
Abstract
Genetic studies of the familial forms of Parkinson’s disease (PD) have identified a number of causative genes with an established role in its pathogenesis. These genes only explain a fraction of the diagnosed cases. The emergence of Next Generation Sequencing (NGS) expanded the scope of rare variants identification in novel PD related genes. In this study we describe whole exome sequencing (WES) genetic findings of 60 PD patients with 125 variants validated in 51 of these cases. We used strict criteria for variant categorization that generated a list of variants in 20 genes. These variants included loss of function and missense changes in 18 genes that were never previously linked to PD (NOTCH4, BCOR, ITM2B, HRH4, CELSR1, SNAP91, FAM174A, BSN, SPG7, MAGI2, HEPHL1, EPRS, PUM1, CLSTN1, PLCB3, CLSTN3, DNAJB9 and NEFH) and 2 genes that were previously associated with PD (EIF4G1 and ATP13A2). These genes either play a critical role in neuronal function and/or have mouse models with disease related phenotypes. We highlight NOTCH4 as an interesting candidate in which we identified a deleterious truncating and a splice variant in 2 patients. Our combined molecular approach provides a comprehensive strategy applicable for complex genetic disorders.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder associated with a host of motor and non-motor symptoms1. These symptoms include muscle rigidity, resting tremor, bradykinesia, and postural instability that maybe accompanied by autonomic dysfunction, sensory symptoms, fatigue, as well as cognitive and behavioral symptoms. Some patients may also develop depression, visual hallucinations, and dementia2. A definitive diagnosis is only possible postmortem and is based on the presence of two hallmarks:(1) Lewy bodies in surviving neurons from various brain regions, including the substantia nigra; and (2) the progressive degeneration of the nigrostriatal system1,3. Even after many decades of research, PD etiology remains largely unknown. However, research continues to demonstrate the strong genetic component of PD, which is important to understand disease mechanisms4,5.
The majority of reported PD cases are sporadic and only 5–10% are regarded as familial. The latter exhibit a monogenic form of the disease with a classical Mendelian mode of inheritance (reviewed in6). Despite the rarity of familial PD, family-based studies have been instrumental in identifying at least six genes with confirmed causal link to PD6,7,8. Point mutations, exon deletions, and copy number variants (CNV) in SNCA (MIM 163890), LRRK2 (MIM 609007), PARKIN (MIM 602544), PINK1 (MIM 608309), and PARK7 (MIM 602533) genes are described in both PD familial and sporadic cases, suggesting that both forms may share the same defective molecular pathways5,9,10,11,12,13,14,15. While linkage analysis led to the identification of high-penetrance disease-causing mutations, genome wide association studies aided the discovery of lower impact common variants with incomplete penetrance that represent risk factors16,17,18,19.
Although substantial progress has been achieved in the field of PD genetics, a large portion of the disease burden could not be explained by the identified mutations and risk variants. This is a reflection of the recognized genetic heterogeneity of PD, as allelic variability has been observed not only across populations but also within families4,13,20,21,22,23,24. Considering the above, it seems that additional genes have yet to be discovered. This has become more attainable with the emergence of whole exome sequencing (WES) that has been successfully applied in the discovery of novel genes in other complex neurological disorders25,26,27,28,29. In PD, WES has led to the identification of more than 40 candidate genes4,30,31,32,33,34. At present, supporting functional evidence for a role in PD pathophysiology is available only for three of the candidate genes (VPS35,TMEM230 and DNAJC13)32,35,36,37,38.
Thus far, the majority of WES findings in PD are derived from familial cases, and when possible validated in larger replication cohort with the same form of the disease4,30,32,34,38. Such an approach is appealing as rare variants with large effect size tend to aggregate in multi-incident families. However, the more common (sporadic) form of PD remains under-investigated apart from a single attempt in which a large cohort of more than 1000 cases (including non-familial) with early onset PD underwent WES assuming recessive inheritance33.
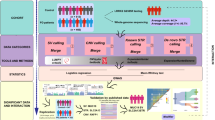
Some analysis strategies for WES data resulted in successful gene discovery in simple Mendelian diseases. However, these strategies are not suited for complex disorders where allelic heterogeneity and oligogenic inheritance are suspected. Here we set out to identify rare variants with likely pathogenic effect representing candidates worthy of further investigation in future studies. To achieve this, we devised a thorough 3-stage analysis strategy to overcome some of the common challenges encountered in gene discovery of complex disorders (Fig. 1). Our stringent prioritization was carried out to increase the likelihood that the candidate variants discovered in this study have a role in PD.

Summary of the 3-stage analysis approach applied in this study. (a) Pre-WES mutation screening of reported genes. (b) WES filtering and validation. (c) Criteria for gene prioritization.
Results
PARKIN exon 3 deletion
A homozygous deletion of PARKIN exon 3 was detected in two early onset cases. One was a familial case (PD-10) with three reported affected siblings, unfortunately; none of the siblings were available for DNA testing. The other case (PD-56) was sporadic with no reported family history of PD. The remaining samples were negative for deletions/duplication in the surveyed genes therefore representing a good sample pool for candidate gene discovery.
Validated rare variants
As our cohort is a mixture of familial and sporadic cases, we searched for homozygous, compound heterozygous and heterozygous putative variants in all samples regardless of the mode of inheritance and consanguinity. At this stage we focused on rare variants in both local and international frequency databases. This inclusive approach offers a number of advantages (1) exploration of interfamilial and intrafamilial heterogeneity, (2) detection of autosomal recessive variants in seemingly “sporadic” cases with uncertain/unknown family history, (3) minimizing variant filtering flaws (inclusion/exclusion) due to inaccurate/incomplete pedigree information or family history and errors in in silico predictions of variant impact.
Our analysis workflow yielded a total of 125 Sanger validated rare variants in 51/60 (85%) cases with 1–6 variants/sample. Of these variants 90 were missense and 34 were loss of function (LoFs) (Fig. 2). Variants were identified in 117 genes, 13 of which were observed in 2 or more cases (NOTCH4, BCOR, FAM174A, EIF4G1, DNAJB9, RABEP1, EPRS, BRINP2, HEPHL1, PUM1, GAMT, SH3TC2 and SEC22A). Two genes were previously reported in PD (ATP13A2 and EIF4G1)39,40,41 (Fig. 3 and Supplementary Table S1).

Breakdown of genetic alterations identified in this study. Pie chart illustrating the type and the number of all the validated genetic alterations (SNVs and CNVs) identified in this study.

Distribution of variants harbouring genes across the genome. Distribution of the identified genes with validated variants across chromosomes.
Among all the identified variants 39 had a pLI ≥ 0.9, 82 had a positive Z score and 34 had both pLI ≥ 0.9 and a positive Z score. While these gene constraint metrics are useful (in combination with other in silico tools) in predicting variant deleteriousness, they are not individually sufficient to infer or exclude pathogenicity42. This is supported by the fact that many of the well-established PD genes have high tolerance for missense and/or LoFs (Supplementary Table S2). Therefore, these metrics were neither considered in the filtering process nor included in the subsequent variant prioritization. On the other hand, CADD and PredictSNP2 prediction results were taken into account for variant prioritization. It is noteworthy, that none of the pre-screened genes in our cohort43 were flagged by our filtering pipeline. This provides reassurance that we did not overlook any candidate or known disease-causing variants in these genes.
In addition to rare variants, we interrogated our PD cohort dataset for shared variants with a deleterious prediction (CADD ≥ 20) these variants were common in the general Saudi population but absent in the international databases. We compiled a list of variants enriched in our cohort and unique to the Saudi population (Supplementary Table S3). Although this list is derived from a small sample, it has the potential to inform variant selection for association studies if empowered with results from a larger cohort in future research.
Prioritised rare variants
Next, we further restricted our list of candidate variants on the basis of fulfilling at least 1 out of 5 proposed conditions (Fig. 1c). Our aim was to discriminate variants that are most likely to contribute to the phenotype. These variants would represent promising candidates that warrant further functional investigation. This strategy produced a list of 22 prioritized single nucleotide variants (SNVs) in 20 genes (Table 1). Five genes carried multiple variants in unrelated patients. A nonsense variant (p.R151X) in FAM174A was detected in two familial cases and their affected relatives (PD-19 and PD-34). Both affected siblings in PD-19 were heterozygous for PARIKIN exon3 deletion43. The index case in PD-34 was homozygous for this variant while his affected siblings and offspring were heterozygous. This is unsurprising, since inter and intrafamilial differences were previously observed in PD cases22,23. A splice-donor variant (c.2591 + 3 G > A) occurring in PUM1 was present in two familial cases (PD-32 and PD-46). The third recurring variant was a missense (p.K715R) within EIF4G1 that was found in two sporadic cases (PD-56 and PD-62) and a familial case (PD-23).
Among the recurrent genes was EPRS, in which two distinct missense variants (p.R838H and p.Y791C) were detected in two sporadic cases (PD-43 and PD-57), respectively. The former case presented with late onset, while the latter had an early onset form. NOTCH4, also harboured two distinct LoF variants (c.2865 + 2T > C and p.Q1257X) in two sporadic cases (PD-1 and PD-64). The former had an early age of onset (41 years) but that of the later was not reported. To our knowledge, none of the shortlisted genes, except for ATP13A2 and EIF4G1, have been previously reported in PD39,40,41,44.
Biological processes and networks over-represented in our genes set
The assessment of the resulting genes list with Ingenuity Pathway Analysis (IPA) discovered “Cell Death and Survival”, “Cellular Assembly and Organization” and “Cell Morphology” to be among the top ranking molecular and cellular functions. As for system level functions, “Nervous System Development and Function”, “Tissue Morphology” and “Embryonic Development” were among the top 5 (Supplementary Table S4). On the other hand, Gene Ontology (GO) analysis revealed significant enrichment in terms pertaining to the nervous system. Terms like “Nervous system development (GO:0007399)”,“Neuron part (GO:0097458)”, “Postsynaptic density (GO:0014069)”, “Postsynaptic specialization (GO:0099572)”, “Asymmetric synapse (GO:0032279)”, and “Neuron to neuron synapse (GO:0098984)”, were all over-represented (Supplementary Table S5). Both approaches identified categories specific or related to the nervous system. In addition, differential brain expression was also determined from public databases (Supplementary Table S6).
Discussion
Next Generation Sequencing (NGS) is widely used to identify disease causing variants in monogenic and complex disorders45,46,47. Both exome and targeted sequencing are used as molecular research tools for gene discovery in neurological disorders25,26,48,49. The success of WES in identifying causative mutations in different disorders has encouraged its application in clinical settings26,50. And with the implementation of precision medicine and through incorporation of molecular screening as part of routine clinical practice, identification of rare variants in complex disorders becomes a research priority.
PD-NGS oriented studies focused on small sets of familial cases or trios, with a few exceptions where large cohorts were used to identify de novo and inherited mutations. Interestingly, the majority of the identified mutations represent private changes restricted to a single family or case, which is expected considering the heterogeneity of the disease4,33. These results suggested that infrequent low penetrant mutations in PD patients could be a major cause of the disease4,51. Hence, searching for a prevalent causative gene/mutation is unreasonable especially that mutations in some of the previously known PD genes with established disease related functional roles are rare21,30,32,35.
Identifying rare variants with minor allele frequencies (MAF) (≤%1) in genes expressed in the brain and/or in molecular pathways linked to a neurological disorder could contribute to our understanding of the genetic basis of PD. We complied a cohort of both familial and sporadic cases that were subjected to WES and analysed the data regardless of consanguinity or mode of inheritance using a strict multistage filtering. The analysis yielded a general list of rare variants in 51/60 cases and prioritized list of potential disease related variants in 25 index cases (Table 1, Supplementary Table S1).
In total 20 genes were shortlisted with potential disease related variants; all these genes were not previously linked to PD except for EIF4G1 and ATP13A2, where there are conflicting reports on their role in familial PD39,41,44. Eleven variants were absent in our local control database and 4 out of these were recorded in international databases, the remaining had a MAF of less than 1% in local and/or international databases. Although the majority of the SNVs identified in this study were unique events in a single family or a sporadic case; our pipeline identified multiple variants in 5 genes. Two different variants were detected in 2 of these genes (NOTCH4 and EPRS), whereas PUM1, FAM174A and EIF4G1 had a single recurrent variant (Table 1, Supplementary Table S1).
All the cases selected for WES in this study were negative for point mutations and confirmed CNV changes in the known genes43, with the exception of one familial and a sporadic case each carried PARKIN exon3 deletion and an additional WES identified variant. The two affected siblings from PD-19 were heterozygous for the PARKIN exon3 deletion and both had a truncating variant in FAM174A, this may represent a case of digenic mechanism for disease progression52. The sporadic case PD-56, homozygous for PARKIN exon3 deletion, had a heterozygous rare missense variant. Although PARKIN exon3 deletion is a confirmed disease-causing mutation13,53,54, these additional variants may play a role in disease progression and may contribute to its phenotypic heterogeneity.
Assigning disease causality of newly identified variants requires rigorous functional assessment and segregation analysis, however, when feasible; replication cohorts provide evidence that may support novel findings4,33. One of the limitations of our study is the lack of unaffected family members or parents for segregation analysis and since we did not have access to a powerful PD disease-related exome dataset; we further investigated the occurrence of our identified variants in Saudi Human Genome Program (SHGP) patient database, which contained cases with overlapping neurodegenerative phenotypes. We found that 7 of our variants were also present in patients with overlapping phenotypes (Supplementary Table S7), which further supports the role of these genes in the development of PD.
To gain an insight into the impact of the identified genes on the central nervous system (CNS), we surveyed the Mouse Genome Informatics (MGI; http://www.informatics.jax.org) database for any available transgenic mice for our candidate genes with a neurological phenotype55. The knockout mouse models of 13 of the prioritized genes presented with different neurological and behavioral phenotypes, including aging related phenotypes, tremors, impaired limb coordination, abnormal gait, abnormal synaptic vascular formation and other specific neurological phenotypes. Of these, NOTCH4 and CLSTN3 showed extensive regional brain anomalies. Tremors and/or abnormal balance were present in SPF7, ATP13A2, CLSTN3 and NEFH transgenic mice, while involuntary movement and limb gasping were observed characteristics in the CELSR1 and SNAP91 transgenic mice, respectively (Supplementary Table S8).
In addition to mining mouse models databases, both GO and IPA analysis of our selected genes list have identified significant enrichment of biological processes/terms related to the nervous system and neuronal function (Supplementary Tables S4 and S5). This enrichment further supports the hypothesis that these genes may influence key neuronal functions and contribute to the disorder.
Taken together, from the 18 genes prioritized here, NOTCH4 is the strongest PD candidate gene with a unique truncation and a splice site variant identified in two sporadic cases; the splice site variant was also present in a familial case in the SHGP pandp (Supplementary Table S7) with an overlapping phenotype. There are conflicting reports about the linkage of NOTCH4 variants to neurological conditions including schizophrenia and Alzheimer’s disease but none linking it to PD56,57,58,59. Several Notch4-allele targeted and knockout mouse models were developed, including reporters and inducible transgenics. Interestingly, one of these transgenes, Tg(tetO-Notch4*)1Rwng (MGI:5502689), contains DNA encoding amino acids 1411–1964 of Notch4 placed under the control of the tetracycline response element and is used to model arteriovenous malformations of the human brain (DOID:0060688). This inducible transgenic mouse exhibits multiple brain abnormalities and increased neuron apoptosis. These mice also present with ataxia and seizures60,61 (Supplementary Table S8). The Notch hetero-oligomer contains 6 characterized domains; a large extracellular domain (ECD), with 10–36 tandem Epidermal Growth Factor (EFG)-like repeats which participate in ligand interactions; a negative regulatory region, containing three cysteine-rich Lin12-Notch Repeats (LNR); a single transmembrane domain (TM); a small intracellular domain (ICD), which includes a RAM (RBPjk-association module); in addition to six ankyrin repeats (ANK), involved in protein-protein interactions; and a PEST domain62,63. The p.Q1257X variant, identified in PD-64 falls in the (LNR) repeat involved in receptor regulation, while the splice variant (c.2865 + 2T > C) affects an exon that falls within Calcium-binding EFG-like domain. It is well established that Notch signalling is an evolutionarily conserved pathway involved in a wide variety of developmental processes, including adult homeostasis, stem cell maintenance, cell proliferation and apoptosis62,64.
In summary, we used a combination of gene dosage and NGS analysis to screen for changes in both familial and sporadic PD cases. Using this approach we identified at least a single potential disease related genetic event in 51/60 cases studied. Our strict filtering and prioritizing criteria retained at least one single rare variant in (26/51) 50% of the cases studied; some of these variants are in strong candidate genes with known brain related functions while others may represent low penetrance risk alleles. Failure to identify potential candidate variants in the remaining cases could be attributed to a number of reasons, including missing variants in poorly covered regions or variants in non-coding or regulatory regions. There is also the possibility of gene dosage alterations existing in genes not included in this study. This approach is suitable for a complex heterogeneous disorder with different molecular mechanisms at play.
Conclusions
We have used a stringent multistage filtering of WES data to identify potential disease related variants in a cohort of both sporadic and familial cases. We identified a number of interesting variants in genes not previously linked to PD. Our data is consistent with previous WES studies where the majority of variants and candidate genes identified represent private events with very low or no rate of replication. The integrative approach employed here generated a useful catalogue of rare potentially deleterious PD candidate variants for further genetic replication and functional assessment studies.
Methods
Patients and samples
We assembled a cohort of 60 Saudi patients (19 familial and 41 sporadic) whom all presented with a consultation of PD symptoms (Supplementary Tables S9, S10 and Fig. S1). Pathogenic point mutations in PD major genes were previously ruled out in these patients43.
Multiplex ligation-dependent probe amplification (MLPA)
Gene dosage alterations were assessed using two commercially available MLPA kits: SALSA MLPA probemix P051-D1 and P052-D1 Parkinson (MRC Holland, The Netherlands) as described65 (Fig. 1a). Together, the probemixes contained MLPA probes covering all exons of the following PD related genes: PARKIN, SNCA, PINK1, PARK7, UCLH1, and GCH1, as well as selected exons of LRRK2, ATP13A2, CAV1, and CAV2. Different probes covering all exons of PARKIN were included in both kits permitting cross verification of any detected changes. Rearrangements detected in PARKIN were verified using P052-D1 MLPA assay and/or by conventional PCR using primers flanking the deleted exon as previously described43.
Whole exome sequencing, data processing and primary analysis
Whole exome sequencing and subsequent data analysis for all samples were performed as previously described48. Briefly, 100 ng of genomic DNA from each sample was sequenced on the Ion Proton platforms using the whole exome AmpliSeq kit (Life Technologies, Carlsbad, CA, USA). A maximum of 17 Gb of DNA sequence was generated for each sequencing run/sample. First, reads were subjected to quality control (QC) checks to eliminate any low quality reads, then were mapped and aligned to UCSC Human reference genome (hg19) (http://genome.ucsc.edu/) using tmap, which is part of the Torrent Suite package. All variants were called using Torrent Suite Variant Caller (Life Technologies, Carlsbad, CA, USA) and annotated with ANNOVAR (http://annovar.openbioinformatics.org).
Filtering and validation
Extensive genetic and allelic heterogeneity, incomplete penetrance and the possible presence of phenocopies, all have often been observed in complex disorders such as PD30,33,34,35,36. With this in mind, we decided to filter for variants with homozygous and heterozygous transmission for both familial and sporadic cases- regardless of the observed/predicted mode of inheritance and consanguinity. This is to allow for interfamilial and intrafamilial heterogeneity and to avoid missing autosomal recessive variants in sporadic cases66,67. Only genes with positive brain expression in publicly available databases (as listed in Gene cards) were selected. Of the selected genes, only functional variants (LoF and missense) were retained before applying the allele frequency filter. We used a stringent minor allele frequency filter of (MAF ≤ 1%) in the SHGP local “ethnically matching” database and/or international (ExAC and 1000 Genomes)42,68. The resulting variants were then validated using Sanger sequencing (Fig. 1b).
Local databases and controls
The SHGP database constitutes NGS data from exomes and 13 targeted gene panels including a panel specific for neurological disorders48,69. At the time of the analysis, the exome database included 2379 local control exomes (termed here SHGP Exdb). The neurological disorders gene panel comprised 1863 patients diagnosed with different neurological disorders including neurodegeneration and neuropathy (this patient database is referred to as SHGP pandb). These databases are from a population with high endogamy and consanguinity and is enriched with rare recessive disease causing mutations, some with founder effect69. We used the SHGP Exdb as the ethnically matched controls and the SHGP pandb as our local replication cohort.
Variant prioritization
Because WES generates a large number of variants even after the initial filtration, we created a list of prioritized variants worthy of further investigation in future studies. It is important to point out that we deliberately avoided using the American College of Medical Genetics (ACMG) variant classification system which is only intended for Mendelian disorders and is considered unsuitable approach for complex disorders70. We therefore, have devised a prioritization strategy in which only variants predicted to be strongly deleterious (CADD > 20 and PredictSNP2 classification of “deleterious” for splice site variants) were considered. Of note, only splice site variants affecting exons not subject to alternative splicing were considered71,72. Candidate variants were further prioritized on the bases of meeting at least 1 of 5 strict criteria (1) presence in a gene that was observed in 2 or more cases, (2) presence in a gene previously associated with PD, (3) the gene harbouring the variant has a mouse model with documented neurological or behavioural deficits, (4) same variant was found in additional affected family members (when available), and finally (5) same variant was observed in our local replication cohort (SHGP pandb) (Fig. 1c).
IPA and GO-enrichment analysis
To identify the functional categories enriched in our genes set (genes containing variants with CADD > 20 and PredictSNP2 classification of “deleterious” for splice site variants), we used two independent web-based applications; Gene Ontology (GO) enrichment analysis (http://geneontology.org/)73 and the Ingenuity Pathway Analysis software core analysis function (IPA®,v01-08,QIAGEN, Redwood City, www.qiagen.com/ingenuity). Our genes set was analyzed for any significant (p < 0.05 and Bonferroni corrected) over-represented GO terms under the three main categories (molecular function, cellular component and biological process). For the IPA core analysis, we first uploaded the genes accession numbers into the software before running either “Expression” or “Variant effect” core analysis (both gave identical results). The analysis was set using the “Ingenuity Knowledge base” as a reference set. The pre- analysis filtering included all “Data sources”, “Tissues and cell lines” and “Mutations findings from the knowledge base”. IPA uses right-tailed Fisher’s exact test to calculate the statistical significance of the resulting functions, pathways and networks. Only the top 5 terms under each category are listed in this study. As for networks, only those with a score >5 were considered.
Ethics, consent and permissions
We declare that informed consents were obtained from all participants in adherence with the declaration of Helsinki and according to KFSHRC IRB and Research Advisory Committee (RAC) rules and regulations under the following approved project (RAC# 2110035).
Data Availability
The data supporting the results of this article are included within the article and its additional files. Additional datasets used and/or analyzed during the current study are available from the corresponding author on request.
References
de Rijk, M. C. et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 54, S21–23 (2000).
Hughes, A. J., Daniel, S. E., Ben-Shlomo, Y. & Lees, A. J. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain: a journal of neurology 125, 861–870 (2002).
Fearnley, J. M. & Lees, A. J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain: a journal of neurology 114(Pt 5), 2283–2301 (1991).
Farlow, J. L. et al. Whole-Exome Sequencing in Familial Parkinson Disease. JAMA neurology 73, 68–75, https://doi.org/10.1001/jamaneurol.2015.3266 (2016).
Kruger, R. et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nature genetics 18, 106–108, https://doi.org/10.1038/ng0298-106 (1998).
Kalinderi, K., Bostantjopoulou, S. & Fidani, L. The genetic background of Parkinson’s disease: current progress and future prospects. Acta neurologica Scandinavica 134, 314–326, https://doi.org/10.1111/ane.12563 (2016).
Gasser, T. et al. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nature genetics 18, 262–265, https://doi.org/10.1038/ng0398-262 (1998).
Jones, A. C. et al. Autosomal recessive juvenile parkinsonism maps to 6q25.2-q27 in four ethnic groups: detailed genetic mapping of the linked region. American journal of human genetics 63, 80–87, https://doi.org/10.1086/301937 (1998).
Chartier-Harlin, M. C. et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet (London, England) 364, 1167–1169, https://doi.org/10.1016/s0140-6736(04)17103-1 (2004).
Khan, N. L. et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain: a journal of neurology 128, 2786–2796, https://doi.org/10.1093/brain/awh667 (2005).
Hatano, Y. et al. Novel PINK1 mutations in early-onset parkinsonism. Annals of neurology 56, 424–427, https://doi.org/10.1002/ana.20251 (2004).
Li, Y. et al. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology 64, 1955–1957, https://doi.org/10.1212/01.wnl.0000164009.36740.4e (2005).
Abbas, N. et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Human molecular genetics 8, 567–574 (1999).
Arias Vasquez, A. et al. A deletion in DJ-1 and the risk of dementia–a population-based survey. Neuroscience letters 372, 196–199, https://doi.org/10.1016/j.neulet.2004.09.040 (2004).
Bonifati, V. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science (New York, N.Y.) 299, 256–259, https://doi.org/10.1126/science.1077209 (2003).
Farrer, M. J. et al. Genomewide association, Parkinson disease, and PARK10. American journal of human genetics 78, 1084–1088; author reply 1092–1084, https://doi.org/10.1086/504728 (2006).
Latourelle, J. C. et al. Genomewide association study for onset age in Parkinson disease. BMC medical genetics 10, 98, https://doi.org/10.1186/1471-2350-10-98 (2009).
Pankratz, N. et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Human genetics 124, 593–605, https://doi.org/10.1007/s00439-008-0582-9 (2009).
Valente, E. M. et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35–p36. American journal of human genetics 68, 895–900 (2001).
Vaughan, J. et al. The alpha-synuclein Ala53Thr mutation is not a common cause of familial Parkinson’s disease: a study of 230 European cases. European Consortium on Genetic Susceptibility in Parkinson’s Disease. Annals of neurology 44, 270–273, https://doi.org/10.1002/ana.410440221 (1998).
Kilarski, L. L. et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 27, 1522–1529, https://doi.org/10.1002/mds.25132 (2012).
Bohlega, S. A. et al. Clinical heterogeneity of PLA2G6-related Parkinsonism: analysis of two Saudi families. BMC research notes 9, 295, https://doi.org/10.1186/s13104-016-2102-7 (2016).
Klein, C., Chuang, R., Marras, C. & Lang, A. E. The curious case of phenocopies in families with genetic Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 26, 1793–1802, https://doi.org/10.1002/mds.23853 (2011).
Nichols, W. C. et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet (London, England) 365, 410–412, https://doi.org/10.1016/s0140-6736(05)17828-3 (2005).
Nemeth, A. H. et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain: a journal of neurology 136, 3106–3118, https://doi.org/10.1093/brain/awt236 (2013).
van de Warrenburg, B. P. et al. Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. European journal of human genetics: EJHG 24, 1460–1466, https://doi.org/10.1038/ejhg.2016.42 (2016).
Jonsson, T. et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99, https://doi.org/10.1038/nature11283 (2012).
Guerreiro, R. et al. TREM2 variants in Alzheimer’s disease. The New England journal of medicine 368, 117–127, https://doi.org/10.1056/NEJMoa1211851 (2013).
Guerreiro, R. et al. Nonsense mutation in PRNP associated with clinical Alzheimer’s disease. Neurobiology of aging 35, 2656 e2613–2656 e2616, https://doi.org/10.1016/j.neurobiolaging.2014.05.013 (2014).
Vilarino-Guell, C. et al. VPS35 mutations in Parkinson disease. American journal of human genetics 89, 162–167, https://doi.org/10.1016/j.ajhg.2011.06.001 (2011).
Zimprich, A. et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. American journal of human genetics 89, 168–175, https://doi.org/10.1016/j.ajhg.2011.06.008 (2011).
Vilarino-Guell, C. et al. DNAJC13 mutations in Parkinson disease. Human molecular genetics 23, 1794–1801, https://doi.org/10.1093/hmg/ddt570 (2014).
Jansen, I. E. et al. Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome biology 18, 22, https://doi.org/10.1186/s13059-017-1147-9 (2017).
Koroglu, C., Baysal, L., Cetinkaya, M., Karasoy, H. & Tolun, A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism & related disorders 19, 320–324, https://doi.org/10.1016/j.parkreldis.2012.11.006 (2013).
Zavodszky, E. et al. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nature communications 5, 3828, https://doi.org/10.1038/ncomms4828 (2014).
Munsie, L. N. et al. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Human molecular genetics 24, 1691–1703, https://doi.org/10.1093/hmg/ddu582 (2015).
Wang, H. S. et al. In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Molecular brain 7, 73, https://doi.org/10.1186/s13041-014-0073-y (2014).
Deng, H. X. et al. Identification of TMEM230 mutations in familial Parkinson’s disease. Nature genetics 48, 733–739, https://doi.org/10.1038/ng.3589 (2016).
Di Fonzo, A. et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68, 1557–1562, https://doi.org/10.1212/01.wnl.0000260963.08711.08 (2007).
Estrada-Cuzcano, A. et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain: a journal of neurology 140, 287–305, https://doi.org/10.1093/brain/aww307 (2017).
Nuytemans, K. et al. Whole exome sequencing of rare variants in EIF4G1 and VPS35 in Parkinson disease. Neurology 80, 982–989, https://doi.org/10.1212/WNL.0b013e31828727d4 (2013).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291, https://doi.org/10.1038/nature19057 (2016).
Al-Mubarak, B. R. et al. Parkinson’s Disease in Saudi Patients: A Genetic Study. PloS one 10, e0135950, https://doi.org/10.1371/journal.pone.0135950 (2015).
Huttenlocher, J. et al. EIF4G1 is neither a strong nor a common risk factor for Parkinson’s disease: evidence from large European cohorts. Journal of medical genetics 52, 37–41, https://doi.org/10.1136/jmedgenet-2014-102570 (2015).
Ng, S. B. et al. Exome sequencing identifies the cause of a mendelian disorder. Nature genetics 42, 30–35, https://doi.org/10.1038/ng.499 (2010).
Rabbani, B., Mahdieh, N., Hosomichi, K., Nakaoka, H. & Inoue, I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. Journal of human genetics 57, 621–632, https://doi.org/10.1038/jhg.2012.91 (2012).
Lalonde, E. et al. Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Human mutation 31, 918–923, https://doi.org/10.1002/humu.21293 (2010).
Al-Mubarak, B. et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: a trio study from Saudi families. Scientific reports 7, 5679, https://doi.org/10.1038/s41598-017-06033-1 (2017).
Lemke, J. R. et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 53, 1387–1398, https://doi.org/10.1111/j.1528-1167.2012.03516.x (2012).
Zech, M. et al. Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up. Movement disorders: official journal of the Movement Disorder Society, https://doi.org/10.1002/mds.26808 (2016).
Pottier, C. et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Molecular psychiatry 17, 875–879, https://doi.org/10.1038/mp.2012.15 (2012).
Schaffer, A. A. Digenic inheritance in medical genetics. Journal of medical genetics 50, 641–652, https://doi.org/10.1136/jmedgenet-2013-101713 (2013).
Kann, M. et al. Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Annals of neurology 51, 621–625, https://doi.org/10.1002/ana.10179 (2002).
Foroud, T. et al. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 60, 796–801 (2003).
Smith, C. L., Blake, J. A., Kadin, J. A., Richardson, J. E. & Bult, C. J. Mouse Genome Database (MGD)-2018: knowledgebase for the laboratory mouse. Nucleic acids research 46, D836–D842, https://doi.org/10.1093/nar/gkx1006 (2018).
Wei, J. & Hemmings, G. P. The NOTCH4 locus is associated with susceptibility to schizophrenia. Nature genetics 25, 376–377, https://doi.org/10.1038/78044 (2000).
Takahashi, S. et al. Family-based association study of the NOTCH4 gene in schizophrenia using Japanese and Chinese samples. Biological psychiatry 54, 129–135 (2003).
Stepanov, V. A. et al. [Replicative association analysis of genetic markers of cognitive traits with Alzheimer’s disease in a Russian population]. Molekuliarnaia biologiia 48, 952–962 (2014).
Shibata, N. et al. Genetic association between Notch4 polymorphisms and Alzheimer’s disease in the Japanese population. The journals of gerontology. Series A, Biological sciences and medical sciences 62, 350–351 (2007).
Murphy, P. A. et al. Endothelial Notch4 signaling induces hallmarks of brain arteriovenous malformations in mice. Proceedings of the National Academy of Sciences of the United States of America 105, 10901–10906, https://doi.org/10.1073/pnas.0802743105 (2008).
Murphy, P. A., Lu, G., Shiah, S., Bollen, A. W. & Wang, R. A. Endothelial Notch signaling is upregulated in human brain arteriovenous malformations and a mouse model of the disease. Laboratory investigation; a journal of technical methods and pathology 89, 971–982, https://doi.org/10.1038/labinvest.2009.62 (2009).
Artavanis-Tsakonas, S., Rand, M. D. & Lake, R. J. Notch signaling: cell fate control and signal integration in development. Science (New York, N.Y.) 284, 770–776 (1999).
Kopan, R. & Ilagan, M. X. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233, https://doi.org/10.1016/j.cell.2009.03.045 (2009).
Venkatesh, K. et al. NOTCH Signaling Is Essential for Maturation, Self-Renewal, and Tri-Differentiation of In Vitro Derived Human NeuralStem Cells. Cellular reprogramming 19, 372–383, https://doi.org/10.1089/cell.2017.0009 (2017).
van der Merwe, C., Carr, J., Glanzmann, B. & Bardien, S. Exonic rearrangements in the known Parkinson’s disease-causing genes are a rare cause of the disease in South African patients. Neuroscience letters 619, 168–171, https://doi.org/10.1016/j.neulet.2016.03.028 (2016).
Tan, E. K. et al. PINK1 mutations in sporadic early-onset Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 21, 789–793, https://doi.org/10.1002/mds.20810 (2006).
Lu, C. S. et al. PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson’s disease. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 159B, 183–191, https://doi.org/10.1002/ajmg.b.32012 (2012).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74, https://doi.org/10.1038/nature15393 (2015).
Saudi Mendeliome Group, S. M. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome biology 16, 134, https://doi.org/10.1186/s13059-015-0693-2 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine: official journal of the American College of Medical Genetics 17, 405–424, https://doi.org/10.1038/gim.2015.30 (2015).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics 46, 310–315, https://doi.org/10.1038/ng.2892 (2014).
Bendl, J. et al. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS computational biology 12, e1004962, https://doi.org/10.1371/journal.pcbi.1004962 (2016).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29, https://doi.org/10.1038/75556 (2000).
Acknowledgements
All NGS library building, sequencing, and bioinformatics analysis was performed by the Saudi Human Genome Program (SHGP) at King Abdulaziz City for Science and Technology (KACST) and King Faisal Specialist Hospital and Research Centre (KFSHRC). In addition, we would like to thank sequencing and genotyping core facilities in the department of Genetics at KFSHRC for performing sequencing. This work was funded and supported by KFSHRC (RAC# 2110035), and KACST grant No. 11-BIO1440-20. The Funding body was not involved in any stage of the study and had no role in the design of the study or collection, analysis and interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
Conception of the study and experiment design: Nada Al Tassan. Whole exome sequencing data analysis and variant validation: Eman Al Yemni, Amna Magrashi, Basma AlAbdulaziz, Jameela Shinwari, Batoul Baz. MLPA experiments and analysis: Abeer Mustafa, Wafa Ali, Mohamed Alhamed and Bashayer Al-Mubarak. Whole exome sequencing: Ewa Goljan, Renad Albar and Amjad Jabaan. Supervision of the whole exome sequencing and Sanger sequencing: Dorota Monies. Bioinformatics data analysis: Mohamed Abouelhoda, Tariq Faquih and Shazia Subhani. Data interpretation: Eman Al Yemni, Bashayer Al-Mubarak and Nada Al Tassan. Patients recruitment and assessment: Thamer Alkhairallah and Saeed Bohlega. Manuscript drafting and editing: Nada AlTassan, Eman Al Yemni, Jameela Shinwari. Batoul Baz and Bashayer Al-Mubarak contributed to manuscript writing.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yemni, E.A., Monies, D., Alkhairallah, T. et al. Integrated Analysis of Whole Exome Sequencing and Copy Number Evaluation in Parkinson’s Disease. Sci Rep 9, 3344 (2019). https://doi.org/10.1038/s41598-019-40102-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40102-x
- Springer Nature Limited
This article is cited by
-
The biology of mammalian multi-copper ferroxidases
BioMetals (2023)
-
A multicenter study of genetic testing for Parkinson’s disease in the clinical setting
npj Parkinson's Disease (2022)
-
Whole-Exome Sequencing Study of Consanguineous Parkinson’s Disease Families and Related Phenotypes: Report of Twelve Novel Variants
Journal of Molecular Neuroscience (2022)
-
Whole exome sequencing in ADHD trios from single and multi-incident families implicates new candidate genes and highlights polygenic transmission
European Journal of Human Genetics (2020)