
Abstract
Streptomycetes are important biotechnological bacteria with complex differentiation. Copper is a well-known positive regulator of differentiation and antibiotic production. However, the specific mechanisms buffering cytosolic copper and the biochemical pathways modulated by copper remain poorly understood. Here, we developed a new methodology to quantify cytosolic copper in single spores which allowed us to propose that cytosolic copper modulates asynchrony of germination. We also characterised the SCO2730/2731 copper chaperone/P-type ATPase export system. A Streptomyces coelicolor strain mutated in SCO2730/2731 shows an important delay in germination, growth and sporulation. Secondary metabolism is heavily enhanced in the mutant which is activating the production of some specific secondary metabolites during its whole developmental cycle, including germination, the exponential growth phase and the stationary stage. Forty per cent of the S. coelicolor secondary metabolite pathways, are activated in the mutant, including several predicted pathways never observed in the lab (cryptic pathways). Cytosolic copper is precisely regulated and has a pleiotropic effect in gene expression. The only way that we know to achieve the optimal concentration for secondary metabolism activation, is the mutagenesis of SCO2730/2731. The SCO2730/2731 genes are highly conserved. Their inactivation in industrial streptomycetes may contribute to enhance bioactive compound discovery and production.
Similar content being viewed by others


Introduction
Streptomycetes are important biotechnological bacteria from which two thirds of the bioactive secondary metabolites used in clinic (mainly antibiotics, but also antitumorals, immunosupressors, etc.) were discovered1,2. They have a complex developmental cycle that makes this bacterium a multicellular prokaryotic model including programmed cell death (PCD) and hyphae differentiation, which leads to aerial mycelium formation and sporulation3,4.
One of the less studied stages of Streptomyces development is spore germination. There are proteomic and transcriptomic works demonstrating that spore germination is highly regulated (reviewed in Bobek et al.5). However, the biomolecular mechanisms controlling germination remain poorly characterised6,7. There are some proteins known to be involved in Streptomyces spore germination: NepA, a structural cell wall protein involved in the maintenance of spore dormancy in S. coelicolor8; SsgA, a protein marking cell-wall sites where germination takes place9; resuscitation-promoting factors (Rpfs), cell wall hydrolases10 controlling germination; OsdR, a Streptomyces orthologue to the M. tuberculosis DevR dormancy regulator, which was demonstrated to be functional in Mycobacterium11; and SCO4439, a D-alanyl-D-alanine carboxypeptidase that controls spore peptidoglycan crosslinking and conditions spore germination12. One of the most intriguing and poorly known aspects of spore germination is asynchrony i.e. some spores germinate early, while others take a long time to germinate or even do not germinate5. To the best of our knowledge, NepA is the only known protein that contributes to this asynchrony8.
Copper has been characterised as a positive regulator of Streptomyces differentiation (aerial mycelium, sporulation) and antibiotic production13,14. By contrast, vegetative growth was described to be unaffected by copper14. At high concentrations (over 750 µM), the positive effect in aerial mycelium development and sporulation becomes a negative effect (delay)15. The mechanisms controlling copper trafficking in Streptomyces remains poorly understood. Worrall and Vijgenboom16 predicted the existence of two copper chaperone/P-type ATPases (CopZ/CopA) modulating copper export (SCO1045/1046, SCO2730/2731). They demonstrated that the expression of the genes encoding these transporters is regulated by CsoR, the master transcriptional repressor modulating copper effects in gene transcription16. This research group also demonstrated that the number of genes responding to copper stress is much higher than those regulated by CsoR. Dwarakanath et al.15 proposed a model in S. lividans describing the copper effect modulating CsoR activity and expression of the SCO2730/2731 and SCO1045/1046 copper chaperones/transporters: under “normal” cytosolic copper concentration, csoR and SCO2730 (copZ) are expressed at a low level; when cytosolic copper concentration increases, CsoR and SCO2730 (CopZ) buffer copper at first, but as soon they become saturated, the CsoR repression of SCO2730/2731 and SCO1045/1046 is unblocked, increasing the expression of these copper secretion transporters that maintain copper homeostasis. Interestingly, CopZ-3079 (the S. lividans SCO2730 orthologue) was demonstrated in vitro to have a 5-fold higher affinity for Cu than CopZ-1317 (the SCO1045 orthologue)17, indicating that its physiological relevance in copper trafficking is higher at low copper concentrations, as proposed in the Dwarakanath et al. model15.
The copper import mechanisms in Streptomyces remain basically unknown. The existence of a membrane-bound periplasmic cupric reductase and a P-type ATPase importing Cu(I) has been postulated18.
In this work we focus on the characterisation of the SCO2730/SCO2731 copper export system. SCO2730 encodes for a putative copper chaperone15, highly conserved in the Streptomyces genus. SCO2731 encodes a conserved putative P-type ATPase, which was predicted to transport the copper carried out by the SCO2730 chaperone15. Our results contribute to the knowledge of complex pleiotropic effects of copper in Streptomyces development, including important effects regulating the asynchrony of germination, vegetative growth and activation of secondary metabolism, which were previously un-described.
Results
SCO2730/2731 are highly conserved in Streptomyces
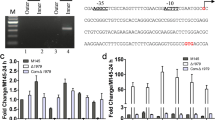
SCO2730 and SCO2731 show an average amino acid similarity of 88.7% and 81% respectively, among S. griseus, S. avermitillis, S. lividans, S. clavuligerus and S. venezuelae. A comparison of SCO2730 orthologues reveal that the SCO2730 ORF probably starts at position 66 (ATG, Met) instead of the GTG (Val), annotated in the StrepDb database (http://strepdb.streptomyces.org.uk/). The S. clavuligerus SCO2731 orthologue is not annotated in the StrepDb database, but is present downstream of SCLAV_1906, the SCO2730 orthologue (data not shown). The SCO2730/2731 orthologues are together in all of the Streptomyces chromosomes analysed. However, the SCO2728–SCO2731 synteny (Fig. 1A) is only present in S. coelicolor and S. lividans.

SCO2730/2731 expression and phenotype of the SCO2730::Tn5062 mutant. (A) Outline of the SCO2728-2731 chromosomal region illustrating the position of the primers used for RT-PCR. Fragments not amplified in the SCO2730::Tn5062 mutant are outlined in red. (B) RT-PCRs demonstrating co-transcription of the following: SCO2728 and SCO2729 (1), SCO2729 and SCO2730 (2), SCO2730 and SCO2731 (3 and 4). Full-length gels are shown in SI Fig. S2. (C) Percentage of germination in the mutant (M) and the S. coelicolor wild-type strain (Wt) at 10 h. (D) Confocal microscope images of the mutant and the wild-type strain stained with SYTO9 (green) and propidium iodide (red) at 10 h. (E) Growth curves (three biological replicates). (F) Undecylprodigiosin production (three biological replicates). (G) Actinorhodin production (three biological replicates). (H) CDA production. (I) Sporulation (grey colour) in SFM medium.
The SCO2730 mutation affects spore germination, antibiotic production and sporulation
Cosmid C46.2.D06 was used to obtain the S. coelicolor SCO2730::Tn5062 mutant harbouring SCO2730 interrupted by Tn506219. RT-PCR demonstrated that SCO2730 and SCO2731 are co-transcribed in the S. coelicolor wild-type strain (amplicons 3 and 4 in Fig. 1A,B). The SCO2731 expression was affected in the SCO2730::Tn5062 mutant (amplicon 4 in Fig. 1B). Surprisingly, amplicon 3 was present in the mutant (Fig. 1B), indicating the existence of a promoter (promoter 4 in Fig. 1A) between the two forward oligonucleotides used for RT-PCR 3 and 4. RT-PCR also revealed that SCO2730 was co-transcribed with SCO2729 (amplicon 2 in Fig. 1B) and that SCO2728 was co-transcribed with SCO2729, suggesting that all of these genes might be transcribed as a single operon.
Spore germination was dramatically delayed in the SCO2730::Tn5062 mutant, compared to the wild-type strain (19.5% in the mutant vs. 100% in the wild-type strain at 10 h in sucrose-free R5A medium) (Fig. 1C). In solid sucrose-free R5A, the S. coelicolor wild-type strain reaches 100% of the germination at approximately 8 hours. However, as germination is highly delayed in the SCO2730::Tn5062 mutant, in this study we show germination at 10 hours. Moreover, there was a high rate of cell death during germination in the SCO2730::Tn5062 mutant (propidium iodide staining, red colour in Fig. 1D, 10 h). Growth was highly delayed in the mutant (Fig. 1E), while undecylprodigiosin and actinorhodin production was doubled (Fig. 1F,G). Calcium dependent antibiotic (CDA) production was slightly increased in the SCO2730::Tn5062 mutant (Fig. 1H). The SCO2730::Tn5062 mutant produced undecylprodigiosin starting with the first time point analysed (24 hours) and indicated that secondary metabolism is accelerated in this mutant (Fig. 1F). Sporulation was highly delayed and reduced in the SCO2730::Tn5062 mutant (Fig. 1I).
The SCO2730::Tn5062 mutant phenotype in germination and antibiotic production depend on the alteration of SCO2730 and SCO2731 expression
Based on the above results, we proceeded to identify the gene/s responsible for the phenotypes detected. We complemented the SCO2730::Tn5062 mutant with different combinations of the three promoters located upstream of SCO2730, combined with the SCO2730 and/or SCO2731 ORFs (strains 1–6 in Fig. 2A). The SCO2730::Tn5062 mutant strains harbouring the different promoters, together with SCO2730 and/or SCO2731, complemented part of the germination (measured at 10 hours) (Fig. 2B) or antibiotic production phenotypes (Fig. 2C–E). Undecylprodigiosin and actinorhodin production was measured at 150 hours, when the maximum production was reached, while CDA was measured in solid plates (see Methods). Only the SCO2730::Tn5062 mutant strain harbouring the three promoters and the SCO2730/2731 ORFs (strain 1) complemented the SCO2730::Tn5062 mutant phenotype in germination (Fig. 2B) and antibiotic production (Fig. 2C–E). Undecylprodigiosin production was restored to the wild-type level. However, the wild-type phenotype was not fully restored. Germination was accelerated in complemented strain 1 compared to the SCO2730::Tn5062 mutant (notice that germination values in Fig. 2B are out of the average ± SD confidence intervals), but strain 1 germination did not reach the wild-type level (Fig. 2B); actinorhodin and CDA production was reduced in strain 1 compared to the mutant (actinorhodin production was out of the average ± SD confidence intervals in Fig. 2D; CDA production was not detectable in strain 1 in Fig. 2E), but the production of both antibiotics was less than that of the wild-type strain.

Complementation of the wild-type phenotype in the SCO2730::Tn5062 mutant. (A) Outline of the SCO2728-2731 chromosomal region and the six complementation constructions created. Promoter localisations are indicated. (B) Percentage of germination at 10 hours. (C) Undecylprodigiosin production at 150 hours. (D) Actinorhodin production at 150 hours. (E) CDA production. (F) SCO2730 expression (qRT-PCR, two biological replicates). (G) SCO2731 expression (qRT-PCR, two biological replicates). Wt: S. coelicolor wild-type strain. Wt*: S. coelicolor harbouring pNG3. Arrows indicate complementation of the phenotype in the SCO2730::Tn5062 mutant harbouring SCO2730/2731 under the control of promoters P1,P2 and P3. M*: SCO2730::Tn5062 mutant harbouring pNG3.
The expression of the SCO2730 and SCO2731 genes was not restored in any of the complemented strains (Fig. 2F,G), indicating the existence of further unknown regulation beyond the three promoters considered in this work and explaining why the wild type phenotype was not restored in the complementation strains. The SCO2730/2731 genes were overexpressed in the mutant strain harbouring the three promoters and the SCO2730/2731 ORFs (strain 1 in Fig. 2F,G). Interestingly, all of the SCO2730::Tn5062 complemented strains that overexpressed SCO2731 (strains 1, 3, 4 and 6) (Fig. 2G) showed an increase in germination (Fig. 2B) compared to the SCO2730::Tn5062 mutant.
SCO2730 and SCO2731 expression correlates with copper secretion during germination
We analysed the cytosolic copper concentration and the expression of the genes encoding the two copper secretion systems described in S. coelicolor (SCO2730/2731 and SCO1045/1046) during development15 (germinated spores, 5–10 h; mycelium, 20–70 h) (Fig. 3A,B). Cytosolic copper (normalised against cytosolic protein) decreases quickly during germination (Fig. 3A). The maximum levels of SCO2730/2731 expression were reached at 10 hours, coinciding with the lowest cytosolic copper concentration and the lowest expression of SCO1045/1046 (Fig. 3B). This result suggests that SCO2730/2731 chaperone/transport system is the main effector in copper secretion during germination.

Cytosolic copper and SCO2730/2731 expression during S. coelicolor development. (A) Cytosolic copper during development in sucrose free R5A liquid cultures. Vegetative growth and secondary metabolism time windows are indicated. (B) SCO2730/2731 and SCO1045/1046 expression during development (qRT-PCRs, two biological replicates). (C) Cytosolic copper in dormant spores (three biological replicates). (D) Cytosolic copper in germinating spores (5- and 10 h) and mycelium (60 h) (three biological replicates). (E) Percentage of germination (three biological replicates) in Wt and Wt80Cu. (F) Macroscopic view of sporulation (grey colour) of the wild-type strain in SFM and SFM amended with 80 µM CuSO4. (G) Histogram showing the cytosolic copper in single spores of the wild type strains obtained in 80 µM CuSO4 copper amended SFM cultures. Wt, wild-type strain. Wt80Cu, spores of the wild-type strain obtained in 80 µM CuSO4 amended SFM cultures.
Next, we analysed cytosolic copper in dormant spores, germinated spores (5 and 10 hours) (Fig. 3C) and mycelium (60 h) of the SCO2730::Tn5062 mutant and the S. coelicolor wild-type strains (Fig. 3D). Dormant spores accumulate high amounts of trehalose and glycogen20 that are not present in germinated spores or mycelia. This makes normalisation and comparison of cytosolic copper concentrations between dormant and germinated spores/mycelia, a challenge. Cytosolic copper was normalised in dormant spores against dry weight, while in germinated spores and mycelium, it was normalised against cytosolic protein (see Methods). Consequently, copper abundances can be compared between dormant spores (Fig. 3C) or between germinated spores and the mycelium (Fig. 3D), but not between dormant spores and germinated spores/mycelium. In the case of the wild-type strain we analysed spores obtained in SFM solid cultures (Wt) and spores obtained in SFM solid cultures amended with 80 µM Cu (Wt80Cu). Cytosolic copper concentration was similar in the SCO2730::Tn5062 mutant and in the Wt dormant spores (11 ± 6 and 8.5 ± 3 ng Cu/mg dry weight respectively) but it was much higher in the Wt80Cu spores (208 ± 30, 24-fold higher than in the wild-type strain) (Fig. 3C). Cytosolic copper dropped dramatically during germination in copper unmodified sucrose-free R5A cultures of the wild-type strain inoculated with Wt or Wt80Cu spores (66-fold and 70- fold drop respectively from 5 to 10 hours) (Fig. 3D), but the reduction in cytosolic copper was lesser in the cultures inoculated with SCO2730::Tn5062 mutant spores (26-fold drop from 5 to 10 hours) (Fig. 3D). Cytosolic copper was higher at 10 hours in the SCO2730::Tn5062 mutant (45.7 ± 5.4 ng Cu/mg protein) compared to the wild-type strain (cultures using Wt or Wt80Cu spores) (18.6 ± 1.6 and 24.2 ± 2.4 ng Cu/mg protein respectively) (Fig. 3D). By contrast, cytosolic copper in the mycelium of the SCO2730::Tn5062 mutant (153 ± 20 ng Cu/mg protein) and the wild-type strains (145.7 ± 18 and 155.4 ± 41 ng Cu/mg protein in Wt and Wt80Cu spores respectively) are comparable (Fig. 3D).
Interestingly, germination was highly accelerated in the Wt80Cu spores compared to the Wt spores (both growing in copper unmodified cultures) (Fig. 3E, 6 hours culture), while growth and antibiotic production were similar (data not shown). Germination starts at comparable time points in the Wt and the Wt80Cu spores (data not shown), but it is much more synchronous in the Wt80Cu than in the Wt, which lead to an acceleration in the time point at which 100% germination is reached (Fig. 3E). Sporulation in the SFM cultures adjusted with 80 µM Cu was delayed compared to the SFM unmodified cultures (Fig. 3F), confirming the effect of copper modulation of sporulation reported before15.
Cytosolic copper quantification in single spores
To analyse cytosolic copper in individual spores, we adapted the method that we developed for the analysis of single eukaryotic cells21 to the study of Streptomyces single spores. Cytosolic copper in the Wt and in the SCO2730::Tn5062 mutant single spores was below the sensitivity of the analysis (see Methods). However, we were able to quantify cytosolic copper in the Wt80Cu spores (Fig. 3G). There was a high variability in the cytosolic copper with a minimum of 0.51 fg Cu per spore and a maximum of 3.83 fg Cu per spore.
Cytosolic copper modulates spore germination and SCO2730/2731–SCO1045/1046 gene expression
The above results suggest an effect of SCO2730 and SCO2731 in the modulation of cytosolic copper concentration and germination (Figs 1 and 3). We next analysed the effect of copper on germination. Germination was delayed in sucrose-free R5A liquid cultures treated with copper concentrations over 40 µM (Fig. 4A). Interestingly, cytosolic copper was not proportional to the copper added to the medium. An increase of 2-fold in the extracellular copper (from 40 to 80 µM) correlated with a 1.3-fold increase of cytosolic copper (from 201.3 ± 7.4 to 272.5 ± 38.8 ng/mg protein) (Fig. 4B).

Extracellular copper effect in germination, cytosolic copper levels and the expression of copper related genes. (A) Variation of the percentage of germination with the extracellular copper concentration in sucrose free R5A solid cultures (three biological replicates). (B) Variation of intracellular copper during germination (6 h) depending on the extracellular copper concentration in sucrose free R5A liquid cultures (three biological replicates). (C) SCO2730/2731, SCO1045/1046 and SCO4136 expression during germination (8 h) in 40 and 80 µM CuSO4 amended cultures (qRT-PCRs, two biological replicates, sucrose free R5A liquid medium).
In order to further study the biomolecular mechanism buffering cytosolic copper during germination, we analysed the expression of the two copper secretion systems reported in Streptomyces (SCO2730/2731, SCO1045/1046)15 during germination in copper modified (by adding 0-, 40- or 80 µM extracellular copper) sucrose-free R5A cultures. The SCO2730/2731 maximum expression (compared to the Wt germinating spores at the 0 µM copper modified medium) was reached at 40 µM extracellular copper, while the maximum expression of the SCO1045/1046 genes was reached at 80 µM copper (Fig. 4C). We further analysed the expression of csoR (SCO4136), the master transcriptional repressor modulating copper effects in gene transcription15. CsoR expression was equally activated during germination at high cytosolic copper concentration, which was reached in the 40 and 80 µM copper modified sucrose-free R5A cultures (Fig. 4C). As reported before15, copper binds to CsoR blocking its interaction with DNA and activating the expression of genes repressed by CsoR, including the csoR gene.
Comparison of the transcriptomes of SCO2730::Tn5062 and S. coelicolor wild-type Cu-amended/non-amended spores, during germination
In order to investigate the molecular mechanisms controlling the phenotypes observed during germination of the SCO2730::Tn5062 mutant and the Wt80Cu spores, we compared their transcriptomes during germination (10 hours) in sucrose-free R5A liquid medium. Data were normalised against the Wt spores (log2 SCO2730::Tn5062 or Wt80Cu/Wt) (SI Table S1). 1790 transcripts showed significant variations (q-value of less than 0.05) in the SCO2730::Tn5062 mutant, 738 in the Wt80Cu and 754 in both, SCO2730::Tn5062 and Wt80Cu (Fig. 5) (SI Table S1).

Transcriptomic analysis of the SCO2730::Tn5062 mutant spores, the S. coelicolor wild-type strain spores obtained in 80 µM CuSO4 amended SFM cultures (Wt80Cu) and in SFM cultures (Wt) during germination (10 h) in sucrose free R5A medium. The Venn diagram shows transcripts related to copper homeostasis/metabolism/regulation, germination and secondary metabolism with significant variations (q-value of less than 0.05) in the SCO2730::Tn5062 mutant (1790 transcripts), the Wt80Cu strain (738 transcripts), or both (754 transcripts), compared to the Wt. Abundance values (average from two biological replicates) are shown as log2 (SCO2730::Tn5062 or Wt80Cu/Wt). Blue bars: transcripts with significant variations in the SCO2730::Tn5062 mutant compared to Wt. Red bars, transcripts with significant variations in the Wt80Cu compared to Wt. Broken bars indicate transcripts not detected in the SCO2730::Tn5062 mutant or the Wt strain. ACT, actinorhodin; RED undecylprodigiosin; CDA, calcium dependent antibiotic. Secondary metabolism cryptic pathways are indicated in italic letters.
The 1790 genes differentially expressed in the SCO2730::Tn5062 mutant compared to the Wt strain, included key secondary metabolism genes, spore germination genes and genes encoding for copper related proteins (copper transporters, proteins using copper as cofactor, proteins whose genes are known to be regulated by copper) (Fig. 5) (Table 1). Most of the genes involved in secondary metabolism were up-regulated in the SCO2730::Tn5062 mutant during germination (see details in the next paragraph). The expression of SCO1046 (CopA)15, SCO4136 (CsoR)15, SCO4586 (ABC transporter involved in multi-copper enzyme maturation, conserved domain database accession COG1277) and SCO3440 (oxidase predicted to bind to copper)16 were up-regulated in the mutant. The expression of SCO4137 and SCO7265 (two genes predicted to be under the control of CsoR)15, SCO7234 (cytochrome c oxidase predicted to use copper as cofactor, PROSITE accession PS00077) and SCO2731 (cation transporter)15, was down-regulated in the mutant. As mentioned above, SCO2731 was expressed in the SCO2730::Tn5062 mutant (amplicon 3 in Fig. 1), from a promoter located inside the SCO2730 ORF, but downstream of the Tn5062 insertion. However, the expression from this promoter is 6.5-fold less than the wild-type strain (Table 1). Interestingly, genes SCO2728 (putative zinc-responsive transcriptional regulator, conserved domain database accession PRK09514) and SCO2729 (putative acetyltransferase), both transcribed together with SCO2730/3731 (Fig. 1A), are 5-fold overexpressed in the mutant (Table 1). Three genes related to spore germination are differentially expressed in the mutant and the wild-type strain: nepA (SCO4002)8, 2-fold up-regulated; rpfC (SCO3098)10, 1.4-fold up-regulated; and rpfE (SCO7458), encoding one of the key peptidoglycan lytic enzymes activated during germination10, whose transcript was absent in the mutant, and perhaps contributing to the delay in germination observed in the mutant (Table 1).
The 738 genes differentially expressed during the germination of the Wt80Cu spores compared to Wt, include key genes involved in secondary metabolism, germination and genes encoding for copper related proteins (Fig. 5) (Table 2). The expression of most of the genes involved in secondary metabolite biosynthesis was down-regulated in the cultures inoculated with Wt80Cu spores (genes involved in actinorhodin, undecylprodigiosin, CDA, 5-hydroxyectoine, hexaketide, deoxysugar and siderophore biosynthesis). Other genes down-regulated in the Wt80Cu cultures were: SCO6710, a putative glycosyl hydrolase supposedly repressed by CsoR15; and SCO3571, a cyclic AMP receptor protein homologue whose mutation was reported to diminish germination22. There were no significant differences in csoR between Wt80Cu and the Wt, indicating that inactivation of the CsoR repressor activity triggered by copper binding previously described15 did not occur at the high cytosolic copper levels present in the Wt80Cu dormant spores (Fig. 3C). Consequently, there remains an unknown CsoR independent mechanism controlling the fast spore germination observed in this strain (Fig. 3E). Interestingly, neither of the two copper chaperones/transporters described in Streptomyces (SCO1045/1046 and SCO2730/2731)15 showed significant variations in the Wt80Cu cultures compared to the Wt cultures (Table 2), indicating that the Wt expression levels present in the Wt80Cu spores are high enough to reduce cytosolic copper during germination to a concentration comparable to that reached in the Wt spores (26 ± 2.4 ng Cu/mg protein, only 1.4- fold higher than the Wt spores at 10-hours) (Fig. 3D). The expression of genes involved in hopene, desferrioxamine, coelichelin and dipeptide biosynthesis, was slightly up-regulated in the Wt80Cu cultures.
The 754 genes differentially expressed during germination in both the SCO2730::Tn5062 mutant and the cultures inoculated with the Wt80Cu spores, compared with the wild-type strain, include secondary metabolism genes, and genes encoding for copper related proteins (Fig. 5) (Table 3). Most of these genes were down-regulated in the cultures inoculated with Wt80Cu spores. Variation in the expression of secondary metabolism genes was discrete (all the abundances were below the 2-fold change, log2 abundance within the +/− 1 interval). The most important variation, was found in the genes encoding proteins related to copper: genes predicted to be modulated by CsoR (SCO1044, SCO3280)15; putative cation-transporting ATPases (SCO0164, SCO0860, SCO6460)15; SCO3281, a cytosolic copper storage protein23; and SCO7657, a putative secreted protein predicted to bind copper16. Interestingly, SCO0164 and SCO7657 were not expressed in the SCO2730::Tn5062 mutant.
Secondary metabolite transcripts with significant variations (q-value < 0.05) in the SCO2730::Tn5062 mutant compared to wild-type strain (Wt)
We next focus on the analysis of the secondary metabolite genes up-regulated in the SCO2730::Tn5062 mutant during germination (10 hours in sucrose-free R5A liquid cultures). The S. coelicolor genome encodes at least 30 predicted secondary metabolite clusters24. The expression of 60 genes belonging to 12 secondary metabolite clusters (40% of the total predicted)24 is up-regulated in the SCO2730::Tn5062 mutant compared to the wild-type strain (Fig. 6) (Tables 1–3, SI Table S1): six well-known secondary metabolite clusters (undecylprodigiosin, hopene, geosmin, actinorhodin, 5-hydroxyectoine, CDA); four predicted cryptic pathways (lantibiotic, hexaketide, dipeptide, aromatic polyketide)24. SCO3800 a gene putatively involved in secondary metabolism biosynthesis (S. coelicolor KEGG pathway map 01110) and SCO1909, a gene putatively involved in antibiotic biosynthesis (pfam03992), also were overexpressed in the mutant.

Heat map illustrating the secondary metabolism transcript abundances. Abundances correspond to log2 SCO2730::Tn5062 / Wt. Only transcripts showing significant variations (q-value < 0.05) in the SCO2730::Tn5062 mutant compared to the wild-type strain (Wt) are shown. S. coelicolor encodes 30 secondary metabolite clusters24: 40% up-regulated in the mutant (red colours), 36% down-regulated (green colours), 24% without variation (not shown in the figure).
Discussion
As introduced above, copper has pleiotropic effects in Streptomyces development and regulates differentiation (aerial mycelium and sporulation) and antibiotic production13,14. In this work we discovered additional copper effects during the vegetative stage, in germination (Fig. 1C) and growth (Fig. 1E).
Based on our results, we propose the model outlined in Fig. 7, which correlates spore germination, vegetative growth, secondary metabolism and sporulation with cytosolic copper and the expression of key genes regulating these processes. In the wild-type strain (outlined in Fig. 7A), germination triggers cytosolic copper secretion thanks to the activation of the SCO2730/2731 copper chaperone/P-type ATPase (Fig. 3A,B). Cytosolic copper reaches its minimum concentration during germination (11.8 ± 0.3 ng Cu/mg protein) which suggests that the SCO2730/2731 secretion system has a higher affinity for copper than does the SCO1045/1046 chaperone/transporter that is activated at the higher cytosolic copper levels reached in the mycelium (Fig. 3A,B). This result agrees with the results of Chaplin et al.17, who demonstrated, in vitro, the higher copper affinity of SCO2730/2731 compared to SCO1045/1046. We postulate that the SCO1045/1046 system is also active during the first stages of copper secretion accompanying germination and until the cytosolic copper concentration reaches the low levels at which the SCO2730/2731 transporter system is activated (Fig. 3B). The low levels of cytosolic copper trigger the expression of rpfE, which is a key peptidoglycan lytic enzyme activated during germination10 (Fig. 5) (Table 1). We postulate that the asynchrony of spore germination is a consequence, at least in part, of the differences in cytosolic copper concentration in single dormant spores (Fig. 3G). The spores with the highest levels of cytosolic copper germinate more slowly than the spores with lower cytosolic copper. Cytosolic copper in wild-type single spores was below the sensitivity of the assay used in this work. However, if we assume that the variability we observed in the Wt80Cu spores (Fig. 3G) is similar to that of the Wt spores and consider an average of cytosolic copper of 1225 ng Cu/mg protein (Fig. 3D), we can expect a range of cytosolic copper in the Wt spores between 625 and 4691 ng Cu/mg of protein (Fig. 7A). During the vegetative stage of the wild-type strain, the expression of SCO2730/2731 genes decreases, while the expression of the SCO1045/1046 genes increases (Fig. 3B) and cytosolic copper increases (Fig. 3A). Secondary metabolism is activated (Fig. 1F,G) at cytosolic copper concentrations between 45 and 200 ng Cu/mg protein (Fig. 3A). During the sporulation stage of wild-type sporulating hyphae, we postulate the activation of a cytosolic copper accumulation mechanism that ends in high cytosolic copper levels in dormant spores (Fig. 3C). The putative zinc-responsive SCO2728 transcriptional regulator (conserved domain database accession PRK09514) might modulate the effect of cytosolic copper in sporulating hyphae. Metal sensitive transcriptional repressors can bind zinc, and also copper, which has been demonstrated, for instance, in the case of CsoR from Bacillus subtilis25. Interestingly, SCO2728 is up-regulated 5.1-fold in the SCO2730::Tn5062 mutant (Table 1), which perhaps indicates autoregulation of its own expression at the high copper concentrations reached in the mutant. As stated above, we postulate that sporulating hyphae accumulate copper, and the high levels of cytosolic copper activate the expression of SCO2728. This autoregulation is comparable to that described for the csoR transcriptional regulator15, but is at a higher copper concentration than csoR, because csoR expression is only slightly increased (1.3-fold) at the copper concentration reached in the SCO2730::Tn5062 mutant (Table 1). Sporulating hyphae activate the expression of nepA (a structural cell wall protein involved in maintenance of spore dormancy)8, which is analogous to what happens during the germination of SCO2730::Tn5062 mutant spores (Table 1), which maintain higher cytosolic levels than does the wild-type strain (Fig. 3D). Interestingly, nepA expression is not regulated by CsoR because nepA lacks the consensus binding sequence for CsoR15. The importance of copper in sporulation has already been reported16 and is also supported by the fact that the BCDA copper chelator represses sporulation in our SFM cultures (data not shown). The positive effect in aerial mycelium development and sporulation becomes a negative effect (delay) at high copper concentrations (over 750 µM)15, which was also observed in this work (Fig. 3F). We postulate that high cytosolic copper concentrations in dormant spores contribute to the inactivation of gene expression.

Model outlining of the effect of cytosolic copper in germination, vegetative growth, secondary metabolism and gene expression in S. coelicolor. (A) Wild-type Strain. (B) SCO2730::Tn5062 mutant. (C) Relationship between cytosolic copper, gene expression and differentiation. Genes down-regulated are highlighted in grey.m
The acceleration in germination observed in the Wt80Cu spores (Fig. 3E) fits with the model outlined in Fig. 7A. In SFM cultures supplemented with 80 µM copper, sporulating hyphae reach the high copper levels of the dormant spores very quickly, which stops gene expression, including that of NepA. Consequently, the Wt80Cu spores harbour less NepA than do the Wt spores obtained in SFM non-modified cultures. The germination of the Wt80Cu spores mimics the phenotype (acceleration in germination) of the NepA mutant8. To further test this hypothesis, we analysed the germination of NepA mutant spores obtained in SFM cultures modified with 80 µM Cu. The copper effect in the germination of NepA mutant spores was much lower than that in the germination of the Wt80Cu spores; there was a 12% increase in germination (SI Fig. S1), compared to a 64% increase observed in Wt80Cu spores (Fig. 3E). This result corroborates that NepA has a major effect on the acceleration of spore germination mediated by copper. However, NepA is not the only effector in this phenotype, as copper still accelerates germination in the NepA mutant (Fig. 3E).
The above model also explains the phenotype of the SCO2730::Tn5062 mutant (Fig. 7B). The absence of SCO2730/2731 led to a higher copper concentration during germination (Fig. 3D). The expression of rpfE was not initiated (Table 1) and secondary metabolism was permanently activated (Figs 1F,G and 5). The overexpression of SCO1046 during the germination of the SCO2730::Tn5062 mutant spores (2.7-fold, Table 1) was unable to reduce cytosolic copper to the low levels reached in the wild-type spores (45.7 ± 8 ng Cu/mg protein in the mutant vs. 11.8 ± 0.3 ng Cu/mg protein in the wild-type strain). SCO1045 is probably also overexpressed in the SCO2730::Tn5062 mutant, but it has a 0.06 q-value that is over the 0.05 threshold used in this work; the abundance of this gene was not included in Table 1 (see SI Table S1). We postulate that the ungerminated dead spores that we observed (Fig. 1D), were those with the highest copper levels (4,552 ng Cu/mg protein and considering a variation in single spores comparable to the Wt80Cu spores). The absence of the SCO2730/2731 copper transporter system in sporulating hyphae did not have a significant effect on the accumulation of cytosolic copper in dormant spores (Fig. 3C), which is not unexpected, considering that the main role of the SCO2730/2731 copper chaperone/transporter is exerted during germination instead of sporulation (Fig. 3B).
The proposed relationship between cytosolic copper, gene expression and differentiation is summarised in Fig. 7C. Cytosolic copper concentrations under 12 ng Cu/mg protein activate the expression of rpfE and germination. Cytosolic copper concentrations under 45 ng Cu/mg of protein are a consequence of SCO2730/2731 copper transporter activity and correspond to vegetative growth. Cytosolic copper concentrations between 45 and 200 ng Cu/mg of protein activate the expression of CsoR and secondary metabolism and are buffered by SCO1045/1046 chaperone/transporter activity. Copper concentrations between 200 and 625 ng Cu/mg of protein modulate sporulation and activate the expression of nepA, csoR and the putative cation-responsive transcriptional regulator SCO2728. Copper concentrations higher than 625 ng Cu/mg protein, contribute to block gene expression in dormant spores.
The expression of SCO2728-SCO2731 genes is complex and controlled by at least four promoters. Promoters 1–4 were recently identified26; promoter 3 was identified as being controlled by the copper dependent CsoR repressor15 (Fig. 1A). The four promoters affect the expression of SCO2730/2731, and complement the SCO2730::Tn5062 mutant phenotypes (Fig. 2B–E), but they do not fully restore the wild-type phenotype (Fig. 2B–E) or the expression of the SCO2729/2730 genes to wild-type levels (Fig. 2F,G). This result suggests the existence of further regulation of the expression of these genes, and perhaps additional uncharacterised promoters or regulatory regions in the DNA regions separating these promoters (Fig. 1A).
SCO2731 is expressed from promoter 4 in the SCO2730::Tn5062 mutant at a much lower amount than in the wild-type strain (Fig. 2G). Interestingly, the SCO2730::Tn5062 complemented strain that overexpressed SCO2731 but lacked SCO2730 (strain 6 in Fig. 2G) showed a moderate increase in germination compared to the SCO2730::Tn5062 mutant (Fig. 2B). This result suggests that the SCO2731 copper transporter ATPase has some transporter activity in the absence of the SCO2730 chaperone. In fact, all of the SCO2730::Tn5062 complemented strains overexpressing SCO2731 (strains 1, 2, 4 and 6) showed an increase in germination (Fig. 2B,G), which is probably due to the SCO2731 copper secretion activity.
Secondary metabolites are usually defined as non-essential compounds produced at the stationary growth phase. However, this is not always the case. There are secondary metabolites that are produced during spore germination, for instance germicidin A or chalcone27, which both show an inhibitory effect in germination27. To the best of our knowledge, the SCO2730::Tn5062 mutant is the first Streptomyces strain reported to enhance the expression of genes encoding some secondary metabolite pathways typical of the stationary growth phase, such as actinorhodin, undecylprodigiosin or CDA27, during the germination stage (10 hours, see above). Forty percent of the S. coelicolor predicted secondary metabolite pathways are overexpressed in the mutant, including several cryptic pathways (Fig. 6). At early time points (10 hours), most of the spores still not germinated in the SCO2730::Tn5062 mutant (Fig. 1C), which makes the secondary metabolite enhancement observed in the mutant even more impressive, since the ungerminated inactive spores reduce the gene expression abundances (Fig. 6) and the secondary metabolite production quantified in the cultures (Fig. 1E,F).
Cations, specially Ca2+, but also Na+, K+, Mg2+, Fe2+, Zn2+, and Cu2+, were reported to be concentrated in dormant spores28. However, the variability of the concentration of these metals in single spores remains unknown. Our methodology to quantify cytosolic copper in single spores, based on a single-cell sample introduction system for triple-quadrupole inductively coupled plasma mass spectrometry (see Methods), can be adapted to quantify the concentration of these and other metals in individual spores.
Overall, in this work we discovered an unexpected pleiotropic effect of cytosolic copper that modulated germination, differentiation and secondary metabolism in S. coelicolor, and we propose a model correlating the copper modulated phenotypes with the expression of key copper homeostasis, and regulatory, genes. The SCO2730/2731 genes are highly conserved in Streptomyces. If, as happens in S. coelicolor, the inactivation of these genes in other Streptomyces can activate or enhance 40% of the secondary metabolite pathways, this knowledge can expand the screening of new secondary metabolites from streptomycetes. It can also contribute to improving the production of already known bioactive compounds.
Methods
Bacterial strains and culture conditions
All Streptomyces and Escherichia coli strains used in this work are listed in Table 4. Spores were harvested from SFM solid plates29 after growth at 30 °C for 12 days. The differentiation analyses were carried out on sucrose-free R5A30 plates covered with cellophane inoculated with 107 spores from a fresh water suspension and cultured at 30 °C. The samples for quantification of germination and actinorhodin and undecylprodigiosin production were obtained from 100 ml sucrose-free R5A30 cultures grown at 30 °C and 200 rpm in 500 ml flasks. Calcium-dependent antibiotic (CDA) production was measured on nutritive agar from Oxoid (Thermo Scientific, UK). Escherichia coli strains were cultured in LB and 2xTY media at 37 °C. The following antibiotics were added to select plasmid-bearing and mutant strains: ampicillin (100 μg/ml), apramycin (100 μg/ml for E. coli, 25 µg/ml for S. coelicolor), chloramphenicol (25 µg/ml), hygromycin (100 μg/ml for E. coli, 200 µg/ml for S. coelicolor), kanamycin (50 μg/ml) and nalidixic acid (25 µg/ml).
DNA and RNA extraction
Genomic DNA isolation was performed following standard methods29. Total RNA samples were isolated as previously described12 using RNeasy Mini spin columns and treated with DNase I (Qiagen). The quantity and integrity of the RNA samples were measured with Nanodrop 2000 (Thermo Scientific) and 2100 Bioanalyzer (Agilent).
Reverse transcription PCR (RT-PCR)
Co-transcription was analysed by RT-PCR. RNA obtained at 48 h sucrose-free R5A liquid cultures. Reverse transcription was performed using the SuperScript one-step RT-PCR system with Platinum Taq DNA polymerase (Invitrogen), using 200 ng of total RNA as template. Chromosomal DNA was used as template in the positive controls. RNA and the DNA polymerase included in the Platinum Taq DNA polymerase (Invitrogen) kit were used in the negative controls. RT-PCR was performed using the primers indicated in Table 4 (outlined in Fig. 1A) as follows. The first-strand complementary DNA (cDNA) synthesis was performed at 55 °C for 30 min followed by an initial denaturation at 94 °C for 2 min. Then, a touchdown was performed in 8 cycles during which the annealing temperature was reduced by 1 °C in each cycle: 94 °C for 15 s, 65 °C (Δ−1 °C) for 30 s and 68 °C for 38 s. For the next 35 cycles (94 °C for 15 s, 55 or 58 °C for 30 s and 68 °C for 38 s), the annealing temperature was set at 55 °C (for primers RT2829F/R) and 58 °C (for primers RT2930F/R, RT3031F/R and RT3031F1/R1). A final extension step was performed at 68 °C for 5 min.
Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
A High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to synthetize cDNA from 0.5 µg of RNA from two biological replicates. Real-Time PCRs were carried out on an ABI PRISM 7900 HT thermocycler (Applied Biosystems). The reactions were performed in triplicate, containing 2 µl of twofold diluted cDNA, 10 µl of SYBR Green PCR Master Mix (Applied Biosystems) and 300 nM of specific primers (listed in Table 4) in a final volume of 20 µl. SCO4758 (amplified using primers q4758F/R)31 was used as a reference since its expression showed no variation between strains in our RNA-seq results (SI Table S1). The DNA contamination and primer dimer amplification were tested in negative controls replacing cDNA by RNA or water. Amplification conditions were as follows: 2 min at 50 °C, 10 min at 90 °C, 40 repetitions of 15 s at 95 °C and 1 min at 60 °C. Primer efficiencies were measured using serial dilutions of genomic DNA as template and the relative quantification of gene expression was performed by the ΔΔCt method32.
The average transcript fold changes and standard deviations were calculated from the two analysed biological replicates. All of the qRT-PCR quantitative data discussed in this work show large differences in the averages, which are out of the average ± SD confidence interval (i.e. error bars are not overlapping).
SCO2730 mutagenesis
The transposon insertion single-gene knockout library created by Professor P. Dyson’s research group19 was used for mutagenesis of SCO2730. Cosmid C46.2.D0619 was used to construct the SCO2730::Tn5062 mutant strain. Gene disruption was carried out by obtaining double cross-overs via conjugation using E. coli ET12567/pUZ8002 as a donor strain and following the protocol described in Kieser et al.29. Mutant strains were confirmed using Southern blotting with chromosomal DNA digested with SalI. Southern hybridization was carried out using established procedures with the digoxigenin-labeled 3442-bp Tn5062 PvuII fragment from plasmid pQM506233 as a probe.
Complementation of SCO2730::Tn5062 mutation
The integrative plasmid pNG334 was used to introduce SCO2730 and/or SCO2731 with different combinations of the three promoters located up-stream of the SCO2730 ORF (promoters P1-3 in Fig. 1A) into the SCO2730::Tn5062 mutant. The specific complementation constructions cloned in pNG3 were: P1P2P3SCO2730/2731, P1SCO2730/2731, P2SCO2730/2731, P3SCO2730/2731, P1P2P3SCO2730, P1P2P3SCO2731.
The synthesis of the next genes was ordered from GeneCust Europe: EcoRV-P1P3SCO2730-XhoI-SpeI (782 bp), XhoI-SCO2731-SpeI (2330 bp), EcoRV-P1P3-XhoI (466 bp), EcoRV-P1P2P3SCO2730-XhoI (919 bp), EcoRV-P2SCO2730-XhoI (485 bp) (SI Table S2). These synthetic genes were provided by GeneCust cloned into pUC57 and were used to create the complementation constructs, including some intermediate forms not shown in Fig. 2.
pNG3-P1P3SCO2730 was created as follows; pUC57-EcoRV-P1P3SCO2730- XhoI-SpeI was digested with EcoRV/SpeI. The EcoRV-P1P3SCO2730-XhoI-SpeI fragment was cloned into pNG3 digested with EcoRV/SpeI.
pNG3-P1P3SCO2730/2731 was constructed digesting pUC57-XhoI-SCO2731-SpeI with XhoI/SpeI and cloning the XhoI-SCO2731-SpeI fragment into pNG3-P1P3SCO2730 digested with the same enzymes.
pNG3-P1P3SCO2731 was created digesting pUC57-EcoRV-P1P3-XhoI with EcoRV/XhoI and cloning the EcoRV-P1P3-XhoI fragment into pNG3-P1P3SCO2730/2731 digested with the same enzymes.
pNG3-P1P2P3SCO2730/2731 was created digesting pUC57-EcoRV-P1P2P3SCO2730-XhoI with EcoRV/XhoI and cloning the EcoRV-P1P2P3SCO2730-XhoI fragment into pNG3.P1P3SCO2730/2731 digested with the same enzymes.
pNG3-P1SCO2730/2731 was created as follows; P1 and SCO2730 were amplified by PCR from pUC57-P1P3P2SCO2730 using primers P1F/R and 2730 F/R (Table 4). Fragments were amplified via PCR using Phusion High-Fidelity DNA Polymerase (Thermo), and were cloned into pCR™-Blunt II-TOPO®. The couple of DNA fragments were combined by overlap extension PCR35 with the primers P1F y 2730 R (Table 4). The PCR product was cloned and sequenced in pCR™-Blunt II-TOPO® using the M13 universal primers. The insert was released with EcoRV/XhoI and cloned into pNG3-P1P3SCO2730/2731 digested with the same enzymes.
pNG3-P2SCO2730/2731 was created digesting pUC57-EcoRV-P2SCO2730-XhoI with EcoRV/XhoI and cloning the EcoRV-P2SCO2730-XhoI fragment into pNG3.P1P3SCO2730/2731 digested with the same enzymes.
pNG3-P3SCO2730/2731 was created as follows; P3-SCO2731 was amplified by PCR from S. coelicolor DNA using primers P3.2731F/R. The Phusion High-Fidelity DNA Polymerase (Thermo) was used; the amplicon was cloned into pCR™-Blunt II-TOPO® and sequenced using the M13 universal primers. The insert was released with EcoRV/SpeI and cloned into pNG3 digested with the same enzymes.
pNG3-P1P2P3SCO2730 was created digesting pNG3-P1P2P3SCO2730.SCO2731 with XhoI/SpeI. The XhoI/SpeI-ends were digested with the S1 Nuclease (Thermo Scientific®) and the plasmid was religated.
pNG3.P1P2P3SCO2731 was constructed as follows. P1P2P3 was amplified by PCR from pNG3-P1P2P3SCO2730.SCO2731 using primers P123F/R (Table 4). The Phusion High-Fidelity DNA Polymerase (Thermo) was used; the amplicon was cloned into pCR™-Blunt II-TOPO® and sequenced using the M13 universal primers. The insert was released with EcoRV/XhoI and cloned into pNG3-P1P3SCO2730/2731 digested with the same enzymes.
Viability staining
Culture samples were obtained and processed for microscopy at various incubation durations, as previously described36. The cells were stained with propidium iodide and SYTO 9 (LIVE/DEAD Bac- Light Bacterial Viability Kit, Invitrogen, L-13152). The samples were observed under a Leica TCS-SP8 confocal laser-scanning microscope at wavelengths of 488 nm and 568 nm excitation and 530 nm (green) or 640 nm (red) emissions36.
Antibiotic production and protein quantification
Undecylprodigiosin and actinorhodin were quantified spectrophotometrically, according to Tsao et al.37 and Bystrykh et al.38. For actinorhodin quantification, KOH was added to the culture samples at a final concentration of 1 N. Cellular pellets were discarded by centrifugation and actinorhodin concentration was spectrophotometrically determined at 640 nm, applying the linear Beer–Lambert relationship (ε640 = 25,320). The culture samples for undecylprodigiosin quantification were vacuum-dried, resuspended in methanol, acidified with 0.5 N HCl and spectrophotometrically assayed at 530 nm, using the Beer–Lambert relationship to estimate concentration (ε530 = 100,500).
Calcium-dependent antibiotic (CDA) production was determined via a bioassay against Bacillus subtilis. Oxoid nutritive agar (ONA) plates (90 mm in diameter) were inoculated with 5 µl of a Streptomyces spore suspension at 1 × 105 spores/ml and incubated at 30 °C. After 2 days, the plates were overlaid with 5 ml of soft ONA (0.75% agar), inoculated with B. subtilis (OD = 0.25) and supplemented with Ca(NO3)2 (60 mM). Negative controls were performed in parallel without adding calcium. Inhibitory halos were measured after 15 h at 30 °C.
Protein quantification
Growth was determined by measuring the protein concentration with the Bradford assay (Biorad) and a bovine serum albumin standard (Sigma). Total protein extracts were obtained mixing a volume of culture with a volume of 1 M NaOH, boiling for 5 min and removing cell debris by centrifugation at 7740 g.
Spore germination
Germination was quantified as previously reported8. Briefly, germination was quantified in solid media with cellophane discs. At different developmental time points, pieces of cellophane discs were cut and processed for confocal microscopy, as described in the previous paragraph. Three biological replicates of the cultures were analysed at different developmental time points. The percentage of germination was assessed from at least 100 spores at each time point. Spores were considered to be germinating when the germ tubes were visible under the confocal microscope.
RNA-seq and bioinformatics analysis
Next-generation sequencing (NGS) was performed by Stab Vida (Caparica, Portugal) from two biological replicates using RNA from SCO2730::Tn5062 and S. coelicolor wild-type Cu-amended/non-amended spores during germination (10 h in sucrose free R5A cultures). Ribosomal RNA was depleted with the Ribo-Zero Bacteria Kit (Illumina), and the cDNA library construction was carried out using the TruSeq Stranded mRNA Library Preparation Kit (Illumina). The DNA was sequenced in the Illumina HiSeq 2500 platform using 100-bp paired-end sequencing reads (at least 20 M reads per sample). Raw data are available via the Gene Expression Omnibus database (accession GSE111126).
Bioinformatics analysis of the sequenced data was performed under the Linux operative system using the following software: FastQC to check the quality of the sequences, Cutadapt for trimming sequences, Bowtie2 for mapping with the Streptomyces coelicolor genome and Cuffdiff for differential expression test analysis39. Variation in transcript abundances was considered significant if the q-value was less than 0.05 (SI Table S1).
The SCO02730/2731 orthologues were obtained from the StrepDB (http://strepdb.streptomyces.org.uk/). SCO2730 orthologues: SLI_3079 (S. lividans), SAV_5332 (S. avermitilis), SVEN_2533 (S. venezuelae), SGR_4828 (S. griseus) and SCLAV_1906 (S. clavuligerus). SCO2731 orthologues: SLI_3080 (S. lividans), SAV_5331 (S. avermitilis), SVEN_2534 (S. venezuelae) and SGR_4827 (S. griseus). The S. clavuligerus SCO2731 orthologue is not annotated in the StrepDB database, but it is located downstream of SCLAV_1906. Amino acid similarities were estimated using the software package Lalign (http://www.ch.embnet.org/software/LALIGN_form.html).
Cytosolic copper quantification in dormant spores, germinated spores and mycelium
The spores or the mycelium were washed 4 times by centrifugation at 12,000 g for 10 min at 4 °C and resuspended in washing buffer (10 mM Tris-HCl pH 7.5; 1 mM EDTA). The samples were washed in washing buffer (10 mM Tris-HCl pH 7.5). For the bulk analysis of Cu in dormant spores, an acid digestion was conducted by resuspending the spores in 65% sub-boiling purified HNO3 at 70 °C for 1 h and then 30% H2O2 for 3 h at the same temperature. For the bulk analysis of Cu in germinated spores and mycelium, the samples were resuspended in rupture buffer (10 mM Tris-HCl pH 7.5, which is the same as the washing buffer described above). The lysis step was made using Fast-Prep (MP™ Biomedicals) with six 20-s force 6.5 cycles and with 1 minute on ice between each run (this method failed to lyse dormant spores; data not shown). Cell debris were eliminated centrifuging samples at 12,000 g for 10 min at 4 °C and discarding pellets. The resulting solutions were finally diluted with water and the total Cu content determined by ICP-MS and referred to the dry mass of the spores (1 mL spores were washed with water, dried at 100 °C to a constant weight on pre-weighted tubes) or protein (measured with the Bradford assay). Cytosolic copper in dormant spores could not be normalised against cytosolic protein, because the nitric acid treatment hydrolysed the proteins.
All the measurements were conducted in the triple quadrupole based ICP-MS Thermo iCAP-TQ (Thermo Fisher Scientific, Bremen, Germany) using the single quad mode and helium as collision gas. For bulk analysis, the ICP was equipped with a Micro Mist nebulizer, a cyclonic spray chamber (both from ESI Elemental Service & Instruments GmbH, Mainz, Germany) and an auto-sampler ASX-560 (Teledyne CETAC Technologies, Omaha, NE, USA).
All solutions were prepared using ultrapure water obtained from a Milli-Q system (Millipore, Bedford, MA, USA). HPLC-grade methanol and hydrogen peroxide for the acid digestions were both obtained from Sigma-Aldrich (Saint Louis, MO, USA). Nitric acid (65%, Suprapur quality) was purchased by Merck Millipore (Darmstad, Germany) and further purified by sub-boiling distillation. External calibrations were carried out with a Cu ICP standard CertiPur® (1000 mg.L−1), purchased from Merck.
Cytosolic copper quantification in single spores
Copper was quantified in individual spores by single-cell-ICP-MS analysis. This methodology allows metal analysis in single cells by introducing diluted suspensions of spores that can be transported intact into the plasma and measured using low integration times. The arrival of several cells within the same integration time needs to be avoided by introducing highly diluted suspensions. For this aim, fresh spore suspensions (108 spores spores·mL−1) were diluted to a final concentration of 105 spores·mL−1 in 10% methanol to ensure the optimal nebulizer performance, according to the manufacturer and previous studies performed by our group21. The samples were pumped at a low flow rate of 10 μl·min−1 using the syringe pump SP101i (Florida, USA) fitted with a 1 mL Hamilton syringe (Nevada, USA). Samples were pumped in the triple quadrupole based ICP-MS Thermo iCAP-TQ (Thermo Fisher Scientific, Bremen, Germany) using the same parameters described above for the bulk Cu measurements. In this case, we used the microflow nebulizer EnyaMist (Burgener, Ontario, Canada) and a self-developed spray chamber, which allows a high transport efficiency of the intact spores to the plasma. Details on this experimental setup and conditions can be found in our previous work21. The data were recorded in the time-resolved analysis mode setting the dwell time to 1 ms and the acquisition time of each run was typically 3 min.
Data Availability
RNA-seq raw data are available via the Gene Expression Omnibus database (accession GSE111126). The datasets generated during the current study are available from the corresponding author on request.
References
Berdy, J. Bioactive microbial metabolites. J Antibiot (Tokyo) 58, 1–26, https://doi.org/10.1038/ja.2005.1 (2005).
Hopwood, D. A. Streptomyces in nature and medicine: the antibiotic makers. (Oxford University Press, 2007).
Flardh, K. & Buttner, M. J. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol 7, 36–49, https://doi.org/10.1038/nrmicro1968 (2009).
Yague, P., Lopez-Garcia, M. T., Rioseras, B., Sanchez, J. & Manteca, A. Pre-sporulation stages of Streptomyces differentiation: state-of-the-art and future perspectives. FEMS Microbiol Lett 342, 79–88, https://doi.org/10.1111/1574-6968.12128 (2013).
Bobek, J., Smidova, K. & Cihak, M. A Waking Review: Old and Novel Insights into the Spore Germination in Streptomyces. Front Microbiol 8, 2205, https://doi.org/10.3389/fmicb.2017.02205 (2017).
Bobek, J., Strakova, E., Zikova, A. & Vohradsky, J. Changes in activity of metabolic and regulatory pathways during germination of S. coelicolor. BMC Genomics 15, 1173, https://doi.org/10.1186/1471-2164-15-1173 (2014).
Strakova, E. et al. Systems insight into the spore germination of Streptomyces coelicolor. J Proteome Res 12, 525–536, https://doi.org/10.1021/pr300980v (2013).
de Jong, W. et al. NepA is a structural cell wall protein involved in maintenance of spore dormancy in Streptomyces coelicolor. Mol Microbiol 71, 1591–1603, https://doi.org/10.1111/j.1365-2958.2009.06633.x (2009).
Noens, E. E. et al. Loss of the controlled localization of growth stage-specific cell-wall synthesis pleiotropically affects developmental gene expression in an ssgA mutant of Streptomyces coelicolor. Mol Microbiol 64, 1244–1259, https://doi.org/10.1111/j.1365-2958.2007.05732.x (2007).
Sexton, D. L. et al. Resuscitation-promoting factors are cell wall-lytic enzymes with important roles in the germination and growth of Streptomyces coelicolor. J Bacteriol 197, 848–860, https://doi.org/10.1128/JB.02464-14 (2015).
Urem, M. et al. OsdR of Streptomyces coelicolor and the Dormancy Regulator DevR of Mycobacterium tuberculosis Control Overlapping Regulons. mSystems 1, https://doi.org/10.1128/mSystems.00014-16 (2016).
Rioseras, B. et al. Characterization of SCO4439, a D-alanyl-D-alanine carboxypeptidase involved in spore cell wall maturation, resistance, and germination in Streptomyces coelicolor. Sci Rep 6, 21659, https://doi.org/10.1038/srep21659 (2016).
Ueda, K., Tomaru, Y., Endoh, K. & Beppu, T. Stimulatory effect of copper on antibiotic production and morphological differentiation in Streptomyces tanashiensis. J Antibiot (Tokyo) 50, 693–695 (1997).
Keijser, B. J., van Wezel, G. P., Canters, G. W., Kieser, T. & Vijgenboom, E. The ram-dependence of Streptomyces lividans differentiation is bypassed by copper. J Mol Microbiol Biotechnol 2, 565–574 (2000).
Dwarakanath, S. et al. Response to copper stress in Streptomyces lividans extends beyond genes under direct control of a copper-sensitive operon repressor protein (CsoR). J Biol Chem 287, 17833–17847, https://doi.org/10.1074/jbc.M112.352740 (2012).
Worrall, J. A. & Vijgenboom, E. Copper mining in Streptomyces: enzymes, natural products and development. Nat Prod Rep 27, 742–756, https://doi.org/10.1039/b804465c (2010).
Chaplin, A. K., Tan, B. G., Vijgenboom, E. & Worrall, J. A. Copper trafficking in the CsoR regulon of Streptomyces lividans. Metallomics 7, 145–155, https://doi.org/10.1039/c4mt00250d (2015).
Vijgenboom, E. & Keijser, B. In Handbook of copper pharmacology and toxicology (ed. Edward J. Massaro) Ch. 31, 503–525 (Humana; Oxford: Blackwell, 2002).
Fernandez-Martinez, L. T. et al. A transposon insertion single-gene knockout library and new ordered cosmid library for the model organism Streptomyces coelicolor A3(2). Antonie Van Leeuwenhoek 99, 515–522, https://doi.org/10.1007/s10482-010-9518-1 (2011).
Brana, A. F., Mendez, C., Diaz, L. A., Manzanal, M. B. & Hardisson, C. Glycogen and trehalose accumulation during colony development in Streptomyces antibioticus. J Gen Microbiol 132, 1319–1326, https://doi.org/10.1099/00221287-132-5-1319 (1986).
Corte Rodriguez, M., Alvarez-Fernandez Garcia, R., Blanco, E., Bettmer, J. & Montes-Bayon, M. Quantitative Evaluation of Cisplatin Uptake in Sensitive and Resistant Individual Cells by Single-Cell ICP-MS (SC-ICP-MS). Anal Chem 89, 11491–11497, https://doi.org/10.1021/acs.analchem.7b02746 (2017).
Derouaux, A. et al. Deletion of a cyclic AMP receptor protein homologue diminishes germination and affects morphological development of Streptomyces coelicolor. J Bacteriol 186, 1893–1897 (2004).
Straw, M. L. et al. A cytosolic copper storage protein provides a second level of copper tolerance in Streptomyces lividans. Metallomics 10, 180–193, https://doi.org/10.1039/c7mt00299h (2018).
Nett, M., Ikeda, H. & Moore, B. S. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep 26, 1362–1384, https://doi.org/10.1039/b817069j (2009).
Ma, Z., Cowart, D. M., Scott, R. A. & Giedroc, D. P. Molecular insights into the metal selectivity of the copper(I)-sensing repressor CsoR from Bacillus subtilis. Biochemistry 48, 3325–3334, https://doi.org/10.1021/bi900115w (2009).
Jeong, Y. et al. The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3(2). Nat Commun 7, 11605, https://doi.org/10.1038/ncomms11605 (2016).
Cihak, M. et al. Secondary Metabolites Produced during the Germination of Streptomyces coelicolor. Front Microbiol 8, 2495, https://doi.org/10.3389/fmicb.2017.02495 (2017).
Salas, J. A., Guijarro, J. A. & Hardisson, C. High calcium content in Streptomyces spores and its release as an early event during spore germination. J Bacteriol 155, 1316–1323 (1983).
Kieser, T. Practical streptomyces genetics. (John Innes Foundation, 2000).
Fernandez, E. et al. Identification of two genes from Streptomyces argillaceus encoding glycosyltransferases involved in transfer of a disaccharide during biosynthesis of the antitumor drug mithramycin. J Bacteriol 180, 4929–4937 (1998).
Li, S., Wang, W., Li, X., Fan, K. & Yang, K. Genome-wide identification and characterization of reference genes with different transcript abundances for Streptomyces coelicolor. Sci. Rep. 5, 15840 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Bishop, A., Fielding, S., Dyson, P. & Herron, P. Systematic insertional mutagenesis of a streptomycete genome: a link between osmoadaptation and antibiotic production. Genome Res 14, 893–900, https://doi.org/10.1101/gr.1710304 (2004).
Gonzalez-Quinonez, N. et al. New PhiBT1 site-specific integrative vectors with neutral phenotype in Streptomyces. Appl Microbiol Biotechnol 100, 2797–2808, https://doi.org/10.1007/s00253-015-7271-0 (2016).
Lee, J., Shin, M. K., Ryu, D. K., Kim, S. & S., R. W. Insertion and deletion mutagenesis by overlap extension PCR. In: Braman J. (eds) In Vitro Mutagenesis Protocols. Methods in Molecular Biology (Methods and Protocols), vol 634. 3rd ed. edn, (Humana, 2010).
Manteca, A., Alvarez, R., Salazar, N., Yague, P. & Sanchez, J. Mycelium differentiation and antibiotic production in submerged cultures of Streptomyces coelicolor. Appl Environ Microbiol 74, 3877–3886, https://doi.org/10.1128/AEM.02715-07 (2008).
Tsao, S. W., Rudd, B. A., He, X. G., Chang, C. J. & Floss, H. G. Identification of a red pigment from Streptomyces coelicolor A3(2) as a mixture of prodigiosin derivatives. J Antibiot (Tokyo) 38, 128–131 (1985).
Bystrykh, L. V. et al. Production of actinorhodin-related “blue pigments” by Streptomyces coelicolor A3(2). J Bacteriol 178, 2238–2244 (1996).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
MacNeil, D. J. et al. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68 (1992).
Flett, F., Mersinias, V. & Smith, C. P. High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol Lett 155, 223–229 (1997).
Acknowledgements
We thank the European Research Council (ERC Starting Grant; Strp-differentiation 280304) and the Spanish “Ministerio de Economía, Industria y Competitividad” (MINECO; BIO2015-65709-R and CTQ2016-80069-C2-1R) for financial support. Nathaly Gonzalez-Quiñonez was funded by a Severo Ochoa fellowship (FICYT, Consejería de Educación y Ciencia, Asturias, Spain). Roberto Álvarez-Fernández García was funded by “Fundación para la Investigación Biosanitaria de Asturias” (FINBA, Asturias, Spain). Thanks to Beatriz Gutierrez Magan (Universidad de Oviedo, Dpto. Biología Funcional, Área de Microbiología) for her laboratory assistance, Paul Dyson and Meirwyn Evans (Swansea University) for providing the disrupted cosmids, Wouter de Jong (University of Groningen, the Netherlands) for providing the S. coelicolor NepA mutant strain, Proof-Reading-Service.com and Nature Publishing Group Language Editing service for proofreading the text.
Author information
Authors and Affiliations
Contributions
N.G., P.Y., B.R., M.T.L.G. and G.F.G. performed the main experiments. M.C.R. and R.A.F.G. performed mass-spectrometry copper quantification. P.Y., A.M. and M.M.B. designed experiments. P.Y. and A.M. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
González-Quiñónez, N., Corte-Rodríguez, M., Álvarez-Fernández-García, R. et al. Cytosolic copper is a major modulator of germination, development and secondary metabolism in Streptomyces coelicolor. Sci Rep 9, 4214 (2019). https://doi.org/10.1038/s41598-019-40876-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40876-0
- Springer Nature Limited
This article is cited by
-
Actinobacterial chalkophores: the biosynthesis of hazimycins
The Journal of Antibiotics (2024)
-
A two-component system gene SACE_0101 regulates copper homeostasis in Saccharopolyspora erythraea
Bioresources and Bioprocessing (2020)
-
The novel ECF56 SigG1-RsfG system modulates morphological differentiation and metal-ion homeostasis in Streptomyces tsukubaensis
Scientific Reports (2020)