
Abstract
Tandem gene duplication has led to an expansion of cone-opsin repertoires in many fish, but the resulting functional advantages have only been conjectured without empirical demonstration. Medaka (Oryzias latipes and O. sakaizumii) have eight (two red, three green, two blue, and one violet) cone opsin genes. Absorbance maxima (λmax) of the proteins vary from 356 nm to 562 nm, but those of the red opsins (long-wavelength sensitive; LWS) are nearly identical, obscuring the necessity of their coexistence. Here, we compared the LWSa and LWSb loci of these sister species and found that the gene duplication occurred long before the latipes–sakaizumii speciation (4–18 million years ago), and the high sequence similarity between the paralogues is the result of at least two events of gene conversion. These repetitive gene conversions would indicate the importance for medaka of retaining two identical LWSs in the genome. However, a newly established medaka mutant with a single LWS showed no defect in LWS expression or behavioural red-light sensitivity, demonstrating functional redundancy of the paralogs. Thus, as with many other genes after whole-genome duplication, the redundant LWS might be on the way to being lost from the current cone opsin repertoire. Thus, non-allelic gene conversion may temporarily provide an easier and more frequent solution than gene loss for reducing genetic diversity, which should be considered when assessing history of gene evolution by phylogenetic analyses.
Similar content being viewed by others


Introduction
Under daylight, vertebrates predominantly receive light by cone cells in the retina. The cones express light-absorbing proteins, the cone opsins, which are classified into four types: red (long-wavelength sensitive; LWS), green (rhodopsin 2; RH2), blue (short-wavelength sensitive 2; SWS2), and violet (SWS1). Vertebrates have various cone opsin repertoires (e.g., birds and fish have all types, whereas mammals [except for monotremes] have only LWS and SWS11), which may reflect visual adaptation in each habitat.
Many fish have increased the number of cone opsin genes by tandem (and whole-genome) duplication and the repertoire often consists of eight or more subtypes2,3,4. The repertoires have been extensively studied in a macro scale, which revealed repetitive gene gains and losses during evolutionary radiation of teleosts2,4. This diversification of cone opsin repertoires is often discussed in relation to various underwater light conditions (e.g., depth, turbidity, or colour of mates5,6,7), but direct empirical evidence supporting these speculations (i.e., what behavioural/ecological advantages the repertoires actually provide) is lacking.
The functional advantage of having two paralogous LWSs (often referred to as LWS and medium-wavelength sensitive [MWS]) is obvious in humans, because a loss of either paralogues causes the red–green colour-blindness8. Similarly, for some monkeys, trichromats have more opportunities for foraging ripe fruits or flowers in woods than dichromats9,10,11. To our knowledge, these are the only examples in which advantages for having cone opsin subtypes have been demonstrated at a behavioural/ecological level.
Medaka, Oryzias latipes and O. sakaizumii, are small freshwater fish native to the Far East. The genus comprises 32 species12 that live in variable habitats ranging from freshwater to brackish water in East/South Asia. Despite their distinctive morphological differences13, O. latipes and O. sakaizumii have been considered to be allopatric subpopulations of the same species until recently (i.e., the Southern and Northern populations of O. latipes, respectively). Their genomic sequences are highly polymorphic (e.g., an SNP rate of 3.42%14), but the species are sexually compatible, which has been one of the best features of this model animal for genetic/genomic experiments such as locus mapping and positional cloning15,16. Although other species in the genus Oryzias are also used for research (e.g., the sex-determining gene17), most studies are performed using these youngest sister species (sometimes without discrimination).
Medaka have two LWS paralogues as in humans. However, unlike the human LWSs, proteins encoded by the medaka LWSa and LWSb are 98.3% (351/357) identical and their absorbance maxima (λmax) are essentially the same at 561 ± 1 and 562 ± 2 nm (mean ± standard error of the mean [sem]), respectively18. This seems to be an exceptional case, considering that the LWS paralogues of other vertebrates (e.g., catarrhine primates, zebrafish, and guppy19,20,21) exhibit mutually distinct λmax. Therefore, it seems to be that either (1) duplication of the medaka LWS occurred very recently, or (2) diversification (neo-functionalization) of the LWS paralogues since an ancient duplication has been suppressed by a mechanism such as reduced mutation rate or gene conversion22.
In this study, we examined nucleotide sequences of the LWSa and LWSb loci of medaka to investigate their evolutionary history. In addition, we established a medaka strain with a single LWS using the CRISPR/Cas9 system and assessed whether the fish has a functional disadvantage because of the decreased copy number23.
Results
Orthologous comparison of the LWS loci between O. latipes and O. sakaizumii
We verified that genomic sequences of LWSa and LWSb of O. sakaizumii (the HNI strain) registered in the GenBank database (a total of 4,320 bp) were perfectly identical to the whole-genome sequence recently available at the UTGB database (http://utgenome.org/medaka_v2/#!Top.md), except for a single A–G mismatch at the 3′ UTR of LWSa (data not shown). We also obtained a corresponding genomic sequence of O. latipes (the Hd-rR strain) from the UTGB database. These genomic sequences (namely, about a 20-kb region between the upstream-neighbouring SWS2b and downstream-neighbouring gnl3-like loci) contained no gap or ambiguous nucleotide. There was no protein-coding gene other than LWSa and LWSb in this region according to the BLASTX program (although at least one microRNA gene does exist24).
As shown in Fig. 1a, these sequences of the sister species were highly similar, except for a few relatively large insertions/deletions in the intergenic regions. In both species, the protein-coding region of LWSa and LWSb consists of 1,071 bp (excluding the stop codon) and is split into six exons with conserved exon–intron boundaries. There are 16 (14 synonymous and two non-synonymous) substitutions in LWSa between the species, whereas there are 10 (all synonymous) substitutions in LWSb (Table 1). Thus, the amino-acid sequences are 99.4% (355/357) and 100.0% (357/357) identical in LWSa and LWSb between the species, respectively.

The medaka LWSa and LWSb loci. (a) Orthologous comparison between O. latipes (the Hd-rR inbred strain) and O. sakaizumii (the HNI inbred strain). Outputs of the mVISTA program using genomic sequences covering the LWSa and LWSb loci are shown at the top and bottom. The two bold black horizontal lines in the middle represent the genomic sequences, and boxes show the positions of exons: light blue, UTR; purple, coding region. Dotted lines indicate relatively large insertions/deletions. The positions of the microRNA (miR-726) gene, which is co-expressed with LWSa24, are shown by arrows. The regions highlighted in yellow and green were used for paralogous comparison in (b). (b) Paralogous comparison of LWSa and LWSb in O. latipes. The mVISTA outputs are shown at the top and bottom. The coding region, the 5′/3′ UTRs, a part of the 5′-upstream region, and the 2nd–5th introns are highly conserved. In spite of the conservation, the CNR-B and CNR-C sequences are shown to be dispensable for expressing LWSa13.
Paralogous comparison of the LWS loci within O. latipes and O. sakaizumii
We then compared the LWSa and LWSb loci of O. latipes (Fig. 1b; a similar graphic was obtained from O. sakaizumii [data not shown], as expected from the high sequence similarity [Fig. 1a]) and found a few intriguing characteristics as explained below.
The coding sequences of LWSa and LWSb were 99.3% (1,064/1,071) identical, and the proteins were 98.9% (353/357) identical. However, the intergenic region was highly divergent, except for a few upstream regions, which may indicate a shared cis-regulatory mechanism between the paralogues24. In terms of the introns, the 1st and the rest (2nd–5th) showed a distinct difference: i.e., whereas the 1st intron was divergent (too different to be aligned as the intergenic regions), sequences of the 2nd–5th introns were 99.4% (351/353) identical between the paralogues (Fig. 1b).
A similar characteristic could be observed also in the coding region: i.e., all the seven nucleotide substitutions in the coding region (see above) were located on the 1st and 2nd exons (a total of 400 bp), whereas the 3rd–6th exons (a total of 671 bp) were 100% identical (Table 1). Thus, the 3′ region (from the 2nd intron to the 6th exon) was more similar between the paralogues than the 5′ region (from the 1st exon to the 2nd exon). This characteristic of the coding region was, however, not very clear in O. sakaizumii: i.e., among a total of 13 nucleotide substitutions, seven and six were located on the 1st–2nd and 3rd–6th exons, respectively (Table 1). The identity of the 2nd–5th introns also drops to 96.9% (344/355) with a 1-bp insertion/deletion.
Further comparison of the four nucleotide sequences (i.e., LWSa of O. latipes, LWSb of O. latipes, LWSa of O. sakaizumii, and LWSb of O. sakaizumii) revealed that the substitutions in the coding region can be classified into three types: (1) conserved between the orthologues (at five sites); (2) conserved between the paralogues (at eight sites); and (3) conserved between three of the four sequences (at 10 sites; Table 2). All the type (1) substitutions were found in the 1st exon and the 5′ half of the 2nd exon, whereas all the type (2) substitutions were found in the 3′ half of the 2nd exon and the downstream exons (Table 2). Furthermore, among a total of 18 substitutions in the highly conserved 2nd–5th introns (Fig. 1b), none and five sites belonged to the types (1) and (2) substitutions, respectively (Table 3).
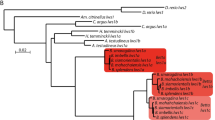
Taken together, using the middle of the 2nd exon as a border, the upstream region is more similar between the orthologues, whereas the downstream region is more similar between the paralogues in the LWS genes of medaka. This conclusion could be further supported by phylogenetic analyses (Fig. 2). The tree constructed using the upstream coding region (positions 1–163) supported sister groups of the orthologues (Fig. 2a), whereas the same analysis using the downstream coding region (positions 297–1,071) supported sister groups of the paralogues (Fig. 2b). We did not use nucleotides at positions 164–296 for these analyses, because the exact border between the upstream and downstream regions is unknown. The tree constructed using the entire coding region (Fig. 2c) supports the topology in Fig. 2b, likely because the downstream region (775 bp) is much longer than the upstream region (163 bp).

Phylogenetic relationships between the LWS genes of O. latipes and O. sakaizumii. (a) The 5′ part which might not undergo gene conversion (nucleotide positions 1–163), (b) the 3′ part which underwent gene conversion (297–1,071), or (c) the entire coding region (1–1,071) were used for the phylogenetic reconstruction by the maximum likelihood method (see Methods for details). Branch supports were shown as bootstrap values in 100 replications. The relationships focused in this study were highlighted by yellow boxes. The position of tilapia fluctuates for unknown reasons, but that of O. melastigma remains stable as an appropriate outgroup of medaka.
Chimeric fusion of the LWSa and LWSb
To assess the functional importance of having two nearly identical LWS genes for medaka, we established a medaka strain with a single LWS using the CRISPR/Cas9 system. Namely, we used gRNA targeting the 2nd exon of LWSa and LWSb25, anticipating that the double cleavages could cause a large deletion and an in-frame fusion of the paralogues (Fig. 3a).

The lwsa:b mutant with a single LWS. (a) Structures of the LWSa and LWSb loci in the wild type (top) and the hybrid LWSa:b locus in the lwsa:b mutant (bottom). Black and white boxes indicate the coding region and 5′/3′ UTRs, respectively. The target sites of the gRNA exist in the 2nd exons, and the genomic region in-between (~7 kb) was deleted in the lwsa:b mutant (dotted lines). Arrowheads show approximate positions of the primers used for genotyping of F2 siblings: white, LWSa-specific forward; black, LWSb-specific forward; grey, LWSb-specific reverse (see the Methods for their sequences). (b) Genotyping of F2 siblings (#1~9) obtained by intercrossing heterozygous F1s. Three genotypes (i.e., wild type, heterozygote, and homozygote) could be distinguished by genomic PCR using two pairs of three primers (see the arrowheads in (a) for their positions). The white–grey pair amplifies a product (~0.6 kb) from the hybrid LWSa:b gene, whereas the black–grey pair amplifies a product from the wild-type LWSb. Thus, No. 1–5 and No. 7 are heterozygous, No. 6 and No. 8 are wild type, and No. 9 is homozygous. A full image of this gel is available as Supplementary Fig. S1. (c) The missense substitution in LWSa:b. The original sequences of LWSa and LWSb are shown at the top with translated amino acids. Black letters show the target sequence of the gRNA6. The sequence of LWSa:b is shown at the bottom. The substituted nucleotide (T > A) and amino acid (Y > N) are highlighted. (d) Comparison of LWSs among vertebrates. Amino-acid sequences were collected from the GenBank database and aligned. MWSs of human and mouse are paralogues of LWS. Amino acids identical to those of the medaka LWSa (top) are shown by dots. The substituted amino acid in LWSa:b is highlighted.
Among 24 adult fish (G0) into which we had microinjected the gRNA and the Cas9 mRNA25, nine possessed the hybrid gene in the caudal fin. From one of these G0s, we obtained 73 F1s, eight of which were heterozygous for the hybrid gene. We intercrossed the heterozygous F1s and obtained 28 F2 adults, whose genotype segregation ratio was wild type:heterozygous:homozygous = 8:16:4 (Fig. 3b; other data not shown). This ratio was not significantly different from that expected (7:14:7; P = 0.424, chi-squared test) and the homozygotes were indistinguishable from other siblings by appearance.
However, the hybrid gene (termed LWSa:b) had a missense substitution (from Y to N) within the target site (Fig. 3c). A mismatch between gRNA and its target site is known to reduce cleavage efficiency in the CRISPR/Cas9 system, particularly when the mismatch is close to the protospacer adjacent motif (PAM)26. Thus, this substitution located at 4-bp upstream of the PAM sequence should efficiently fix the LWSa:b in the G0 embryos, whereas other hybrid genes without such substitution would be repetitively cleaved until the gRNA or Cas9 protein lost their activity. Although we looked for hybrid genes without any substitution or with a silent substitution, none were found (the screening could not be completed because of accidental loss of other G0s and F1s during breeding).
The substituted tyrosine is widely, but not completely, conserved between vertebrates (Fig. 3d), which may indicate a replaceable role of this amino acid. The substituted tyrosine is the 128th residue from the N terminus (which corresponds to the 131st residue in the human LWS). This site does not correspond to any of “the tuning sites”27 that have been known so far to crucially affect λmax (i.e., 180th, 197th, 277th, 285th, and 308th residues in the human LWS, which correspond to 164th, 181st, 261st, 269th, and 292nd residues in the bovine RH1).
Transcription of LWSa, LWSb, and LWSa:b
Genomic background of the host strains used in the above genetic engineering was color interfere (ci)28,29 (see the Methods section for more details about the strains). The ci might have originated from the latipes–sakaizumii hybrid population30, but its coding sequences of LWSa and LWSb were identical to those of O. sakaizumii (see Fig. 4b; other data not shown).

Expression of the LWS genes in the eyes of adult medaka. (a) Semi-quantitative reverse transcriptase PCR. Results of two fish from each genotype (wild type, LWSa:b heterozygote, LWSa:b homozygote, and lws+2a+5b mutant25) are shown as representatives. The number of PCR cycles was 24 (the amplification was stopped before plateau; see Supplementary Fig. S2 for results of stepwise PCR) in both LWS and β-actin (Actb). Whereas the reduction in the lws+2a+5b mutants, which has double frameshift mutations on LWSa and LWSb25, is apparent, expression in the lwsa:b mutants is equivalent to that in the wild type, despite the copy-number difference. (b) Direct sequencing of the reverse transcriptase PCR products. All the 14 nucleotide substitutions among the LWS genes (i.e., 13 substitutions between LWSa and LWSb [Table 1] and the missense substitution in LWSa:b [Fig. 3c]) are summarized at the top. The numbers indicate the positions of nucleotides from the translation-initiation site. Electropherograms at five sites (arrows) are shown as representatives. Note that (1) signals from LWSb are generally higher than those from LWSa in the wild type; (2) the homozygote expresses only LWSa:b; and (3) using the missense substitution at the position 382 as a border, signals from LWSa in the heterozygote are stronger or weaker than those in the wild type at the upstream or downstream region, respectively.
Using a pair of primers that commonly amplify the entire coding regions of LWSa, LWSb, and LWSa:b, we assessed expression of the mRNAs in the eyes of adult fish by semi-quantitative reverse transcription PCR. As shown in Fig. 4a, no apparent difference could be detected among the wild type, heterozygote, and homozygote, in spite of the copy-number difference. In terms of the lws+2a+5b mutant, which has double frameshift mutations on both LWSa and LWSb25, the expression appears to be reduced, likely because of the nonsense-mediated mRNA decay (NMD).
Direct sequencing of these qPCR products revealed that the wild type expresses both LWSa and LWSb, with higher expression in LWSb (Fig. 4b, top). The homozygotes express only the hybrid LWSa:b (Fig. 4b, bottom). The signals (the peak intensity of electropherogram) from LWSa are stronger in the heterozygotes in comparison with those in the wild type at the upstream region from the CRISPR/Cas9 target site (e.g., the G and T signals at the positions 163 and 279), whereas the opposite is the case at the downstream region (e.g., the A signals at the positions 465 and 531; Fig. 4b, middle). This result could be explained by the LWSa:LWSb ratio in the heterozygote: i.e., the heterozygote has three copies of LWS (LWSa, LWSb, and LWSa:b), and the ratio is 2:1 and 1:2 at upstream and downstream of the target site, respectively.
Spectral sensitivity of the lws a:b mutant
Lastly, we investigated behavioural red-light sensitivity of the homozygotes of LWSa:b (termed the lwsa:b mutant; n = 6) based on the optomotor response (OMR). At each wavelength we tested (i.e., 720 nm, 750 nm, and every 10 nm between 800 and 850 nm), the lwsa:b mutants responded to the rotating black-and-white stripes as quickly as the wild type (n = 5; Fig. 5a; P > 0.05, Student’s t test without correction). The duration of OMR (Fig. 5b) and a total distance of swimming (Fig. 5c) during the test were also similar between the wild type and the lwsa:b mutant at each wavelength, except for 720 nm and 750 nm (P < 0.05). Results of the OMR test are not always stable, because sometimes fish intentionally ignore or go against the moving stripes31. If it is the case, the OMR could be recovered at longer wavelengths (e.g., at λ = 810 nm; Fig. 5), which was an important criterion for judging whether fish do not or cannot see the stripes.

Optomotor response (OMR) of the lwsa:b mutant under monochromatic red light. The lwsa:b mutants (n = 6) were individually tested for OMR at every 10 nm in λ = 720–750 nm and 800–850 nm (light-grey bars). Dark-grey bars indicate the wild type (n = 5)31. Mean and standard error of the mean are shown. An asterisk indicates a significant difference between the wild type and mutant (P < 0.05; Student’s t test without correction). Different letters in the bars (a–d in the wild type and w–z in the mutant) indicate a significant difference according to a one-way ANOVA and the Tukey post hoc HSD test (P < 0.05). The OMR was evaluated by three parameters: (a) the average time (s) when fish started OMR (delay), (b) the average period (%) fish continued the OMR (duration), and (c) the total distance of swimming (round) towards the direction of the rotating stripes (distance). The fish occasionally showed no interest in the rotating stripes, which leads to values supporting low OMR (e.g., at λ = 730 nm of the lwsa:b mutants). However, such low OMR could be recovered at longer wavelength (e.g., λ = 810 nm), indicating that the low OMR was because of not decreased photosensitivity but decreased interests in the rotating stripes.
Significant attenuation of the OMR (i.e., gradual reduction in photosensitivity) could be detected in all three parameters described above (i.e., increase in the delay and decreases in the duration and distance) at λ ≥ 830 or 840 nm in both strains (P < 0.05, one-way ANOVA and the Tukey post hoc honestly significant difference [HSD] test). Thus, unlike the lws+2a+5b medaka, which exhibited the significant attenuation of OMR at much shorter wavelengths (i.e., λ ≥ 740 or 750 nm25,31), the behavioural red-light sensitivity of the lwsa:b mutant seems to be equivalent to that of the wild type, despite the copy-number difference.
Discussion
Evolutionary history of the medaka LWS genes
In this study, we compared four genomic sequences of the LWS loci: i.e., LWSa and LWSb of O. latipes and O. sakaizumii. The latipes–sakaizumii speciation was estimated to be about 4–18 million years ago32,33. Entire sequences of the LWS loci are consistent with the hypothesis that the LWSa–LWSb duplication preceded this speciation, because the genomic sequences were much more divergent between the duplicates (i.e., paralogues; Fig. 1b) than between the species (i.e., orthologues; Fig. 1a).
However, a part of the LWS genes was more similar between the paralogues than between the orthologues (Tables 2 and 3: Fig. 2b). This extraordinary similarity could be explained by non-allelic (ectopic) gene conversion22, which seems to frequently occur during the cone opsin evolution in vertebrates34,35. To explain all the nucleotide substitutions summarized in Tables 1–3, we propose that the sister species could undergo at least two gene conversions, one of which occurred after the latipes–sakaizumii speciation, for the following reasons.
First, the 13 nucleotides conserved between the paralogues, but not between the orthologues (Tables 2 and 3), could most plausibly be explained by a gene conversion after the speciation. Otherwise, identical substitutions at identical sites of the paralogues (e.g., from C to T substitutions at the 318th nucleotide of LWSa and LWSb of O. latipes) need to occur 13 times, which seems highly unlikely. Second, the region involved in the gene conversion (i.e., from the 3′ half of the 2nd exon to the 6th exon) is much more conserved between the paralogues in O. latipes (a total of two substitutions in the 2nd intron) than in O. sakaizumii (a total of 17 substitutions [six in the exons and 11 in the introns] and a 1-bp insertion/deletion in the 3rd intron; see also Table 1), indicating that the timing of the latest gene conversion is different between O. sakaizumii and O. latipes (i.e., earlier in O. sakaizumii).
Taken together, one gene conversion occurred very recently in O. latipes after the latipes–sakaizumii speciation. The timing of another gene conversion for O. sakaizumii could be close to the timing of the speciation, because the number of nucleotide substitutions is similar between the orthologues and between the paralogues (Table 1; Fig. 2b), but the actual timing could not be specified in this study.
We suspect that even more gene conversion might affect the 1st and the 5′ half of the 2nd exons, if Beloniformes (e.g., medaka and flying fish) and Cyprinodontiformes (e.g., guppy and platy) had shared the same duplication event4. This is because; (1) the branch length of the phylogenetic tree in Fig. 2a is apparently shorter in medaka than in guppy (i.e., 6–7 substitutions between the medaka paralogues and 6–25 substitutions between the guppy paralogues), and (2) even this region fails to show orthologous relationships between LWSa/b of medaka and LWS1–4 of guppy (Fig. 2a). Alternatively, these fish might not share the same duplication event (Lin et al. [2005] used only O. latipes as a representative of Beloniforms4). A chance for assessing the true timing of gene duplication has anyway been lost, if gene conversion has involved the entire loci.
Considering that at least two gene conversions could occur during speciation of sister species, non-allelic gene conversion between tandemly duplicated genes would be rather more frequent events than previously supposed36. This notification is important, because molecular-phylogenetic analyses alone could mislead the history of gene evolution (i.e., the orthologous/paralogous relationship). Indeed, from the phylogenetic tree using the entire cDNA sequences (Fig. 2c), the medaka LWSs seem to undergo very recent and lineage-specific gene duplications, which is highly unlikely according to the present study (Fig. 1; Tables 1–3).
Functional importance for retaining two nearly identical LWS genes
Four paralogous LWSs of guppy have undergone distinct diversification (neo-functionalization) showing divergent λmax20 (see also Fig. 2). Although the LWSs that medaka currently have are nearly identical in sequence and λmax18, a trace of such diversification seems to remain as four non-synonymous substitutions between the paralogues in the 1st and 2nd exons (Table 1). Thus, we suspect that λmax of the medaka LWSa and LWSb were previously different, as LWSs of other vertebrates19,20,21. Alternatively, diversification of the medaka LWSs might be suppressed even prior to the conversions (e.g., by reduced mutation rates). Whichever the case is, the following questions need to be addressed. (1) How were the converted alleles fixed in medaka (e.g., by selection or genetic drift)? (2) Why do medaka continue to retain the very similar duplicates, instead of losing either?
After the 3 R whole-genome duplication which occurred at a common ancestor of teleosts, 70–80% of the duplicated genes were lost taking 60 million years, most likely because of functional redundancy37. The fact that wild-type medaka show no apparent advantage in red-light sensitivity in comparison with the lwsa:b mutant (Fig. 5) suggests that the paralogues are indeed redundant and either could be dispensable. In addition, the alternative expression system of the LWS (and other cone opsin) paralogues known in human and zebrafish (i.e., a shared upstream enhancer competitively accessed by the LWS promoters38,39,40), which medaka may also have24, seems to be nonsense, if the alternates are identical. Considering these discrepancies, we propose that the current cone opsin repertoire of medaka is at a provisional state and about to reduce the copy number of LWS. Medaka may have retained the duplicates because gene conversion is a quicker and more frequent34,35 solution than gene loss for reducing genetic diversity.
However, it is also possible that the alternative expression of the nearly identical (i.e., different) LWSs is not necessary for medaka in the laboratory (Fig. 5), but is in nature. From this standpoint, a recent population genetic study using stickleback, which demonstrated a dramatic change of allele frequency in SWS2 after 19 years of habituation to a new habitat41, is intriguing. Similar trans-generation experiments using the wild-type and lwsa:b medaka (and a knock-in medaka with divergent LWSs) would be worthwhile to elucidate which cone opsin repertoire could dominate the population. Whatever is the result, however, the process (i.e., whether or not the repertoire actually provides behavioural/ecological advantages to the fish) would remain unknown (i.e., natural/sexual selection vs genetic drift). To reveal the actual driving forces of the cone opsin diversifications, establishment and behavioural phenotyping of cone opsin mutants, as shown here, would also be necessary.
Methods
Ethical issues
All the experiments presented here were conducted in accordance with the Animal Experiment Committees of JWU and NIBB. All the experimental protocols were approved by the same committees.
Sequence analysis in silico
We obtained genomic sequences encompassing the LWSa and LWSb loci of O. latipes (the Hd-rR strain) and O. sakaizumii (the HNI strain) from the UTGB database (version 2.2.4; http://utgenome.org/medaka_v2/#!Top.md). We manually annotated the sequences according to the LWS sequences in GenBank (LWSa: AB223051 and LWSb: AB223052) and also by homology search using the BLASTX program. The annotated sequences were manually trimmed (e.g., between the 3′ and 5′ ends of neighbouring genes) and aligned using the LAGAN program at the mVISTA website (http://genome.lbl.gov/vista/mvista/submit.shtml). For more detailed local comparisons, we used the Genetyx-Mac (ver. 16) software.
Phylogenetic reconstruction was performed by the PhyML42 at the ATGC website (http://www.atgc-montpellier.fr) using the default settings; i.e., a substitution model was automatically selected by the smart model selection (SMS) based on the Akaike’s information criterion (AIC)43, and a starting tree was constructed by the BIONJ44. For an outgroup, we used LWS of O. melastigma (XM_024269264), together with LWS1–4 of guppy (Poecilia reticulata; AB748985, LC127183, LC127184, and LC127185), LWS of tilapia (Oreochromis niloticus; AF247128), and LWS1/2 of zebrafish (Danio rerio; AB087803 and AB087804).
Establishment of a medaka strain with a single LWS gene; the lws a:b mutant
As described elsewhere25, we microinjected Cas9 mRNA and gRNA into fertilized eggs of control medaka: the color interfere (ci) and Actb–SLα:GFP strains28,29. ci lacks a fish-specific hormone, somatolactin alpha (SLα) and Actb–SLα:GFP overexpresses SLα28,29. Neither strain exhibits defect in viability or fertility in comparison with wild type45. We have been using these strains for establishing/characterizing colour-blind lines25,31,46, because of their unique behaviours showing colour-dependent mate choice47,48,49.
The gRNA targets the 2nd exons of the LWSa and LWSb loci (5′-GCGTGTTTGAGGGCTATGTGG-3′) which are tightly linked on chromosome 515,18. Whether a deletion of the approximately 7-kb region sandwiched by the target sites occurred was detected by genomic PCR and agarose-gel electrophoresis using two combinations of three primers: LWSa-specific forward: 5′-GGCAGAAAAGTTGGTTGGAT–3′, LWSb-specific forward: 5′-TTGTTTCCCAGATCCCTTTG-3′, and LWSb-specific reverse: 5′-CAATTCTAGTGATTCAAGACTCATTTATAAAAG-3′, whose positions are shown in Fig. 3a (see figure legend for the rational). PCR conditions were 60 s at 94 °C for initial denaturation, 40 cycles of 20 s at 98 °C, 1 min at 60 °C, 1 min at 72 °C, and 10 min at 72 °C for final extension. The amplified products in the gel were visualized by ethidium-bromide staining and UV irradiation.
Semi-quantitative reverse transcription PCR
We extracted total RNA from the eyes of adult medaka (under 1 year old) using ISOGEN II (Nippon Gene). The RNA was treated with deoxyribonuclease (RT Grade) for Heat Stop (Nippon Gene) and used for reverse transcription (RT) using ReverTra Ace (TOYOBO) and polyT primer. Using the RT products (cDNAs) as templates, we performed PCR using the following primers, which were designed to commonly amplify the LWSa, LWSb, and hybrid LWSa:b cDNAs; forward: 5′-GGCAGAGSAGTGGGGAAAACAGG-3′ and reverse: 5′-TATGCAGGAGCCACAGAGGAGACC-3′. For a control, we amplified the β-actin (Actb) cDNA using the following primers; forward: 5′-GATTCCCTTGAAACGAAAAGCC-3′ and reverse: 5′-CAGGGCTGTTGAAAGTCTCAAAC-3′. PCR conditions were 60 s at 94 °C for initial denaturation, 16–26 cycles of 20 s at 98 °C, 1 min at 58 °C, 1 min at 72 °C, and 10 min at 72 °C for final extension. All the products were visualized by agarose-gel electrophoresis, ethidium-bromide staining, and UV irradiation.
Optomotor response (OMR) test using the Okazaki large spectrograph (OLS)
As previously described31, we performed quantitative OMR tests using the lwsa:b mutants (n = 6), which we outline here briefly.
The OLS generates a series of monochromatic lights (λ = 250–1,000 nm) of about 10-m width using a xenon arc lamp and a diffraction grating50. The light intensity (photon density) varies depending on wavelength, but was 1.4–7.5 × 1015 photons/cm2/s at λ = 720–850 nm31. Using light at an intended wavelength, we illuminated from the top a cylindrical glass tank and an electronic apparatus for rotating black-and-white stripes around the tank. We placed individual medaka into the tank, started the OLS illumination, waited for 30 s, and rotated the stripes in a clockwise, counter-clockwise, and clockwise direction for 30 s each for an OMR test. The behaviour of the fish was recorded using an infra-red (IR)-sensitive video camera (A10FHDIR; Kenko) and the position of the fish in each frame (x–y coordinates) was detected by the UMATracker software51.
Using the series of x–y coordinates, we calculated (1) the average time (s) in the three rotations until the fish started the OMR (delay), (2) the average period (%) of the fish showing the OMR in the tree rotations (duration), and (3) the total distance (round) the fish swam towards the direction of the rotating stripes during the test (distance). The data were averaged at each wavelength and compared between the wild type and lwsa:b mutant by Student’s t test. We also compared the averages in each strain at different wavelengths using one-way analysis of variance (ANOVA) and the Tukey post hoc HSD test. The data for the wild-type fish (n = 5) in Fig. 5 are those previously reported31.
References
Bowmaker, J. K. Evolution of colour vision in vertebrates. Eye 12, 541–547 (1998).
Rennison, D. J., Owens, G. L. & Taylor, J. S. Opsin gene duplication and divergence in ray-finned fish. Mol. Phylogenet. Evol. 62, 986–1008 (2012).
Bowmaker, J. K. Evolution of vertebrate visual pigments. Vision Res. 48, 2022–2041 (2008).
Lin, J. J., Wang, F. Y., Li, W. H. & Wang, T. Y. The rises and falls of opsin genes in 59 ray-finned fish genomes and their implications for environmental adaptation. Sci. Rep. 7, 15568 (2017).
Valen, R., Edvardsen, R. B., Søviknes, A. M., Drivenes, Ø. & Helvik, J. V. Molecular evidence that only two opsin subfamilies, the blue light- (SWS2) and green light-sensitive (RH2), drive color vision in Atlantic Cod (Gadus morhua). PLoS One, https://doi.org/10.1371/journal.pone.0115436 (2014).
Escobar-Camacho, D., Ramos, E., Martins, C. & Carleton, K. L. The opsin genes of amazonian cichlids. Mol. Ecol. 26, 1343–1356 (2017).
Wang, F. Y., Chung, W. S., Yan, H. Y. & Tzeng, C. S. Adaptive evolution of cone opsin genes in two colorful cyprinids, Opsariichthys pachycephalus and Candidia barbatus. Vision Res. https://doi.org/10.1016/j.visres.2008.04.026 (2008).
Cole, B. L. Protan colour vision deficiency and road accidents. Clin. Exp. Optom. 85, 246–253 (2002).
Melin, A. D. et al. Trichromacy increases fruit intake rates of wild capuchins (Cebus capucinus imitator). Proc. Natl. Acad. Sci. 114, 201705957 (2017).
Veilleux, C. C. et al. Group benefit associated with polymorphic trichromacy in a Malagasy primate (Propithecus verreauxi). Sci. Rep. https://doi.org/10.1038/srep38418 (2016).
Hogan, J. D., Fedigan, L. M., Hiramatsu, C., Kawamura, S. & Melin, A. D. Trichromatic perception of flower colour improves resource detection among New World monkeys. Sci. Rep. 8, 4–10 (2018).
Mokodongan, D. F. et al. Phylogenomics reveals habitat-associated body shape divergence in Oryzias woworae species group (Teleostei: Adrianichthyidae). Mol. Phylogenet. Evol. 118, 194–203 (2018).
Asai, T., Senou, H. & Hosoya, K. Oryzias sakaizumii, a new ricefish from northern Japan (Teleostei: Adrianichthyidae). Ichthyol. Explor. Freshwaters 22, 289–299 (2011).
Kasahara, M. et al. The medaka draft genome and insights into vertebrate genome evolution. Nature 447, 714–719 (2007).
Naruse, K. et al. A detailed linkage map of medaka, Oryzias latipes: comparative genomics and genome evolution. Genetics 154, 1773–84 (2000).
Fukamachi, S., Shimada, A. & Shima, A. Mutations in the gene encoding B, a novel transporter protein, reduce melanin content in medaka. Nat. Genet. 28, 381–385 (2001).
Matsuda, M. & Sakaizumi, M. Evolution of the sex-determining gene in the teleostean genus Oryzias. Gen. Comp. Endocrinol. https://doi.org/10.1016/j.ygcen.2015.10.004 (2016).
Matsumoto, Y., Fukamachi, S., Mitani, H. & Kawamura, S. Functional characterization of visual opsin repertoire in Medaka (Oryzias latipes). Gene 371, 268–278 (2006).
Chinen, A., Hamaoka, T., Yamada, Y. & Kawamura, S. Gene duplication and spectral diversification of cone visual pigments of zebrafish. Genetics 163, 663–675 (2003).
Kawamura, S. et al. Spectral sensitivity of guppy visual pigments reconstituted in vitro to resolve association of opsins with cone cell types. Vision Res. 127, 67–73 (2016).
Kawamura, S. Color vision diversity and significance in primates inferred from genetic and field studies. Genes and Genomics 38, 779–791 (2016).
Hastings, P. J. Mechanisms of Ectopic Gene conversion. Genes (Basel). 1, 427–439 (2010).
Ewen-Campen, B., Mohr, S. E., Hu, Y. & Perrimon, N. Accessing the Phenotype Gap: Enabling Systematic Investigation of Paralog Functional Complexity with CRISPR. Developmental Cell, https://doi.org/10.1016/j.devcel.2017.09.020 (2017).
Daido, Y., Hamanishi, S. & Kusakabe, T. G. Transcriptional co-regulation of evolutionarily conserved microRNA/cone opsin gene pairs: Implications for photoreceptor subtype specification. Dev. Biol. 392, 117–129 (2014).
Homma, N., Harada, Y., Uchikawa, T., Kamei, Y. & Fukamachi, S. Protanopia (red color-blindness) in medaka: A simple system for producing color-blind fish and testing their spectral sensitivity. BMC Genet. 18, 10 (2017).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Yokoyama, S., Yang, H. & Starmer, W. T. Molecular basis of spectral tuning in the red- and green-sensitive (M/LWS) pigments in vertebrates. Genetics 179, 2037–2043 (2008).
Fukamachi, S., Sugimoto, M., Mitani, H. & Shima, A. Somatolactin selectively regulates proliferation and morphogenesis of neural-crest derived pigment cells in medaka. Proc. Natl. Acad. Sci. 101, 10661–10666 (2004).
Fukamachi, S., Yada, T., Meyer, A. & Kinoshita, M. Effects of constitutive expression of somatolactin alpha on skin pigmentation in medaka. Gene 442, 81–87 (2009).
Takehana, Y. et al. Origin of Boundary Populations in Medaka (Oryzias latipes Species Complex). Zoolog. Sci. 33, 125–131 (2016).
Matsuo, M., Ando, Y., Kamei, Y. & Fukamachi, S. A semi-automatic and quantitative method to evaluate behavioral photosensitivity in animals based on the optomotor response (OMR). Biol. Open 7, bio033175 (2018).
Takehana, Y., Nagai, N., Matsuda, M., Tsuchiya, K. & Sakaizumi, M. Geographic Variation and Diversity of the Cytochrome b Gene in Japanese Wild Populations of Medaka, Oryzias latipes. Zoolog. Sci. 20, 1279–1291 (2003).
Setiamarga, D. H. E. et al. Divergence time of the two regional medaka populations in Japan as a new time scale for comparative genomics of vertebrates. Biol. Lett. 5, 812–816 (2009).
Hiwatashi, T. et al. Gene conversion and purifying selection shape nucleotide variation in gibbon L/M opsin genes. BMC Evol. Biol. 11, 312 (2011).
Cortesi, F. et al. Ancestral duplications and highly dynamic opsin gene evolution in percomorph fishes. Proc. Natl. Acad. Sci. 112, 1493–1498 (2015).
Harpak, A., Lan, X., Gao, Z. & Pritchard, J. K. Frequent nonallelic gene conversion on the human lineage and its effect on the divergence of gene duplicates. Proc. Natl. Acad. Sci. 114, 201708151 (2017).
Inoue, J., Sato, Y., Sinclair, R., Tsukamoto, K. & Nishida, M. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proc. Natl. Acad. Sci. https://doi.org/10.1073/pnas.1507669112 (2015).
Takechi, M. Temporal and spatial changes in the expression pattern of multiple red and green subtype opsin genes during zebrafish development. J. Exp. Biol. 208, 1337–1345 (2005).
Tsujimura, T., Hosoya, T. & Kawamura, S. A single enhancer regulating the differential expression of duplicated red-sensitive opsin genes in Zebrafish. PLoS Genet. 6, 1–10 (2010).
Tsujimura, T., Chinen, A. & Kawamura, S. Identification of a locus control region for quadruplicated green-sensitive opsin genes in zebrafish. Proc. Natl. Acad. Sci. 104, 12813–12818 (2007).
Marques, D. A. et al. Convergent evolution of SWS2 opsin facilitates adaptive radiation of threespine stickleback into different light environments. PLoS Biol. 15, e2001627 (2017).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Lefort, V., Longueville, J. E. & Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol., https://doi.org/10.1093/molbev/msx149 (2017).
Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Komine, R. et al. Transgenic medaka that overexpress growth hormone have a skin color that does not indicate the activation or inhibition of somatolactin-α signal. Gene 584, 38–46 (2016).
Kamijo, M., Kawamura, M. & Fukamachi, S. Loss of red opsin genes relaxes sexual isolation between skin-colour variants of medaka. Behav. Processes 150, 25–28 (2018).
Fukamachi, S. et al. Dual control by a single gene of secondary sexual characters and mating preferences in medaka. BMC Biol. 7, 64 (2009).
Ikawa, M., Ohya, E., Shimada, H., Kamijo, M. & Fukamachi, S. Establishment and maintenance of sexual preferences that cause a reproductive isolation between medaka strains in close association. Biol. Open 6, 244–251 (2017).
Utagawa, U., Higashi, S., Kamei, Y. & Fukamachi, S. Characterization of assortative mating in medaka: Mate discrimination cues and factors that bias sexual preference. Horm. Behav. 84, 9–17 (2016).
Watanabe, M. et al. Design and performance of the Okazaki Large Spectrograph for photobiological research. Photochem Photobiol 36, 491–498 (1982).
Yamanaka, O. & Takeuchi, R. UMATracker: an intuitive image-based tracking platform. J. Exp. Biol. jeb. 182469, https://doi.org/10.1242/jeb.182469 (2018).
Acknowledgements
We thank Shigehiro Kuraku at RIKEN for the helpful discussions about gene conversion and Mizuki Sano at JWU for assistance with the OMR test. This study was supported by a Grant-in-Aid for Scientific Research (C) (#17K07506) from the Japan Society for the Promotion of Science and research funds from JWU to S.F. The study was also supported as a Priority Collaborative Research Project (#16–101) at NIBB.
Author information
Authors and Affiliations
Contributions
Y.H. established and M.M. characterized the lwsa:b mutant. Y.K. supported the OMR tests using the OLS. M.G. contributed to the mutant characterization. S.F. designed and supervised the experiments, analysed the nucleotide sequences, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Harada, Y., Matsuo, M., Kamei, Y. et al. Evolutionary history of the medaka long-wavelength sensitive genes and effects of artificial regression by gene loss on behavioural photosensitivity. Sci Rep 9, 2726 (2019). https://doi.org/10.1038/s41598-019-39978-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39978-6
- Springer Nature Limited
This article is cited by
-
Knockout of lws1 in zebrafish (Danio rerio) reveals its role in regulating feeding and vision-guided behavior
Functional & Integrative Genomics (2024)
-
Behavioral photosensitivity of multi-color-blind medaka: enhanced response under ultraviolet light in the absence of short-wavelength-sensitive opsins
BMC Neuroscience (2023)