
Abstract
Spontaneous polarization (Ps) in ferroelectrics has provided the impetus to develop piezoelectric devices such as sensors, actuators and diagnostic imaging transducers. Widely used lead-based perovskites exhibit a composition-driven phase diagram involving a transition region, known as a morphotropic phase boundary, where the ferroelectric structure changes dramatically and the piezoelectric activity is maximal. In some perovskites, ferroic polarization coexists with nonpolar rotations of octahedra, suggesting an unprecedented phase diagram. Here, we show morphotropic phase boundaries, where ‘ferrielectric’ appears as a bridging phase between ferroelectrics with rhombohedral and tetragonal symmetries in Bi1/2Na1/2TiO3-based perovskites. Neutron diffraction analysis demonstrates that the intermediate ferrielectric displays a small Ps resulting from up and down polarizations coupled with an in-phase TiO6 rotation. Our ab initio calculations indicate that a staggered Bi-O conformation at an appropriate chemical pressure delivers the ferrielectric-mediated phase boundaries, which provides a promising platform for (multi)ferroic materials with enhanced physical properties.
Similar content being viewed by others


Introduction
Because functions of ferroelectrics are governed by spontaneous polarization (Ps) in response to external stimuli, the majority of studies have focused on controlling polar lattice distortions1,2,3. In lead-based perovskites, e.g., Pb(Zr, Ti)O3, a composition-driven transition region, known as a morphotropic phase boundary (MPB), separates tetragonal and rhombohedral ferroelectrics4,5, between which an intermediated monoclinic appears as a bridging phase6,7,8,9. In the vicinity of the MPBs, a piezoelectric response is markedly enhanced because of the symmetry-allowed polarization rotation10,11,12,13. The nature of these phase boundaries where ferroelectric instabilities compete with each other can be explained by the intrinsic high-pressure MPBs in PbTiO3 tuned through a chemical pressure9.
For perovskite oxides and related materials with two-dimensional interfaces, a polar atomic configuration is triggered by the condensation of rotation instabilities of oxygen octahedra14,15,16,17,18. In a wide range of simple perovskites, octahedral rotations are ubiquitous; however, a system with multiple instabilities is relatively rare19,20. In some perovskites, such as the Bi1/2Na1/2TiO3–BaTiO3 system21, ferroic polarization coexists with nonpolar modes of TiO6 octahedra, suggesting a distinct phase diagram where ferroelectrics with rhombohedral R3c (or monoclinic Cc) and tetragonal P4mm are mediated by a ferroic phase involving TiO6 rotations22,23,24,25. Despite several decades of intensive research, it remains challenging to identify clear, unambiguous MPBs in which the subtle but important distortions participate because of a structural complexity partially owing to compositional inhomogeneity and/or of a detection limit of analytical methods21,23,24,25,26,27,28,29,30, see Supplementary Table 1. Moreover, there has been no research on the electronic origin of MPBs where abrupt changes in crystal structure, symmetry and atomic reconstruction are derived from orbital interactions.
Composition-driven phase diagram
Figure 1 displays the lattice parameters as a function of the Ba composition (x) along with the crystal structures and their atomic displacements. The evolution of the neutron powder diffraction (NPD) patterns and the refinement results, along with the structural data, are shown in Supplementary Figs 1 and 2 and Supplementary Tables 2 and 3. In addition to the fundamental hkl reflections, superlattice reflections distinct to the constituent phases appear: 1/2{o o o} for x ≤ 4% and 1/2{o o e} for x = 6% and 7% (o is an odd number, and e is an even number). The 1/2{o o o} reflection arises from an out-of-phase TiO6 rotation about [111]pc (a−a−a− in Glazer notation31) typical for the rhombohedral R3c while the 1/2{o o e} reflection is attributed to an in-phase TiO6 rotation about [001] (a0a0c+) of the tetragonal P4bm22,25,26,27,30. We assign x ≥ 8% to the tetragonal P4mm, as identified by a clear splitting of the 200 and 002 reflections resulting from an apparent tetragonality (c/a)32,33. The variation in the superlattice reflections are consistent with our synchrotron-radiation X-ray diffraction analysis on single crystals25, where extrinsic factors such as ferroelectric and/or ferroelastic domain structures play no role.

Composition-driven structure evolution. (a) Lattice parameters, cell deformation, cell volume (V) and rotation angle (ω) of TiO6 octahedra determined by the high-resolution NPD Rietveld analysis as a function of Ba composition (x) in (1 − x)Bi1/2Na1/2TiO3–xBaTiO3. Note that two morphotropic phase boundaries (MPBs) exists, where the tetragonal P4bm appears as a bridging phase between the ferroelectric phases with rhombohedral R3c and tetragonal P4mm. The pseudocubic (pc) parameters are adopted; for the rhombohedral R3c, α and a are the angle and parameter, respectively, σ represents the cell deformation along [111]pc expressed by \(\sigma =\sqrt{({\rm{1}}+{\rm{2cos}}{\alpha }_{{\rm{pc}}})/(1-\,\cos \,{\alpha }_{{\rm{pc}}})}\), and ω is the rotation angle about [111]pc (//hexagonal c axis, chexa); for the tetragonal P4bm, apc is a/√2, and ω is the rotation angle about [001]. Tetragonality (c/a) is plotted as a cell deformation for the P4bm and P4mm phases. (b–g) Crystal structures and atomic displacements; the left panels show the structures with thermal ellipsoids of the constituent atoms; the light panels exhibit the off-center displacements (Δz) along [111]pc (R3c) and [001] (P4bm and P4mm) from the hypothetical paraelectric positions. Oc and Oa denote the apical and planner O atoms, respectively, for the tetragonal lattices. Note that the tetragonal P4bm has ferrielectricity arising from up (Ti) and down (the A-site atoms) displacements associated with a small but significant Ps along [001].
We note that the two distinct MPBs exist at x ~ 5% and ~ 7.5%: the Ba-poor rhombohedral R3c with a relatively large ferroelectric distortion (σ) of ~1%, the intermediate tetragonal P4bm with an extremely small tetragonality (c/apc ≈ 1.00), and the Ba-rich tetragonal P4mm with a tetragonality (c/a) of ~1.02. We did not find a phase coexistence around the MPBs reported in the previous reports26,27,30. Across the first MPB at x ~ 5%, the mode and magnitude of the TiO6 rotation change; a rotation angle (ω) of the rhombohedral R3c is as large as ~8 deg., while that of the tetragonal P4bm is ~3 deg. (Fig. 1a). The polar atomic displacements (Δz) are schematized in Fig. 1e–g and the electric dipole moments (p = Δz × Zeff*) are depicted in Supplementary Fig. 3. The rhombohedral R3c features a cooperative displacement of the A-site atoms and Ti along the hexagonal chexa axis (//[111]pc). We found that the tetragonal P4bm displays a ferrielectric polar configuration that up and down dipole moments give rise to nonzero Ps. Beyond the second MPB at x~7.5%, the tetragonal symmetry changes to P4mm, where the A-site atoms and Ti exhibit a robust cooperative off-centring along [001]. We think that the maximal piezoelectric properties reported for the ceramics with around x = 6–7%21,23,27,34,35 are directly related to the ferrielectric-mediated MPBs.
First morphotropic phase boundary (1st MPB)
Our NPD analysis for the solid solutions provides an averaged structure regarding the A-site atoms but reveals the precise conformations of TiO6 octahedra. In the DFT calculations to investigate the 1st MPB for the Bi1/2Na1/2TiO3 cell (Supplementary Fig. 4), the experimentally determined TiO6 structure is adopted; the data of x = 4% are used for the rhombohedral R3c and those of x = 6% for the tetragonal P4bm, because an essential local structure of TiO6 does not show a significant change, regardless of the Ba composition36. The optimization of the positions of Bi and Na under the constraint of the fixed fractional coordinates of TiO6 octahedra yields information on the free energy and the chemical bonding in the lattice environments similar to the real crystal.
Free energy and Bi-O lengths
Figure 2 exhibits the relations between free energy G, cell volume V and pressure p (see the total energy U(V) curves in Supplementary Fig. 5a). Using the fitting parameters in Eq. 1, we obtain GR3c for the rhombohedral R3c and GP4bm for the tetragonal P4bm along with their difference, which is expressed as ΔG = GR3c − GP4bm. We identified the 1st phase-boundary p (p1st) at ~−1 GPa. In the higher-p region, ΔG becomes negative; the rhombohedral R3c is stabilized at a smaller V (Fig. 2c). In contrast, in the lower-p region, ΔG becomes positive; the P4bm phase is lower in free energy at a larger V. In the rhombohedral R3c, e.g., at p ~ 0.4 GPa, the bond valence sum (BVS) (Supplementary Fig. 5b) is estimated to be 1.12 for Na and 4.18 for Ti, which are close to their respective formal valences (Na+ and Ti4+). The BVS of Bi is yet 2.65, which is much smaller than the formal valence (Bi3+). With decreasing p and beyond p1st, the BVS of Bi recovers to ~2.73 in the tetragonal P4bm, whereas those of Na and Ti do not exhibit an anomaly.

Nature of first morphotropic phase boundary. The Bi1/2Na1/2TiO3 cells (Supplementary Fig. 4) are adopted for the DFT calculations of the rhombohedral R3c and the tetragonal P4bm. (a) Free-energy difference ΔG = GR3c − GP4bm and (b), cell volume V per ABO3 unit cell as a function of pressure p. From the fitting analysis of the total energy U(V) shown in Supplementary Fig. 5a, we obtain GR3c for the R3c phase and GP4bm for the P4bm one. The first phase-boundary p (p1st) separating the R3c and P4bm phases is present at ~−1 GPa. (c) Bi-O bond lengths as a function of p, where asterisk (*) denotes longer bond owing to the off-centring of Bi. Wavefunctions of the rhombohedral R3c at the Γ point whose energy levels are (d) −4.81 eV and e −5.20 eV, where the optimized structure at p ≈ 0.4 GPa is adopted. In (d), the bonding interaction between Bi-6p and O1-2p is seen not only for the short Bi-O1 but also for the long Bi-O1*. In (e), the orbital mixing of Bi-6p and O-2p is seen for the short Bi1-O2, where the interaction of the short Bi1-O1 has a minor role. With decreasing p, Bi-O1* and Bi-O2 are lengthened, and thereby these states are lower in energy, leading to a destabilization of the rhombohedral R3c.
The length of Bi-O as a function of p (Fig. 2c) provides insight into the 1st MPB. In the centrosymmetric structure, Bi is surrounded by twelve O atoms. In the rhombohedral R3c, Bi is displaced along [111]pc, leading to four different lengths: the shortest Bi-O1 (×3), followed by Bi-O2 (×3), Bi-O2* (×3) and Bi-O1* (×3), where asterisk (*) denotes a longer bond. Essentially, Bi-O1 is independent of p at ~0.240 nm, whose length is in good agreement with the experiments37. In contrast, Bi-O2 is lengthened when p decreases, leading to a smaller BVS of Bi. In the tetragonal P4bm, the displacement of Bi along [001] results in three different lengths: the shorter four, the intermediate four (Bi-O1) and the longer four. Moreover, the in-plane TiO6 rotation enables Bi to form the shortest Bi-O2 (×2) and the next Bi-O3 (×2). The tetragonal P4bm accommodates the markedly short Bi-O2 of ~0.225 nm, followed by the comparably short Bi-O2 of ~0.243 nm with respect to Bi-O1 in the rhombohedral R3c. The in-phase TiO6 rotation is accompanied by the short Bi-O2 even though the Δz of Bi is relatively small.
Electronic origin
We investigate the electronic structures of the rhombohedral R3c at p ≈ 0.4 GPa and the tetragonal P4bm at p ≈ −2.8 GPa (see Supplementary Figs 6 and 7). In the rhombohedral R3c, the majority of the occupied Bi-6p states is higher in energy from −5.6–−4.4 eV at p ≈ 9 GPa to −5.2–−4.2 eV at p ≈ 0 GPa. The minimum of the valence band is formed by the wave function composed mainly of the p states of Bi, O1, and O2 (Fig. 2e); its energy increases from −5.6 eV at p ≈ 9 GPa to −5.2 eV at p ≈ 0.4 GPa at the Γ point, which is attributed to a lengthened Bi-O2. The next-lowest band is formed by the hybridized orbital of Bi-6p and O1-2p (Fig. 2d). Even though Bi-O1 remains unchanged, its energy increases from −5.3 eV at p ≈ 9 GPa to −4.8 eV p ≈ 0.4 GPa at the Γ point because Bi-O1* substantially increases in length. The rhombohedral instability caused by the elongated Bi-O2 and Bi-O1* near p1st stems from the shift of the Bi-6p-derived states to higher energy. The details of the tetragonal P4bm are described later.
Second morphotropic phase boundary (2nd MPB)
Across the 2nd MPB, the phase changes from the tetragonal P4bm to the tetragonal P4mm. Because the P4bm phase has an in-plane TiO6 rotation, it is natural to consider that the rotation distortion is suppressed near the 2nd MPB, as observed in the experiments (Fig. 1a). Taking into account the evolution of the TiO6 rotation along with the atomic displacements, we performed the structural optimization of the Ba2/8Bi3/8Na3/8TiO3 cell (Supplementary Fig. 8) under the constraint of the experimentally determined tetragonality of cpc/apc = 1.0001 (x = 6%) for the P4bm phase and cpc/apc = 1.0218 (x = 12%) for the P4mm phase.
Free energy and reconstruction of Bi-O bonds
Figure 3 shows the relation between ΔG = GP4bm − GP4mm, V and p, along with the Bi-O lengths, as a function of p. The U(V) curves are presented in Supplementary Fig. 9. We found the 2nd phase-boundary p (p2nd) at ~2 GPa: ΔG becomes negative in the higher-p region; the P4bm phase is stabilized at a smaller V, whereas ΔG becomes positive in the lower-p region; and the P4mm phase emerges at a larger V.

Nature of second morphotropic phase boundary. The Ba2/8Bi3/8Na3/8TiO3 cells (Supplementary Fig. 8) are adopted for the DFT calculations of the tetragonal P4bm and the tetragonal P4mm. (a) Free-energy difference ΔG = GP4bm − GP4mm and (b), cell volume V per ABO3 unit cell as a function of pressure p. From the fitting analysis of the total energy U(V) shown in Supplementary Fig. 9a, we obtain GP4bm for the P4bm phase and GP4mm for the P4mm one. The second phase-boundary p (p2nd) separating the P4bm and P4mm phases is present at ~ ~2 GPa. Bi-O bond lengths as a function of p for (c) Bi1-O, (d) Bi2-O and (e) Bi3-O. Note that a reconstruction was found for Bi1-O and Bi3-O at p2nd, whereas Bi2-O presents a monotonic tendency. (f) Staggered Bi-O conformation composed of the short Bi1-O3 and Bi3-O6 bonds in the tetragonal P4bm leading to a robust in-phase TiO6 rotation, where the Ps vector is parallel to the dipole moment derived from the Bi-Na and Bi-Ba layers.
Our DFT calculations shed light on the structural origin of the in-phase TiO6 rotation. The octahedral rotation angle (ω) as a function of p is shown in Supplementary Fig. 10. The ω of Ti1-O6 sandwiched between the Bi-Ba and Bi-Na layers is as large as ~12 deg., where its rotation axis is parallel not only to the dipole moment derived from Na and Ba but also to the Ps vector. We note that a staggered conformation of Bi-O is formed around Ti1-O6 by the shortened Bi1-O3 and Bi3-O6 bonds (Fig. 3f), which stems from its large ω. The ω of Ti1-O6 is significantly reduced when approaching p2nd, suggesting that the instability of the P4bm phase is closely related to the suppression of the Ti1-O6 rotation. In reality, for the lattice of the solid solution where Bi, Ba, and Na have a quasi-random distribution28, we consider the following structural feature: the Ti1-O6 unit with the staggered Bi-O bonds drives a coherent in-phase rotation about the polar axis in the entire crystal; thus, the resultant ω is averaged to 2–3 deg., as observed in Fig. 1.
Although we did not find a prominent feature in the BVSs as a function of p (Supplementary Fig. 11), it is noticeable that Bi-O shows a reconstruction across p2nd; Bi1-O and Bi3-O display discontinuous changes in length caused by the Ti1-O6 rotation, whereas Bi2-O exhibits a monotonic tendency. The off-centring of Bi along [001] in the P4mm phase results in a short bond with four equivalent O atoms, i.e., Bi1-O3, Bi2-O7, and Bi3-O8, while each of these degenerated bonds splits into two different lengths in the P4bm phase. Interestingly, Bi1-O exhibits the most striking feature across p2nd: Bi1-O3 in the P4mm phase is divided into the shortest Bi1-O3 and the longest Bi1-O6 in the P4bm phase, which results from the Ti1-O6 rotation. Given that p decreases and approaches p2nd, Bi-O6 is substantially shortened, whereas Bi1-O3 remains unchanged. In addition, a decrease in p elongates Bi1-O4 and Bi1-O5, which leads to a smaller BVS of Bi1 to 2.54 near p2nd.
Electronic origin
First, we describe the relation between the crystal structure and the electronic states of the P4mm phase (see Supplementary Fig. 12b). Two common features appear in the orbital interactions regarding Bi-6p, which constitutes most of the lower part of the valence band. The first is the hybridization in the O-Bi(6px + py)-O-Ti unit, e.g., Bi1 has low-lying 6px + py states at ~−5.3 eV (Bi1-6pz is higher in energy at ~−4.5 eV). In the O1-Bi1-O3-Ti1 unit, as seen in the wavefunction at the X point (Fig. 4d), Bi1-6px + py is mixed not only with the nearest O3-2p but also with the adjacent Ti1-3d, in addition to a significant contribution from the next-nearest O1-2p. A similar feature is seen also in the O5-Bi2-O7-Ti3 unit, which constitutes the minimum valence band. The second is the hybridization in the O-Bi(6pz)-O unit. In the O1-Bi1-O3 unit, Bi1-6pz forms the bonding states with O1-2p and O3-2p, as found in the wavefunction at the X point (Fig. 4c). Because the displacements of Bi2 and Bi3 along [001] are much larger than that of Bi1 (Bi2-O7 and Bi3-O8 become short), their bonding orbitals in the O-Bi(6pz)-O units are lower in energy, as seen in the density of states (DOS).

Wave functions. The orbital interactions in the Ba2/8Bi3/8Na3/8TiO3 cells (Supplementary Fig. 8) that stabilize polar and rotation distortions: for the tetragonal P4bm (p ≈ 9.8 GPa), the orbitals at the Γ point whose energy levels are (a) −3.86 eV and (b) −5.20 eV; for the tetragonal P4mm (p ≈ 0.9 GPa), those at (c) the X point (−4.46 eV) and d the X point (−5.30 eV). For both phases, the hybridizations of c the out-of-plane Bi(6pz)-O(2p) and (d) the in-plane Bi(6px + py)-O(2p) give rise to the bonding states at the bottom of the valence bands, leading to the Bi displacement along [001]. In addition, the P4bm phase is stabilized by the staggered Bi(6pz)-O(2p) hybridizations (a,b) associated with the short Bi1-O3 and Bi3-O6 bonds, which drives a robust Ti1O6 rotation. In (b) (lower panel), solid blue lines representing mirror plane and closed ellipses denoting twofold axis are depicted. These symmetry elements are present also in (a) (lower panel).
In addition to the orbital interactions described above, distinct hybridizations emerge in the P4bm phase, where Bi-6pz plays a crucial role. According to the wave function at the Γ point in Fig. 4a, Bi1-6pz is hybridized with O3-2p, in which O4-2p and O5-2p have a minor contribution whereas O6 does not participate. The energy of this state at p ~ 2.2 GPa is −3.86 eV and that of the other Bi1-6pz-derived state is −4.67 eV at the Y point (Supplementary Fig. 13). An increase in p lowers these energies by 0.1–0.2 eV, which is partly attributed to the lower GP4bm. In addition, the orbital mixing of Bi3-6pz and O6-2p (Fig. 4b) leads to the short Bi3-O6, which contributes to the large ω of Ti1-O6. We found that the in-phase TiO6 rotation stabilizing the P4bm phase originates from the staggered Bi-O bonds (Fig. 3f) derived from the Bi-6pz and O-2p interaction.
Discussion
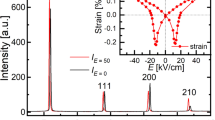
We demonstrate the ferrielectric-mediated MPBs where the tetragonal P4bm appears as a bridging phase. The ferrielectric P4bm has two order parameters: polarization (p) and octahedral rotation (ω); their coupling energy can be expressed as Gc = κ(p2ω2 + p4ω2), where κ is constant25. Given that Gc exceeds a certain threshold, the energy landscape has an extremely flat valley, along which an application of electric field (E) displaces the P4bm phase substantially from the ground state. It is worth noting that the single crystal in the P4bm phase displays an intrinsic piezoelectric strain constant (d33) of as high as 1,000 pm/V (ref.25), which is much larger than that of conventional Pb(Zr, Ti)O3 ceramics4. Moreover, the P4bm phase undergoes an E-induced phase transition to the P4mm phase even at a small E of 20 kV/cm, which is accompanied by an extremely large strain increase by ∼0.6%. By exploiting the intrinsic response superimposed on the field-induced phase transition, we could achieve high-performance Bi-based piezoelectric materials.
The ferrielectric P4bm is expected to emerge in the solid solution with multiferroic BiFeO3, since BiFeO3 has the same symmetry with Bi1/2Na1/2TiO3 (rhombohedral R3c with a−a−a−)38, raising the possibility of a novel multiferroic nature with ferrielectric polarization and (anti)ferromagnetic order. Moreover, an electric control of magnetization could be dramatically enhanced when an E-induced ferri- to ferroelectric transition takes place, because ∂P/∂E of the phase transition is extraordinary, ∂P/∂E > 10,000ε0 (ε0 is the permittivity of vacuum), which is several orders of magnitude larger than that of the conventional ferroelectrics39. We could design other (multi)ferroic materials as well based on the staggered chemical bonds at an appropriate chemical pressure, here that we have demonstrated the existence and electronic origin of the ferrielectric-mediated MPBs in Bi-based polar perovskites.
Methods
Sample preparation
Powders of (1 − x)Bi1/2Na1/2TiO3–xBaTiO3 (x = 0, 4%, 6%, 7%, 8%, 9%, 10%, and 12%) were prepared by solid-state reaction of the raw materials of Bi2O3 (99.99%), TiO2 (99.99%), Na2CO3 (99.99%), and BaCO3 (99.99%). These starting materials were thoroughly mixed by ball milling using beads as small as 100 μm and then calcined at 1,223 K for 4 h. The obtained powders were crushed by ball milling and then calcined again at 1,423 K for 4 h in order to achieve a homogeneous solid solution.
Neutron powder diffraction analysis
Time of flight (TOF) NPD data were collected using a high-resolution neutron powder diffractometer iMateria (BL20)40 at Japan Proton Accelerator Research Complex (J-PARC). Using the collected data in the d range of 0.05 < d/nm < 0.25 with a resolution Δd/d = 0.16%, the crystal structure was refined by the Rietveld method using a computer software Z-Rietveld41. When required, the pseudocubic (pc) notation is adopted to denote the Miller indices (hkl) and the crystal orientation. Recent studies24,29,37,42,43 indicate that BNT-BT with a low Ba composition (x) at room temperature belongs to space group Cc with a small monoclinic distortion from rhombohedral R3c structure. Since the monoclinic distortion is less than 0.1% and we focus on the marked change in the crystal structure across phase boundaries, we regard the room-temperature phase at a small x as rhombohedral R3c for simplicity throughout this paper.
Ab initio Density functional theory (DFT) calculations
DFT calculations were performed via the generalized gradient approximation44 with a plane wave basis set. The projector-augmented wave method45 was applied by the Vienna ab initio simulation package (VASP)46. We employed the gradient-corrected exchange-correlation functional of the Perdew-Burke-Ernzerhof revised for solids (PBEsol)47 and a plane-wave cut-off energy of 520 eV. The adopted mesh size of the k-point sampling grid was less than 5 nm‒1 for structural optimization, 2.5 nm−1 for density-functional perturbation theory (DFPT) calculations, 1 nm−1 for density of states (DOS) and band-structure calculations.
To obtain the Born effective charges, the atomic positions of Bi and Na were optimized in the (Bi1/2Na1/2TiO3)6 cells with an A-site ordering (described below) under the constraints of the fixed TiO6 octahedral structures determined by the NPD analysis. Similarly, that of Ba in the BaTiO3 cells were optimized in the experimental cell size with the fixed octahedral configurations. Then, we performed the DFPT calculations and obtained the Born effective charges of all the constituent atoms. Adopting a weighted average (mol %) of the Born effective charges (Zeff*) obtained in their respective Bi1/2Na1/2TiO3 and BaTiO3 cells, we estimated the averaged Zeff* of each atom in the solid solutions, as listed in Supplementary Table 4.
To study the nature of the composition-driven phase boundaries, we have to take into account the following two factors: the arrangement of the A-site atoms and the expansion of the cell volume. For Ba-poor compositions, it is reasonable to adopt the supercell of Bi1/2Na1/2TiO3 according to the experimentally determined crystal structure. In contrast, for Ba-rich compositions, it is necessary to consider the supercell that involves Ba because the tetragonal P4mm is the feature of BaTiO3. Therefore, we performed the DFT calculations to investigate the 1st phase boundary for the (Bi1/2Na1/2TiO3)6 cell and the 2nd phase boundary for the (Ba2/8Bi3/8Na3/8TiO3)8 cell. We confirmed that the influence of partial Ba occupation on the A site can be regarded as an expansion of unit cell volume in our calculations, the details of which are described in Supplementary Fig. 14.
Gröting et al.48,49,50 have reported that the A-site arrangement largely influences the phase stability, which is also confirmed in our preliminary calculations. Through comparing several A-site atomic arrangements for investigating how the stable phase varies as experimentally observed, we adopted the rock-salt A-site ordering25,51 that provides a reasonable structure for the P4bm phase having a small Ps compared with the P4mm phase25. The arrangement of the A-site atoms lowers the symmetry, e.g., the Bi and Na ordering in the rhombohedral changes space group from R3c to R3. For simplicity, the higher symmetry is used to denote the space group throughout this paper.
We calculated the total energy (U) per ABO3 unit cell as a function of the cell volume (V) and then analyzed by the Murnaghan equation of state52:
where E0, B0, B0′, and V0 are the total energy, the bulk modulus and its first derivative with respect to the hydrostatic pressure (p) and V at p = 0. Since the free energy (G) is expressed as G = U + pV, we can obtain the relation between G and p using the fitting parameters in Eq. 1.
Data Availability
The data that support the findings of this study are available upon request from the corresponding authors.
References
Rabe, K. M., Ahn, C. H. & Triscone, J.-M. Physics of Ferroelectrics. (Springer Berlin Heidelberg, 2007).
Scott, J. F. Applications of Modern Ferroelectrics. Science 315, 954–959 (2007).
Catalan, G., Seidel, J., Ramesh, R. & Scott, J. F. Domain wall nanoelectronics. Rev. Mod. Phys. 84, 119–156 (2012).
Jaffe, B., Cook, W. R. Jr. & Jaffe, H. Piezoelectric Ceramics. (Academic Press, 1971).
Park, S.-E. & Shrout, T. R. Ultrahigh strain and piezoelectric behavior in relaxor based ferroelectric single crystals. J. Appl. Phys. 82, 1804–1811 (1997).
Noheda, B. et al. A monoclinic ferroelectric phase in the Pb(Zr1−xTix)O3 solid solution. Appl. Phys. Lett. 74, 2059–2061 (1999).
Guo, R. et al. Origin of the High Piezoelectric Response in PbZr1−xTixO3. Phys. Rev. Lett. 84, 5423–5426 (2000).
Grinberg, I., Cooper, V. R. & Rappe, A. M. Relationship between local structure and phase transitions of a disordered solid solution. Nature 419, 909–911 (2002).
Ahart, M. et al. Origin of morphotropic phase boundaries in ferroelectrics. Nature 451, 545–548 (2008).
Fu, H. & Cohen, R. E. Polarization rotation mechanism for ultrahigh electromechanical response in single-crystal piezoelectrics. Nature 403, 281–283 (2000).
Noheda, B. Structure and high-piezoelectricity in lead oxide solid solutions. Curr. Opin. Solid State Mater. Sci. 6, 27–34 (2002).
Damjanovic, D. A morphotropic phase boundary system based on polarization rotation and polarization extension. Appl. Phys. Lett. 97, 62906 (2010).
Kitanaka, Y. et al. Synchrotron radiation analyses of lattice strain behaviors for rhombohedral Pb(Zn1/3Nb2/3)–PbTiO3 single crystals under electric fields. J. Ceram. Soc. Japan 121, 632–637 (2013).
Bousquet, E. et al. Improper ferroelectricity in perovskite oxide artificial superlattices. Nature 452, 732–736 (2008).
Benedek, N. A. & Fennie, C. J. Hybrid Improper Ferroelectricity: A Mechanism for Controllable Polarization-Magnetization Coupling. Phys. Rev. Lett. 106, 107204 (2011).
Pitcher, M. J. et al. Tilt engineering of spontaneous polarization and magnetization above 300 K in a bulk layered perovskite. Science 347, 420–424 (2015).
Rondinelli, J. M. & Fennie, C. J. Octahedral rotation-induced ferroelectricity in cation ordered perovskites. Adv. Mater. 24, 1961–1968 (2012).
Mulder, A. T., Benedek, N. A., Rondinelli, J. M. & Fennie, C. J. Turning ABO3 Antiferroelectrics into Ferroelectrics: Design Rules for Practical Rotation-Driven Ferroelectricity in Double Perovskites and A3B2O7 Ruddlesden-Popper Compounds. Adv. Funct. Mater. 23, 4810–4820 (2013).
Benedek, N. A. & Fennie, C. J. Why Are There So Few Perovskite Ferroelectrics? J. Phys. Chem. C 117, 13339–13349 (2013).
Acosta, M. et al. Piezoelectricity and rotostriction through polar and non-polar coupled instabilities in bismuth-based piezoceramics. Sci. Rep. 6, 28742 (2016).
Takenaka, T., Maruyama, K. & Sakata, K. Bi1/2Na1/2)TiO3-BaTiO3 System for Lead-Free Piezoelectric Ceramics. Jpn. J. Appl. Phys. 30, 2236–2239 (1991).
Jones, G. O. & Thomas, P. A. Investigation of the structure and phase transitions in the novel A-site substituted distorted perovskite compound Na0.5Bi0.5TiO3. Acta Crystallogr., Sect. B 58, 168–178 (2002).
Jo, W. et al. Evolving morphotropic phase boundary in lead-free (Bi1/2Na1/2)TiO3–BaTiO3 piezoceramics. J. Appl. Phys. 109, 14110 (2011).
Ma, C., Guo, H. & Tan, X. A New Phase Boundary in (Bi1/2Na1/2)TiO3−BaTiO3 Revealed via a Novel Method of Electron Diffraction Analysis. Adv. Funct. Mater. 23, 5261–5266 (2013).
Kitanaka, Y. et al. Polarization twist in perovskite ferrielectrics. Sci. Rep. 6, 32216 (2016).
Jo, W. et al. On the phase identity and its thermal evolution of lead free (Bi1/2Na1/2)TiO3-6 mol% BaTiO3. J. Appl. Phys. 110, 74106 (2011).
Ma, C., Guo, H., Beckman, S. P. & Tan, X. moco and Destruction of Morphotropic Phase Boundaries through Electrical Poling: A Case Study of Lead-Free (Bi1/2Na1/2)TiO3-BaTiO3 Piezoelectrics. Phys. Rev. Lett. 109, 107602 (2012).
Maurya, D. et al. Origin of high piezoelectric response in A-site disordered morphotropic phase boundary composition of lead-free piezoelectric 0.93(Na0.5Bi0.5)TiO3–0.07BaTiO3. J. Appl. Phys. 113, 114101 (2013).
Garg, R., Rao, B. N., Senyshyn, A., Krishna, P. S. R. & Ranjan, R. Lead-free piezoelectric system (Na0.5Bi0.5)TiO3-BaTiO3: Equilibrium structures and irreversible structural transformations driven by electric field and mechanical impact. Phys. Rev. B 88, 14103 (2013).
Ge, W. et al. Evolution of structure in Na0.5Bi0.5TiO3 single crystals with BaTiO3. Appl. Phys. Lett. 105, 162913 (2014).
Glazer, A. M. The classification of tilted octahedra in perovskites. Acta Crystallogr., Sect. B 28, 3384–3392 (1972).
Onozuka, H., Kitanaka, Y., Noguchi, Y. & Miyayama, M. Crystal Growth and Characterization of (Bi0.5Na0.5)TiO3–BaTiO3 Single Crystals Obtained by a Top-Seeded Solution Growth Method under High-Pressure Oxygen Atmosphere. Jpn. J. Appl. Phys. 50, 09NE07 (2011).
Rao, B. N., Khatua, D. K., Garg, R., Senyshyn, A. & Ranjan, R. Structural crossover from nonmodulated to long-period modulated tetragonal phase and anomalous change in ferroelectric properties in the lead-free piezoelectric Na1/2Bi1/2TiO3-BaTiO3. Phys. Rev. B 91, 214116 (2015).
Chiang, Y.-M., Farrey, G. W. & Soukhojak, A. N. Lead-free high-strain single-crystal piezoelectrics in the alkaline–bismuth–titanate perovskite family. Appl. Phys. Lett. 73, 3683–3685 (1998).
Pforr, F. et al. Relaxation of dynamically disordered tetragonal platelets in the relaxor ferroelectric 0.964Na1/2Bi1/2TiO3-0.036BaTiO3. Phys. Rev. B 96, 184107 (2017).
Groszewicz, P. B., Breitzke, H., Jo, W., Rödel, J. & Buntkowsky, G. Local structure of the B-site in BNT-xBT investigated by 47,49 Ti NMR: Effect of barium content. J. Appl. Phys. 121, 114104 (2017).
Aksel, E. et al. Local atomic structure deviation from average structure of Na0.5Bi0.5TiO3: Combined x-ray and neutron total scattering study. Phys. Rev. B 87, 104113 (2013).
Fischer, P., Polomska, M., Sosnowska, I. & Szymanski, M. Temperature dependence of the crystal and magnetic structures of BiFeO3. J. Phys. C Solid State Phys. 13, 1931–1940 (1980).
Moulson, A. J. & Herbert, J. M. Electroceramics. (John Wiley & Sons, Ltd, 2003).
Ishigaki, T. et al. IBARAKI materials design diffractometer (iMATERIA)—Versatile neutron diffractometer at J-PARC. Nucl. Instrum. Methods Phys. Res., Sect. A 600, 189–191 (2009).
Oishi, R. et al. Rietveld analysis software for J-PARC. Nucl. Instrum. Methods Phys. Res., Sect. A 600, 94–96 (2009).
Gorfman, S. & Thomas, P. A. Evidence for a non-rhombohedral average structure in the lead-free piezoelectric material Na0.5Bi0.5TiO3. J. Appl. Crystallogr. 43, 1409–1414 (2010).
Ogino, M. et al. Polarization Rotation and Monoclinic Distortion in Ferroelectric (Bi0.5Na0.5)TiO3–BaTiO3 Single Crystals under Electric Fields. Crystals 4, 273–295 (2014).
Langreth, D. C. & Perdew, J. P. Theory of nonuniform electronic systems. I. Analysis of the gradient approximation and a generalization that works. Phys. Rev. B 21, 5469–5493 (1980).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Perdew, J. P. et al. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Gröting, M., Hayn, S. & Albe, K. Chemical order and local structure of the lead-free relaxor ferroelectric. J. Solid State Chem. 184, 2041–2046 (2011).
Gröting, M., Kornev, I., Dkhil, B. & Albe, K. Pressure-induced phase transitions and structure of chemically ordered nanoregions in the lead-free relaxor ferroelectric Na1/2Bi1/2TiO3. Phys. Rev. B 86, 134118 (2012).
Gröting, M. & Albe, K. Theoretical prediction of morphotropic compositions in Na1/2Bi1/2TiO3-based solid solutions from transition pressures. Phys. Rev. B 89, 54105 (2014).
Groszewicz, P. B. et al. Reconciling Local Structure Disorder and the Relaxor State in (Bi1/2Na1/2)TiO3-BaTiO3. Sci. Rep. 6, 31739 (2016).
Murnaghan, F. D. Finite Deformation of an Elastic Solid. (Wiley, 1951).
Acknowledgements
We acknowledge Prof. T. Ishigaki and Prof. A. Hoshikawa for technical assistance for neutron powder diffraction measurements at the Japan Proton Accelerator Research Complex (2014A0282, 2014B0068). We thank Mr. M. Ogino for sample preparations. This research is partly supported by JSPS KAKENHI Grant Numbers 26249094 and 17H06239.
Author information
Authors and Affiliations
Contributions
Y.N. conceived and initiated the project. M.M. directed the research. Y.N. and Y.K. carried out the theoretical study. Y.K. carried out the structural analysis. Y.N. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kitanaka, Y., Miyayama, M. & Noguchi, Y. Ferrielectric-mediated morphotropic phase boundaries in Bi-based polar perovskites. Sci Rep 9, 4087 (2019). https://doi.org/10.1038/s41598-019-40724-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40724-1
- Springer Nature Limited