic parents and young offspring, may mediate phenological shifts at least in early life stages, with likely carry-over consequences in adulthood, independently of any micro-evolutionary change in population genetic composition at clock.
Concluding remarks
In conclusion, we found extensive evidence for differential variation in methylation levels according to sex and age, whose physiolocal and ecological causes remain to be elucidated. In addition, we also showed extensive, yet locus-dependent parent-offspring resemblance in methylation levels, with a hint that, for fathers, resemblance with the offspring is more strongly mediated by variation in germline methylation, genetic variation in susceptibility to methylation or sperm factors rather than by extrinsic environmental factors during early life stages. Such intergenerational resemblance in methylation has obvious implications for estimates of genetic heritability of traits that depend on clock genes. There is widespread evidence that rapid phenological changes have occurred in animal populations in response to climate change, and the fast pace of such changes has proven difficult to be reconciled with micro-evolutionary changes in populations. Epigenetic effects can rapidly generate phenotypic plasticity in phenological and other traits. The association between methylation at clock and phenological traits, in combination with intergenerational resemblance in methylation levels can thus provide a mechanistic basis to interpret the rapid phenological changes that populations have recently undergone in response to climate change.
Methods
In spring 1999–2002 and 2010–2016 we captured all adult breeding barn swallows at a total of 12 colonies (=farms) located near Milan (Northern Italy) over nine years. Swallows were sexed, individually marked, subjected to blood sampling for methylation and genetic parentage analyses, and released. The breeding birds were assigned to their nest by observation of color rings. The nests were inspected every 2–5 days. When nestlings were 6–12 days old, they were also individually marked and a blood sample was collected for methylation and genetic paternity analyses. In all years, all adults at the breeding colonies were captured to identify the marked offspring born in the previous year that were recruited as 1-year-old breeding adults. These recruits were also subject to blood sampling for methylation analyses. Thus, for each breeding pair that generated one (or more; see below) recruited offspring, we measured methylation for the male and the female mates, and for their offspring both as nestlings in the year of birth and in the year following that of birth, when they reached sexual maturity.
Because in the years preceding those of blood sampling for methylation analyses all breeding adults were captured in the study colonies and barn swallows have extremely high breeding philopatry, in each sampling year we could assume that adult individuals that had not been captured in the previous year were ca. 1-year-old individuals, immigrating from other colonies (unless they were local recruits)41. Thus, in each year, breeding parents could be assigned to either of two age classes: yearling (1-year old) or ‘older’ (two or more years old). All the recruits thus were in the ‘yearling’ age class.
Breeding date (expressed as Julian date of laying of the first egg in the first clutch) was known for all the parents and also for part of their offspring that were later recruited as breeders.
Paternity analyses
Genetic paternity analyses allowed us to assess if the parental father of the recruited offspring was its biological (hereafter ‘genetic’) father or it rather was the non-genetic (hereafter ‘social’) father because the offspring was sired by another male.
Genetic paternity analyses were performed according to previously published protocols41,45. Full methodological details for paternity analyses are reported in the Supplementary Material (SOM).
Methylation analyses
We analyzed a total of six loci at five clock genes: clock, per2, per3, cry1, and timeless. When we started the present work no barn swallow genome sequence was available. We therefore decided to exploit the synteny between passerine birds to isolate the barn swallow genomic sequences of interest. Mainly, the genomes of Ficedula albicollis, Phylloscopus trochilus and Pseudopodoces humilis, were used. Except for clock, where PCR primers for the poly-Q exon and 5′-UTR region of the gene, specific for barn swallow, were already available69, we concentrated our attention on the promoter region of the other selected genes. A 5,000 bp region upstream of the predicted ATG start codon was identified in the different species, sequences were aligned and the portions conserved in all species were examined to identify candidate CpG methylation sites, flanked by DNA sequences suitable for PCR primer design. For each specific gene the primers were designed to cover the greatest possible number of CpG sites within the promoter region, taking into account the necessary length of the PCR amplicon, length of the target sequence, and primers that avoided CpGs. Sequences were designed using PyroMark Assay Design software (Qiagen, Germany). The identified primers were used to amplify the homologous sequence present in the barn swallow genome. Those fragments were purified (Wizard SV Gel and PCR clean-up System, Promega, Madison, Wi, USA) and sequenced (BMR genomics, Padua, Italy) to confirm their homology and to design specific PCR primers required for bisulfite pyrosequencing. Primer sequences are reported in SOM (Table 2S).
One µg DNA (concentration 50 ng/µl) was treated using EZ DNA Methylation-Gold™ Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. Final elution was performed with 30 µl of M-Elution Buffer. Bisulfite-treated DNA was stored at −20 °C and used shortly after treatment. Analysis of DNA methylation was performed using previously published methods37,69, with minor modifications. Briefly, a 50 µl PCR was carried out in 25 µl of GoTaq Green Master mix (Promega, Madison, WI, USA), 1 pmol of the forward primer, 1 pmol of the biotinylated reverse primer, 50 ng of bisulfite-treated genomic DNA and water. The biotin-labelled primers were used to purify the final PCR product using Sepharose beads. The PCR product was bound to Streptavidin Sepharose HP (Amersham Biosciences, Uppsala, Sweden) and the Sepharose beads containing the immobilized PCR product were purified, washed, denatured using a 0.2 M NaOH solution, and washed again using the Pyrosequencing Vacuum Prep Tool (Pyrosequencing, Inc., Westborough, MA), as recommended by the manufacturer. Then, 0.3 µΜ pyrosequencing primer was annealed to the purified single-stranded PCR product and pyrosequencing was performed using the PyroMark MD System (Pyrosequencing, Inc.). The degree of methylation was expressed as percentage of methylated cytosines divided by the sum of methylated and unmethylated cytosines (%5mC). Every sample was measured three times and the average of the replicates was used in statistical analyses. Assays which did not result in good quality pyrograms were repeated. If after 3 replicates the pyrogram quality was still not satisfactory according to pyrosequencing quality controls, the measure resulted in a missing value (see below and SOM for sample sizes). Each assay also included a bisulfite conversion check to verify full conversion of the DNA.
The mean coefficients of variation of methylation levels for the different loci was: clock 5′-UTR: 1.0%; clock poly-Q: 1.3%; cry1: 18.5%; per2: 1.0%; per3: 19%; timeless: 1.0%. The frequency distributions of methylation levels at the six loci is reported in Fig. 1S in SOM
Statistical analyses
The complete version with full details of the Statistical analyses section is reported in the SOM. For clarity, the description of the statistical analyses is here organized so to correspond to the main sub-sections of the Results.
Because methylation variables are proportions, dependent methylation variables were always arcsin√x-transformed.
Methylation in pair members
We tested if methylation was correlated between males and females forming the same social breeding pair in linear mixed models (LMM) assuming a Gaussian error distribution with methylation of females as the dependent variable, methylation of the male mates as the independent variable, and year and colony as random factors.
Methylation in relation to sex and life stage
Variation in methylation according to sex and life stage was also analyzed in LMMs. For each locus, each methylation datum of parents and offspring was classified according to sex (male: 1; female: 2) and life stage (parent and 1-year-old offspring: 1; offspring at the nestling stage: 2). In the models, methylation was considered as the dependent variable, sex and life stage as fixed effect factors (with their two-way interaction), and year and colony as random effects. In addition, we also included a random factor family, i.e. a factor where the father (whether genetic or social), the mother, and their offspring at both the nestling and the adult stage were assigned the same code. The effect of the sex by life stage interaction is always presented in the Results, but when it was non-significant it was excluded from the model including the main effects. In these analyses, we also tested for the (random) effect of year and colony on methylation, by comparing the model including both random effects with reduced models that contained only the year or, respectively, the colony effects, by likelihood ratio tests.
Methylation at the nestling and 1-year-old recruit stage
The relationship between methylation at the 1-year-old stage (dependent variable) and at the nestling stage (independent variable) was analyzed in LMMs with year and colony as random effects.
Methylation in parents and offspring
The relationship between methylation at the nestling stage or at the 1-year-old recruit stage and methylation of the father and mother was analyzed in LMMs where we included methylation of the nestling as the dependent variable and methylation of the father and of the mother as independent variables. In addition, we included a factor ‘paternity’ to account for the fact that the father was the genetic or, respectively, the social, non-genetic father of the offspring. The interaction between paternity and methylation of the father allowed us to test whether the relationship between nestling’s and father’s methylation differed between genetic and social fathers. Finally, we also included a factor offspring sex to account for variation in methylation between male and female offspring and its interaction with maternal methylation, to account for any differential effect of methylation of mothers on methylation of sons as compared to daughters. The interaction between paternal methylation and offspring sex was not tested because of admixture of genetic and social fathers (see also SOM). The effect of sex as well as the effect of the interaction between paternity and paternal methylation were removed from final models when statistically non-significant. However, even when the paternity by father’s methylation was statistically non-significant, we still reported and inspected the within-paternity group relationships because these could provide information as to whether the strength (not the slope) of the relationship between nestling and father differed according to paternity (see also SOM).
As an exception to the above modelling approach, because of very high collinearity between mates at clock 5′-UTR (r = 0.648, n = 64, P < 0.0001; see also Results), for this locus the relationships with the mother or the father were tested in separate models.
Breeding date in relation to methylation
We tested whether methylation at the six loci statistically predicted breeding date. We thus designed LMM with sex and age (yearling vs older) as fixed effects and methylation, and their two-way interactions as predictors. In the models we also included year as a random factor and, in addition, a factor ‘family’ which accounted for the fact that in the dataset pairs of mates and, in some cases, trios (two parents plus one breeding offspring) were included. In the analyses on the effect of methylation at the two clock loci, where we also considered information on methylation and breeding date already reported in a study based in both Italy and Switzerland37, we also included a random factor ‘study area’ (Italy or Switzerland). Because the mean and the variances of the clock 5′-UTR and clock poly-Q between the present methylation data and those previously reported37 differed, methylation data were standardized to a mean of 0 and a variance of 1 within the two datasets.
Because repeated tests were run on the same individuals at the six loci, the results of the statistical tests were corrected according to the false discovery rate (FDR) procedure (see also SOM). Throughout the Results, uncorrected P values are presented and those that remained significant after FDR correction are marked with a ‘†’.
The analyses were run using SAS 9.3 statistical package.
Sample sizes
We sampled 58 pairs of breeding adults, and their recruited offspring both as nestlings and as 1-year-old recruits. Of these breeding pairs, 5 had two recruited offspring and 1 had three recruits included in the sample (65 offspring in total). The 65 offspring comprised 54 males (39 WPO and 15 EPO) and 11 females (8 WPO and 3 EPO). This sex-bias in recruitment is typical of the barn swallow and other birds due to female-biased natal dispersal47.
The size of the sample for each parental sex, life stage and individual locus, after exclusion of the outliers (i.e. observations that deviated more than 3 standard deviations from the mean), is reported in details in the SOM.
Information on breeding date was available also for for 37 offspring that were recruited as yearling breeders. In addition, we considered breeding date and methylation data at the two clock loci for 58 males and 26 females that were already included in a previous study37.
The Regione Lombardia administration gave permission for this study (permits no. 2959 and 11316; see also Compliance with guidelines and regulations section at the bottom of the main text).
All experiments were performed in accordance with relevant guidelines and regulations. All experimental protocols were approved by the Regione Lombardia administration (permits no. 2959 and 11316) following approval by the Istituto Superiore per la Protezione e la Ricerca Ambientale (ISPRA).
Data Availability
The dataset supporting the conclusions of this article will be made available by the corresponding author upon request.
References
Waddington, C. H. Evolutionary systems-animal and human. Eugen. Rev. 52, 23–29 (1960).
Jablonka, E. & Raz, G. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 84, 131–176 (2009).
Wang, Y., Liu, H. & Sun, Z. Lamarck rises from his grave: Parental environment-induced epigenetic inheritance in model organisms and humans. Biol. Rev. Camb. Philos. Soc. 92, 2084–2111 (2017).
Jablonka, E. & Lamb, M. J. The changing concept of epigenetics. Ann. N. Y. Acad. Sci. 981, 82–96 (2002).
Haig, D. The (dual) origin of epigenetics. Cold Spring Harb. Symp. Quant. Biol. 69, 67–70 (2004).
Holliday, R. Epigenetics: a historical overview. Epigenetics 1, 76–80 (2006).
Epigenetics. (Cold Spring Harbor Laboratory Press 2015).
Szyf, M. Lamarck revisited: epigenetic inheritance of ancestral odor fear conditioning. Nat. Neurosci. 17, 2–4 (2014).
Szyf, M. Nongenetic inheritance and transgenerational epigenetics. Trends Mol. Med. 21, 134–144 (2015).
Sharma, R. et al. Effects of increased paternal age on sperm quality, reproductive outcome and associated epigenetic risks to offspring. Reprod. Biol. Endocrinol. 13, 35 (2015).
Hanson, M. A. & Skinner, M. K. Developmental origins of epigenetic transgenerational inheritance. Environ Epigenet 2 (2016).
Vaiserman, A. M., Koliada, A. K. & Jirtle, R. L. Non-genomic transmission of longevity between generations: potential mechanisms and evidence across species. Epigenetics Chromatin 10, 38 (2017).
Jensen, P. Transgenerational epigenetic effects on animal behaviour. Prog. Biophys. Mol. Biol. 113, 447–454 (2013).
Dias, B. G. & Ressler, K. J. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat. Neurosci. 17, 89–96 (2014).
Trerotola, M., Relli, V., Simeone, P. & Alberti, S. Epigenetic inheritance and the missing heritability. Hum. Genomics 9, 17 (2015).
van Otterdijk, S. D. & Michels, K. B. Transgenerational epigenetic inheritance in mammals: how good is the evidence? FASEB J. 30, 2457–2465 (2016).
Richards, E. J. Inherited epigenetic variation—revisiting soft inheritance. Nat. Rev. Genet. 7, 395 (2006).
Jensen, P. Behaviour epigenetics–the connection between environment, stress and welfare. Appl. Anim. Behav. Sci. 157, 1–7 (2014).
Tsankova, N., Renthal, W., Kumar, A. & Nestler, E. J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367 (2007).
Laine, V. N. et al. Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 7, 10474 (2016).
Verhulst, E. C. et al. Evidence from pyrosequencing indicates that natural variation in animal personality is associated with DRD 4 DNA methylation. Mol. Ecol. 25, 1801–1811 (2016).
Riyahi, S., Sánchez-Delgado, M., Calafell, F., Monk, D. & Senar, J. C. Combined epigenetic and intraspecific variation of the DRD4 and SERT genes influence novelty seeking behavior in great tit Parus major. Epigenetics 10, 516–525 (2015).
Soulsbury, C. D. et al. Age-and quality-dependent DNA methylation correlate with melanin-based coloration in a wild bird. Ecol. Evol (2018).
André, S. M. & Markowski, V. P. Learning deficits expressed as delayed extinction of a conditioned running response following perinatal exposure to vinclozolin. Neurotoxicol. Teratol. 28, 482–488 (2006).
Skinner, M. K., Anway, M. D., Savenkova, M. I., Gore, A. C. & Crews, D. Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS One 3, e3745 (2008).
Leroux, S. et al. Embryonic environment and transgenerational effects in quail. Genet. Sel. Evol. 49, 14 (2017).
Heard, E. & Martienssen, R. A. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109 (2014).
Lane, M., Robker, R. L. & Robertson, S. A. Parenting from before conception. Science 345, 756–760 (2014).
Kappeler, L. & Meaney, M. J. Epigenetics and parental effects. Bioessays 32, 818–827 (2010).
Curley, J. P., Mashoodh, R. & Champagne, F. A. Epigenetics and the origins of paternal effects. Horm. Behav. 59, 306–314 (2011).
Nelson, V. R. & Nadeau, J. H. Transgenerational genetic effects. Epigenomics 2, 797–806 (2010).
Barlow, D. P. & Bartolomei, M. S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 6 (2014).
Gu, T.-P. et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610 (2011).
Jiang, Y., Bolnick, D. I. & Kirkpatrick, M. Assortative mating in animals. Am. Nat. 181, E125–38 (2013).
Potok, M. E., Nix, D. A., Parnell, T. J. & Cairns, B. R. Germline epigenetics, and reprogramming in zebrafish early embryos. Epigenetics Chromatin 6, O23 (2013).
Rubenstein, D. R. et al. Sex-specific fitness effects of unpredictable early life conditions are associated with DNA methylation in the avian glucocorticoid receptor. Mol. Ecol. 25, 1714–1728 (2016).
Saino, N. et al. Migration phenology and breeding success are predicted by methylation of a photoperiodic gene in the barn swallow. Sci. Rep. 7, 45412 (2017).
Bell-Pedersen, D. et al. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 6, 544–556 (2005).
Eckel-Mahan, K. & Sassone-Corsi, P. Metabolism and the circadian clock converge. Physiol. Rev. 93, 107–135 (2013).
Altman, R. B. Avian Medicine and Surgery. (Saunders, 1997).
Costanzo, A. et al. Lifetime reproductive success, selection on lifespan, and multiple sexual ornaments in male European barn swallows. Evolution 71, 2457–2468 (2017).
Romano, A., Costanzo, A., Rubolini, D., Saino, N. & Møller, A. P. Geographical and seasonal variation in the intensity of sexual selection in the barn swallow Hirundo rustica: a meta-analysis. Biol. Rev. Camb. Philos. Soc. 92, 1582–1600 (2017).
Møller, A. P. Intraspecific nest parasitism and anti-parasite behaviour in swallows, Hirundo rustica. Anim. Behav. 35, 247–254 (1987).
Møller, A. P., Brohede, J., Cuervo, J. J., de Lope, F. & Primmer, C. Extrapair paternity in relation to sexual ornamentation, arrival date, and condition in a migratory bird. Behav. Ecol. 14, 707–712 (2003).
Costanzo, A. et al. Extrapair fertilizations vary with female traits and pair composition, besides male attractiveness, in barn swallows. Anim. Behav. 134, 183–191 (2017).
Yom-Tov, Y. An updated list and some comments on the occurrence of intraspecific nest parasitism in birds. Ibis 143, 133–143 (2008).
Møller, A. P. Sexual selection and the Barn Swallow. Model Systems in Behavioral Ecology: Integrating Conceptual, Theoretical, and Empirical Approaches 359–380 (1994).
Turner, A. The barn swallow. (T & AD Poyser, London, 2006).
Staff, E. Go forth and replicate. Nature 536, 373 (2016).
Liedvogel, M., Szulkin, M., Knowles, S. C. L., Wood, M. J. & Sheldon, B. C. Phenotypic correlates of Clock gene variation in a wild blue tit population: evidence for a role in seasonal timing of reproduction. Mol. Ecol. 18, 2444–2456 (2009).
Caprioli, M. et al. Clock gene variation is associated with breeding phenology and maybe under directional selection in the migratory barn swallow. PLoS One 7, e35140 (2012).
Jones, M. J., Goodman, S. J. & Kobor, M. S. DNA methylation and healthy human aging. Aging Cell 14, 924–932 (2015).
Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 (2002).
Paoli-Iseppi, D. et al. Measuring animal age with DNA methylation: From humans to wild animals. Front. Genet. 8, 106 (2017).
Hall, E. et al. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 15, 522 (2014).
Rancourt, R. C., Schellong, K., Tzschentke, B., Henrich, W. & Plagemann, A. DNA methylation and expression of proopiomelanocortin (POMC) gene in the hypothalamus of three-week-old chickens show sex-specific differences. FEBS Open Bio (2018).
Liechti, F. et al. Timing of migration and residence areas during the non-breeding period of barn swallows Hirundo rustica in relation to sex and population. J. Avian Biol. 46, 254–265 (2015).
Singer, Z. S. et al. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol. Cell 55, 319–331 (2014).
Chen, S. et al. Consistency and Variability of DNA Methylation in Women During Puberty, Young Adulthood, and Pregnancy. Genet. Epigenet. 9, 1179237X17721540 (2017).
Champroux, A., Cocquet, J., Henry-Berger, J., Drevet, J. R. & Kocer, A. A Decade of Exploring the Mammalian Sperm Epigenome: Paternal Epigenetic and Transgenerational Inheritance. Front Cell Dev Biol 6, 50 (2018).
Brooked, M. G., Rowley, I., Adams, M. & Baverstock, P. R. Promiscuity: an inbreeding avoidance mechanism in a socially monogamous species? Behav. Ecol. Sociobiol. 26, 191–199 (1990).
Westneat, D. F., Sherman, P. W. & Ml., M. The ecology and evolution of extra-pair copulations in birds. Curr Ornithol 7, 331–369 (1990).
Schmoll, T. A review and perspective on context-dependent genetic effects of extra-pair mating in birds. J. Ornithol. 152, 265–277 (2011).
O’Malley, K. G., Camara, M. D. & Banks, M. A. Candidate loci reveal genetic differentiation between temporally divergent migratory runs of Chinook salmon (Oncorhynchus tshawytscha). Mol. Ecol. 16, 4930–4941 (2007).
Chakarov, N., Jonker, R. M., Boerner, M., Hoffman, J. I. & Krüger, O. Variation at phenological candidate genes correlates with timing of dispersal and plumage morph in a sedentary bird of prey. Mol. Ecol. 22, 5430–5440 (2013).
Romano, A. et al. Circadian genes polymorphism and breeding phenology in a resident bird, the yellow-legged gull. J. Zool. 304, 117–123 (2018).
Møller, A. P., Fiedler, W. & Berthold, P. Effects of Climate Change on Birds. (OUP Oxford, 2010).
Charmantier, A. & Gienapp, P. Climate change and timing of avian breeding and migration: evolutionary versus plastic changes. Evol. Appl. 7, 15–28 (2014).
Romano, A. et al. Methylation of the circadian Clock gene in the offspring of a free-living passerine bird increases with maternal and individual exposure to PM10. Environ. Pollut. 220, 29–37 (2017).
Acknowledgements
We are grateful to all the students who helped during field work and to farm owners who allowed us to enter their properties. No ad hoc funding was available for the present study.
Author information
Authors and Affiliations
Contributions
V.B., N.S. conceived the study; R.A., A.C., M.C., M.P., A.R., D.R., N.S. collected field data; B.A., A.C., M.C., L.G., J.M., M.P. performed laboratory analyses; R.A., N.S. performed the statistical analyses; V.B., A.C., G.F., L.G., A.R., N.S. drafted the manuscript. All authors gave final approval for publication.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saino, N., Albetti, B., Ambrosini, R. et al. Inter-generational resemblance of methylation levels at circadian genes and associations with phenology in the barn swallow. Sci Rep 9, 6505 (2019). https://doi.org/10.1038/s41598-019-42798-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42798-3
- Springer Nature Limited
This article is cited by
-
Epigenetics and seasonal timing in animals: a concise review
Journal of Comparative Physiology A (2024)
-
Birds of a feather flock together: a dataset for Clock and Adcyap1 genes from migration genetics studies
Scientific Data (2023)