
Abstract
HPV genomic variability and chromosomal integration are important in the HPV-induced carcinogenic process. To uncover these genomic events in an HPV infection, we have developed an innovative and cost-effective sequencing approach named TaME-seq (tagmentation-assisted multiplex PCR enrichment sequencing). TaME-seq combines tagmentation and multiplex PCR enrichment for simultaneous analysis of HPV variation and chromosomal integration, and it can also be adapted to other viruses. For method validation, cell lines (n = 4), plasmids (n = 3), and HPV16, 18, 31, 33 and 45 positive clinical samples (n = 21) were analysed. Our results showed deep HPV genome-wide sequencing coverage. Chromosomal integration breakpoints and large deletions were identified in HPV positive cell lines and in one clinical sample. HPV genomic variability was observed in all samples allowing identification of low frequency variants. In contrast to other approaches, TaME-seq proved to be highly efficient in HPV target enrichment, leading to reduced sequencing costs. Comprehensive studies on HPV intra-host variability generated during a persistent infection will improve our understanding of viral carcinogenesis. Efficient identification of both HPV variability and integration sites will be important for the study of HPV evolution and adaptability and may be an important tool for use in cervical cancer diagnostics.
Similar content being viewed by others


Introduction
Human papillomavirus (HPV) is the main cause of cervical cancer1, one of the most common cancers in women worldwide, causing more than 200,000 deaths each year2,3. A persistent infection with HPV high-risk genotypes is recognised as a necessary cause of cancer development4. Of the 13 carcinogenic high-risk types, HPV16 and 18 are associated with about 70% of all cervical cancers5,6. HPV infection is also associated with cancer in penis, vulva, vagina, anus, and head and neck7. However, only a small fraction of HPV infections at any site will progress to cancer8. This indicates that in addition to HPV infection, additional factors such as HPV genomic variability and integration, could contribute to the HPV-induced carcinogenic process. An appropriate sequencing approach is needed to uncover these genomic events during a persistent HPV infection.
HPV contains an approximately 7.9 kb circular double-stranded DNA genome, consisting of early region (E1, E2, E4-7) genes, late region (L1, L2) genes and an upstream regulatory region (URR)9. To date, more than 200 HPV types have been identified10. Each individual HPV type shares at least 90% sequence identity in the conserved L1 open reading frame (ORF) nucleotide sequence. Isolates of the same HPV types that differ by 1–10% or 0.5–1% across the genome are referred to as variant lineages or sublineages, respectively11,12.
Despite phylogenetic relatedness, HPV variant lineages can differ in their carcinogenic potential13,14,15,16. Traditionally, studies have focused on cancer risk of main variants. However, recent studies have revealed variability below the level of variant lineages that may be evidence of intra-host viral evolution and adaptation17,18,19,20. In contrast to a limited number of studies on HPV variability, HPV integration into the host genome has been more widely studied and is regarded as a determining event in cervical carcinogenesis21,22,23. Upon integration, disruption or complete deletion of the E1 or E2 gene is often observed, resulting in constitutive expression of the E6 and E7 oncogenes24,25,26, inactivation of cell cycle checkpoints and genetic instability23. Viral integration may also lead to modified expression of cellular genes nearby, disruption of genes, as well as genomic amplifications that may promote oncogenesis23,27. The finding of certain chromosomal clusters of integration in precancerous lesions and cancers28 also suggests a selective advantage of specific HPV integrations. Still, several important questions remain for HPV integration and more comprehensive analyses of integration sites are needed in order to expand our understanding of HPV pathogenesis.
The development of next generation sequencing (NGS) technologies has provided new tools for viral genomic research. During the recent years, a few studies have described different NGS based approaches to study HPV variability and integration in the human genome. The most common approaches used in HPV genomic analyses are based on target enrichment using highly multiplexed degenerate primers29, enrichment by multiplex PCR using HPV16 forward primers30, bead-based target capture31,32,33, and rolling circle amplification34 followed by NGS. These methods are however designed to detect either HPV integration or HPV variability. In addition, target capture methods poorly enrich HPV and remain expensive due to high probe cost and off-target sequencing.
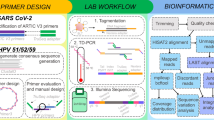
In order to contribute to the understanding of the role of intra-host HPV genomic variability and chromosomal integration in carcinogenesis, we have developed an innovative library preparation strategy followed by an in-house bioinformatics pipeline named TaME-seq (tagmentation-assisted multiplex PCR enrichment sequencing). TaME-seq combines tagmentation and multiplex PCR enrichment, allowing simultaneous HPV genomic variability and integration analysis (Fig. 1). TaME-seq, with highly efficient target enrichment and reduced sequencing cost, enables deep sequencing analysis in order to find low frequency variants and rare integration events. Here, we present the results of HPV integration and genomic variability analysis in HPV16, 18, 31, 33 and 45 positive clinical samples and cell lines. The method described here provides an important tool for comprehensive studies of HPV genomic variability and chromosomal integration, and it can also be adapted to studies on other viruses such as retroviruses, adeno-associated viruses and integrating human herpesviruses.

Primer design, laboratory and bioinformatics workflows of the TaME-seq method.
Results
Read mapping analysis and genome coverage
Table 1 summarises liquid-based cytology (LBC) samples (n = 21), cell lines (n = 4) and plasmid samples (n = 3) included in the analysis. The samples generated 154.8 million raw reads of which 72.5 million reads (47%) mapped to the target HPV reference genomes. Only a small fraction (0.08%) of the reads mapped to other HPV types than those reported positive by HPV genotyping. The mean coverage ranged from 303 to 273898, while the fraction of the genome covered by minimum 10 × ranged from 0.35 to 1, and the fraction of the genome covered by minimum 100 × ranged from 0.33 to 1 (Table 1). HPV genome sequencing coverage aligned to the target HPV genomes with the location of HPV genomic regions and primers is visualised for CaSki, HeLa, LBC34, LBC11 and MS751 (Fig. 2). Overall, the samples showed varying HPV genome coverage profiles (Supplementary Figs S1–S5). Totally, 10 HPV positive samples were excluded from further analysis due to poor sequencing coverage (Supplementary Table S1). Sequencing of the HPV negative control samples resulted in no or negligible amount (<500) of reads mapped to target HPV genomes (Supplementary Table S2). The MS751 cell line was confirmed not to contain HPV18 sequences (Supplementary Table S1)35.

HPV genome sequencing coverage in HPV positive samples. The coverage plots of (a) CaSki, (b) HeLa, (c) LBC34, (d) LBC11, and (e) MS751 are aligned to the respective target HPV genomes. The location of early (E1, E2, E4-7), late (L1, L2) genes, URR, and forward (red arrows) and reverse (blue arrows) HPV primers is indicated below the genomic positions.
Deletions in HPV genomes
The method enables identification of regions covered with very few or no sequencing reads, interpreted as large HPV genomic deletions. Cell lines HeLa and MS751 are known to contain partial HPV genomes due to deletions of 2.5 kb and 5 kb, respectively35,36, which was confirmed by our method (Fig. 2). A large deletion of 4.8 kb was revealed in the clinical sample LBC105, indicating partial or complete deletion of HPV18 genes E1, E2, E4, E5, L1 and L2 (Supplementary Fig. S2).
HPV-human integration sites
A two-step strategy was applied to detect possible integration sites (Fig. 3). A total of 27 integration sites were detected in cell lines CaSki, SiHa, HeLa and MS571 (Table 2). For CaSki, 16 previously reported integration sites30,32,37 were confirmed. In addition, three novel sites were identified. These mapped to HPV16 E6, E2 and L1 genes. One was located in an intronic region of the gene BRSK1; two were located more than 50 kb from annotated genes (Table 2). Three sites, including one previously reported site as a control30,37, were subjected to Sanger sequencing to confirm the integration sites (Supplementary Table S3). Integration sites identified in SiHa, HeLa and MS751 were consistent with previous studies31,35,36,37,38,39 and were not subjected to validation by Sanger sequencing. Additionally, two integration sites were detected in the clinical sample LBC105 (Table 2). The integration breakpoints were mapped to the HPV E1 and L1 genes flanking the deleted region (Supplementary Fig. S2) and they were located in intronic regions of the gene GTF2IRD1 (Table 2). Both integration sites were confirmed by Sanger sequencing (Supplementary Table S3).

An IGV visualisation of HISAT2 and LAST alignments to find HPV-human integration breakpoints. All the reads were first mapped with HISAT2 and then the unmapped reads were remapped with LAST. (a) SiHa reads mapping to chromosome 13 (GRCh38/hg38). Light blue HISAT2 reads have pairs mapping to HPV16 reference genome. Multi-coloured parts of the LAST reads are mismatched bases that map to HPV16 (not visualised). (b) SiHa reads mapping to HPV16 reference genome. Orange HISAT2 reads have pairs mapping to chromosome 13 (GRCh38/hg38). Multi-coloured parts of the LAST reads are mismatched bases that map to chromosome 13 (not visualised). Red arrows point to the exact breakpoint positions.
Evaluation of variant calling using SiHa technical replicates
Sequencing libraries of the SiHa cell line served as technical replicates to assess the variant calling performance. In both SiHa-1 and SiHa-2, more variable sites were detected with higher mean coverage (Fig. 4). Number of variable sites in SiHa-1 ranged from 477 to 809 and mean coverage ranged from 2554 to 17561. Number of variable sites in SiHa-2 ranged from 257 to 522 and mean coverage ranged from 646 to 5609 (Fig. 4; Supplementary Table S4). First, reproducibility of variant calling was assessed within the same SiHa sequencing library. Concordance rate of variable sites was calculated using HiSeq 2500 result as the reference value. The concordance rates varied from 92% (HiSeq downsampled 90%) to 45% (MiSeq) in SiHa-1 and from 89% (HiSeq downsampled 90%) to 27% (MiSeq) in SiHa-2 (Supplementary Table S4). Concordance rates of variants, including low frequency variation, between replicates (different library, same sequencing platform) were calculated to evaluate the effect of library preparation steps on the number of variable sites found in each sample. Concordance rates were 21% and 19% in SiHa-1 and SiHa-2, respectively (Supplementary Table S5).

Number of variable sites in SiHa replicates. SiHa-1 (red dots) and SiHa-2 (blue dots) served as technical replicates to assess the variant calling performance. In SiHa libraries, sequenced on MiSeq and HiSeq 2500 platforms, increasing number of variable sites were detected with higher mean coverage.
HPV genomic variability
Variability was analysed in cell lines and LBC samples. Samples had variable sites (variant allele frequency >0.2% and coverage ≥100×) in all genes with the exception of regions that were deleted or had low sequencing coverage. The number of variable sites was normalised by the length of each HPV genomic region. Genomic regions had varying percentages of variable sites (0–28%) in each of the samples. Overall, there were samples within each HPV type that had >15% variable sites in at least one HPV gene (Fig. 5). Principally, samples with higher mean coverage had more variable sites (Supplementary Table S6), which is in line with the results from the variant analysis done on SiHa replicates (Fig. 4). CaSki had most variable sites (1017) of the cell lines and LBC54 had most variable sites (1641) of the clinical samples (Supplementary Table S6). A variant profile with variable site positions and variant allele frequency (VAF) is shown for CaSki and LBC54 (Fig. 6). Overall, the results show considerable variability in the samples throughout the HPV genome (Fig. 5, Supplementary Figs S6–S10).

Proportion of variable sites in HPV genes in HPV positive samples. The number of variable sites was normalised by the length of each HPV gene. Gradient green (0% variable sites) to red (30% variable sites) color-coding of the results is shown to present the considerable variability in the samples throughout the HPV genome.

HPV nucleotide variation observed in two samples. The plots showing variable sites and variant allele frequency (%) in (a) CaSki, and (b) LBC54 are aligned to the respective target HPV genomes. The location of genes and URR is indicated below the genomic positions. The red line indicates the variant calling threshold value of 0.2%.
Discussion
Here, we present a novel cost-efficient approach, TaME-seq, for the simultaneous analysis of HPV variation and chromosomal integration. Previous methods have been less effective and/or limited to either one of the two analyses29,30,31,32,33,34. To demonstrate the performance of TaME-seq, we employed HPV16, 18, 31, 33 and 45 positive clinical samples, HPV positive cell lines and HPV plasmids. With 47% of the total of 154.8 million raw reads mapped on the target HPV reference genomes, TaME-seq proved to be highly efficient in HPV target enrichment. Other approaches for HPV target enrichment have reported much lower HPV mapping ratios32,40, requiring more sequencing and therefore at a higher sequencing cost. TaME-seq currently covers HPV16, 18, 31, 33 and 45, being the most common HPV genotypes in cervical cancer5. TaME-seq can be extended to cover additional HPV types, as well as other viruses, by implementing new primers to the method.
The ability of TaME-seq to detect chromosomal integration sites has been shown for the HPV positive cervical cancer cell lines CaSki, SiHa, HeLa and MS751. CaSki cells contain a high copy number (~600 copies/cell) of integrated full-length HPV16 arranged in concatemers41,42. SiHa (1–2 HPV16 copies/cell)39,41 and HeLa (10–50 HPV18 copies/cell)43 cells harbour integrated HPV genomes. MS751 cells contains integrated HPV4535, but in contrast to the product specification sheet (ATCC, Manassas, VA) no HPV18, which was verified in our analyses. For CaSki, 16 previously reported integration sites30,32,37 were detected by our method. In addition, three novel integration sites were identified. Known integration sites in SiHa31,37,39, HeLa31,36 and MS75135, as well as large deletions demonstrated in HeLa36 and MS75135, were confirmed by the TaME-seq method. Of the 21 LBC samples, HPV integration sites could only be detected in one sample, being in line with previous studies reporting no or few HPV integration events in LSIL/ASC-US samples44,45. However, other studies report integration events also in LSIL samples32,46. The detection of integrated forms of the virus is also dependent on the amount of episomes in the sample; low copy integration sites may remain undetected against a high background of episomal HPV.
The high sequencing coverage throughout the HPV genome enables detection of low frequency variants. Variant calling was evaluated using SiHa replicates to set the variant calling threshold. Previous studies have used variant calling thresholds of 0.5% or 1%17,34. With the high coverage provided by the TaME-seq method there is potential for detecting very low frequency variation. We have therefore analysed the variation using 0.2% as the variant calling threshold. Multiple and stringent filtering steps was included to filter out non-reliable variants, as we are approaching the inherent error rate profile of the PCR amplification and Illumina sequencing47. However, the threshold for variant calling is dependent on experimental and analytical basis and must be set according to the study aims.
The results from the SiHa analysis indicate that calling ultra-low frequency variants is dependent on the sequencing coverage. Lower sequencing coverage results in the detection of fewer variants and less concordance between sample replicates. In order to find ultra-low frequency variants, high sequencing coverage is required. Figure 4 shows that at the mean coverage of 12000×, the number of variants in SiHa-1 is approaching saturation. This indicates that more variants are not likely to be found even with higher sequencing coverage. Finally, differences in sequencing coverage affect the number of variable sites found, but also experimental approaches due to stochastic sampling and variant calling can fail to reveal low frequency variants. Overall, our results uncover low frequency variants in the samples, potentially introduced by DNA repair mechanisms and APOBEC enzyme mediated DNA editing48,49,50, although some bias may be introduced by PCR and sequencing. Variable sites are present in all genes of the studied HPV types. Traditionally, studies have focused on sequence variation on a viral sublineage level13,14,15,16 or the high variability has been interpreted as HPV variant co-infections29. The development of NGS technologies has provided comprehensive tools for the study of HPV genomic variability. Recent studies have reported high HPV variability that may be evidence of intra-host viral evolution and adaptation generated during a chronic HPV infection17,18,19,20.
Our study has some limitations. Firstly, TaME-seq is not intended for determining HPV genotypes and we recommend it for analyses of HPV variability and integration events in samples with known HPV status. Secondly, due to variation in amplification efficacy, an uneven coverage is seen for different genomic regions. Sudden drops in the coverage, that are not genomic deletions, may be due to suboptimal primer performance or poor alignment against the reference genomes. This issue can be solved partly by designing new primers covering these regions and optimising the primer performance. Also, the read alignment step can be further optimised. Alternatively, alignment could be performed by de novo assembly to create consensus sequences for the alignment. Thirdly, enough viral DNA and good dsDNA quality are important for achieving consistent tagmentation results in the Nextera protocol51. Sample preparation of the excluded LBC samples failed likely due to very low viral load in the samples, which was not quantified separately.
In summary, we have developed a NGS approach that allows the simultaneous study of HPV genomic variability and chromosomal integration. TaME-seq is applicable to large sample cohorts due to its highly efficient target enrichment, leading to less off-target sequences and therefore reduced sequencing cost. Comprehensive studies on HPV intra-host variability generated during a persistent infection will improve our understanding of viral carcinogenesis. Efficient identification of HPV genomic variability and integration sites will be important both for the study of HPV evolution, adaptability and may be a useful tool for cervical cancer diagnostics.
Methods
Samples
Anonymised LBC samples from routine cervical cancer screening were included in the study, comprising cases of atypical squamous cells of undetermined significance (ASC-US) and low-grade squamous intraepithelial lesions (LSIL). HPV positive samples with the cobas 4800 HPV test (Roche Molecular Diagnostics, Pleasanton, CA) were extracted for DNA using the automated system NucliSENS easyMAG (BioMerieux Inc., France) with off-board lysis. The samples were HPV genotyped using the modified GP5+/6+ PCR protocol (MGP)52, followed by HPV type-specific hybridisation using Luminex suspension array technology53 or the Anyplex™ II HPV28 assay (Seegene, Inc., Seoul, Korea). LBC samples (n = 31) were positive for HPV16, 18, 31, 33 or 45 alone, or had multiple infections including at least one of the five types. DNA extracted from the HPV positive cervical carcinoma cell lines CaSki, SiHa, HeLa and MS751 (ATCC, Manassas, VA) served as positive controls. WHO international standards for HPV 16 (1st WHO International Standard for Human Papillomavirus Type 16 DNA, NIBSC code: 06/202) and 18 (1st WHO International Standard for Human Papillomavirus Type 18 DNA, NIBSC code: 06/206)(NIBSC, Potters Bar, Hertfordshire, UK) and a plasmid containing the strain HPV3354 were used as additional positive controls. Laboratory-grade water and DNA from an HPV negative human sample were included as negative controls. DNA was quantified by the fluorescence-based Qubit dsDNA HS assay (Thermo Fisher Scientific Inc.,Waltham, MA, USA).
Primer design
HPV16, 18, 31, 33, and 45 whole genome reference and variant sequences were obtained from the PapillomaVirus Episteme (PaVE) database55. All the available reference and variant sequences within an HPV type were aligned using the multiple sequence alignment tool ClustalO56. The sequence alignment was converted to a consensus sequence for each HPV type in CLC Sequence viewer version 7.7.1 (QIAGEN Aarhus A/S). TaME-seq HPV primers were designed using Primer357 and HPV consensus sequences as the source sequence. Finally, primers were modified by adding an Illumina TruSeq-compatible adapter tail (5′-AGACGTGTGCTCTTCCGATCT-3′) to the 5′-end and then synthesised by Thermo Fisher Scientific, Inc. (Waltham, MA).
Library preparation and sequencing
Primer pools for each HPV type were prepared by combining primers separately in equal volumes. Samples were subjected to tagmentation using Nextera DNA library prep kit (Illumina, Inc., San Diego, CA). Tagmented DNA was purified using DNA Clean & Concentrator™-5 columns (Zymo Research, Irvine, CA) according the manufacturer’s instructions or ZR-96 DNA Clean & Concentrator™-5 plates (Zymo Research, Irvine, CA) according to the Nextera® DNA Library Prep Reference Guide (15027987 v01) before PCR amplification for target enrichment. Amplification was performed using Qiagen Multiplex PCR Master mix (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For each sample, two PCR reactions were performed separately with 0.75 µM of HPV primer pools, 0.5 µM of i7 index primers (adapted from Kozich et al.58) and 1 µl of i5 index primers from the Nextera index kit (Illumina, Inc., San Diego, CA). The cycling conditions were as follows: initial denaturation and hot start at 95 °C for 5 minutes; 30 cycles at 95 °C for 30 seconds, at 58 °C for 90 seconds and at 72 °C for 20 seconds; final extension at 68 °C for 10 minutes. Following amplification, libraries were pooled in equal volumes and the final sample pool was purified with Agencourt® AMPure® XP beads (Beckman Coulter, Brea, CA). The quality and quantity of the pooled libraries were assessed on Agilent 2100 Bioanalyzer using Agilent High Sensitivity DNA Kit (Agilent Technologies Inc., Santa Clara, CA) and by qPCR using KAPA DNA library quantification kit (Kapa Biosystems, Wilmington, MA). Sequencing was performed on the MiSeq platform (Illumina, Inc., San Diego, CA) or on the HiSeq 2500 platform (Illumina, Inc., San Diego, CA). Samples were sequenced as 151 bp paired-end reads and two 8 bp index reads.
Sequence alignment
Raw paired-end reads were trimmed for adapters, HPV primers, quality (-q 20) and finally for minimum length (-m 50) using cutadapt (v1.10)59. Trimmed reads were mapped to human (GRCh38/hg38) and HPV16, 18, 31, 33 and 45 reference genomes obtained from the PaVE database55 using HISAT2 (v2.1.0)60. Mapping statistics and sequencing coverage were calculated using the Pysam package61 with an in-house Python (v3.5.4) script. Downstream analysis was performed using an in-house R (v3.4.4) script. Results from both reactions of the same sample were combined and method performance was then evaluated based on the percentage of obtained reads mapped to the HPV reference genome, mean sequencing coverage and percentage of HPV reference genome coverage for each sample. Further analysis was performed when a sample had >20000 reads mapped to the target HPV reference genome. The target HPV genomes correspond to the HPV types for which the samples were reported positive by HPV genotyping.
Detecting HPV-human integration sites
The paired-end reads that mapped (HISAT2) with one end to a human chromosome and the other end to the target HPV reference genome were identified as discordant read pairs. If a specific position had ≥2 read pairs with unique start or end coordinates, it was considered as a potential integration site. To determine the exact position of HPV-human integration breakpoints, previously unmapped reads were remapped to human and HPV reference genomes (as above) using the LAST (v876) aligner (options -M -C2)62. Positions covered by ≥3 junction reads, with unique start or end coordinates, were considered as potential integration breakpoints. Integration site detection was not based on reads sharing the same start and end coordinates as these reads were considered as potential PCR duplicates. Selected HPV integration breakpoints were confirmed by PCR amplification and Sanger sequencing.
Sequence variation analysis
Mapped nucleotide counts over HPV reference genomes and average mapping quality values of each nucleotide were retrieved from BAM files and variant calling was performed using an in-house R script. To reduce the effects of PCR amplification and sequencing artefacts in the variation analysis, filtering was applied before the variant calling. Nucleotides seen ≤2 times in each position and nucleotides with mean Phred quality score of <20 were filtered out. Nucleotide counts from both reactions of the same sample were combined and variant allele frequencies (VAF) of the three minor alleles in each position were calculated. If results from either of the reaction showed >5 times larger VAF with <20% of the total coverage, it was discarded from variant calling. Finally, variants were called if VAF was >0.2% and coverage was ≥100×.
Two sequencing libraries of SiHa cell line served as technical replicates to assess the variant calling performance. The technical replicates were sequenced on the MiSeq platform or on the HiSeq 2500 platform. In addition, HiSeq raw sequencing data was downsampled randomly and defined portions (90%, 75%, 50% and 25%) of the original reads were further analysed. Reproducibility of calling variants in the replicates was assessed by calculating concordance rate. The concordance rate (Rc) between duplicates was defined as follows:
where Nc was the number of concordant variants between a pair of replicate samples, and N1 and N2 were the total number of variants detected in each of the duplicated sample.
Ethical approval
This study was approved by the regional committee for medical and health research ethics, Oslo, Norway [2017/447] and we confirm that all experiments were performed in accordance with the committee’s guidelines and regulations.
Data Availability
Sequence data from cell lines will be available at European Nucleotide Archive (ENA) accession number ERP111061. Plasmids are third party property and requests must be made to International Human Papillomavirus Reference Center and Institut Pasteur. Sequencing data from clinical samples will be available from the authors upon request with obtained ethical approval. Clinical sequence data may be deposited at the European Genome-phenome Archive (EGA) (ethical and legal assessments are on-going).
References
Walboomers, J. M. et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189, 12–19, 10.1002/(sici)1096-9896(199909)189:1<12::aid-path431>3.0.co;2-f (1999).
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–386, https://doi.org/10.1002/ijc.29210 (2015).
Fitzmaurice, C. et al. The Global Burden of Cancer 2013. JAMA Oncol 1, 505–527, https://doi.org/10.1001/jamaoncol.2015.0735 (2015).
Bosch, F. X., Lorincz, A., Munoz, N., Meijer, C. J. & Shah, K. V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 55, 244–265 (2002).
de Sanjose, S. et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. The Lancet Oncology 11, 1048–1056, https://doi.org/10.1016/s1470-2045(10)70230-8 (2010).
Crosbie, E. J., Einstein, M. H., Franceschi, S. & Kitchener, H. C. Human papillomavirus and cervical cancer. The Lancet 382, 889–899, https://doi.org/10.1016/s0140-6736(13)60022-7 (2013).
Forman, D. et al. Global burden of human papillomavirus and related diseases. Vaccine 30(Suppl 5), F12–23, https://doi.org/10.1016/j.vaccine.2012.07.055 (2012).
Moscicki, A. B. et al. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 30(Suppl 5), F24–33, https://doi.org/10.1016/j.vaccine.2012.05.089 (2012).
Bernard, H. U. Taxonomy and phylogeny of papillomaviruses: an overview and recent developments. Infect. Genet. Evol. 18, 357–361, https://doi.org/10.1016/j.meegid.2013.03.011 (2013).
Bzhalava, D., Eklund, C. & Dillner, J. International standardization and classification of human papillomavirus types. Virology 476, 341–344, https://doi.org/10.1016/j.virol.2014.12.028 (2015).
Bernard, H. U. et al. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401, 70–79, https://doi.org/10.1016/j.virol.2010.02.002 (2010).
Burk, R. D., Harari, A. & Chen, Z. Human papillomavirus genome variants. Virology 445, 232–243, https://doi.org/10.1016/j.virol.2013.07.018 (2013).
Cornet, I. et al. HPV16 genetic variation and the development of cervical cancer worldwide. Br. J. Cancer 108, 240–244, https://doi.org/10.1038/bjc.2012.508 (2013).
Mirabello, L. et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J. Natl. Cancer Inst. 108, https://doi.org/10.1093/jnci/djw100 (2016).
Chan, P. K. et al. Geographical distribution and oncogenic risk association of human papillomavirus type 58 E6 and E7 sequence variations. Int. J. Cancer 132, 2528–2536, https://doi.org/10.1002/ijc.27932 (2013).
Chen, A. A., Gheit, T., Franceschi, S., Tommasino, M. & Clifford, G. M. Human Papillomavirus 18 Genetic Variation and Cervical Cancer Risk Worldwide. J. Virol. 89, 10680–10687, https://doi.org/10.1128/jvi.01747-15 (2015).
de Oliveira, C. M. et al. High-level of viral genomic diversity in cervical cancers: A Brazilian study on human papillomavirus type 16. Infect. Genet. Evol. 34, 44–51, https://doi.org/10.1016/j.meegid.2015.07.002 (2015).
Mirabello, L. et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 170, 1164–1174 e1166, https://doi.org/10.1016/j.cell.2017.08.001 (2017).
Hirose, Y. et al. Within-Host Variations of Human Papillomavirus Reveal APOBEC-Signature Mutagenesis in the Viral Genome. J. Virol. https://doi.org/10.1128/jvi.00017-18 (2018).
Dube Mandishora, R. S. et al. Intra-host sequence variability in human papillomavirus. Papillomavirus Res, https://doi.org/10.1016/j.pvr.2018.04.006 (2018).
Zur Hausen, H. Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer 2, 342–350, https://doi.org/10.1038/nrc798 (2002).
Pett, M. & Coleman, N. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? J. Pathol. 212, 356–367, https://doi.org/10.1002/path.2192 (2007).
McBride, A. A. & Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 13, e1006211, https://doi.org/10.1371/journal.ppat.1006211 (2017).
Jeon, S., Allen-Hoffmann, B. L. & Lambert, P. F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 69, 2989–2997 (1995).
Doorbar, J., Egawa, N., Griffin, H., Kranjec, C. & Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 25(Suppl 1), 2–23, https://doi.org/10.1002/rmv.1822 (2015).
Ziegert, C. et al. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 22, 3977–3984, https://doi.org/10.1038/sj.onc.1206629 (2003).
Peter, M. et al. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J. Pathol. 221, 320–330, https://doi.org/10.1002/path.2713 (2010).
Kraus, I. et al. The Majority of Viral-Cellular Fusion Transcripts in Cervical Carcinomas Cotranscribe Cellular Sequences of Known or Predicted Genes. Cancer Res. 68, 2514–2522, https://doi.org/10.1158/0008-5472.Can-07-2776 (2008).
Cullen, M. et al. Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res 1, 3–11, https://doi.org/10.1016/j.pvr.2015.05.004 (2015).
Xu, B. et al. Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS One 8, e66693, https://doi.org/10.1371/journal.pone.0066693 (2013).
Liu, Y., Lu, Z., Xu, R. & Ke, Y. Comprehensive mapping of the human papillomavirus (HPV) DNA integration sites in cervical carcinomas by HPV capture technology. Oncotarget 7, 5852–5864, https://doi.org/10.18632/oncotarget.6809 (2016).
Hu, Z. et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 47, 158–163, https://doi.org/10.1038/ng.3178 (2015).
Holmes, A. et al. Mechanistic signatures of HPV insertions in cervical carcinomas. npj Genomic Medicine 1, https://doi.org/10.1038/npjgenmed.2016.4 (2016).
Kukimoto, I. et al. Genetic variation of human papillomavirus type 16 in individual clinical specimens revealed by deep sequencing. PLoS One 8, e80583, https://doi.org/10.1371/journal.pone.0080583 (2013).
Geisbill, J., Osmers, U. & Durst, M. Detection and characterization of human papillomavirus type 45 DNA in the cervical carcinoma cell line MS751. J. Gen. Virol. 78(Pt 3), 655–658, https://doi.org/10.1099/0022-1317-78-3-655 (1997).
Adey, A. et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500, 207–211, https://doi.org/10.1038/nature12064 (2013).
Akagi, K. et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 24, 185–199, https://doi.org/10.1101/gr.164806.113 (2014).
Mincheva, A., Gissmann, L. & zur Hausen, H. Chromosomal integration sites of human papillomavirus DNA in three cervical cancer cell lines mapped by in situ hybridization. Med. Microbiol. Immunol. 176, 245–256 (1987).
el Awady, M. K., Kaplan, J. B., O’Brien, S. J. & Burk, R. D. Molecular analysis of integrated human papillomavirus 16 sequences in the cervical cancer cell line SiHa. Virology 159, 389–398 (1987).
Li, T. et al. Universal Human Papillomavirus Typing Assay: Whole-Genome Sequencing following Target Enrichment. J. Clin. Microbiol. 55, 811–823, https://doi.org/10.1128/JCM.02132-16 (2017).
Baker, C. C. et al. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 61, 962–971 (1987).
Yee, C., Krishnan-Hewlett, I., Baker, C. C., Schlegel, R. & Howley, P. M. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 119, 361–366 (1985).
Meissner, J. D. Nucleotide sequences and further characterization of human papillomavirus DNA present in the CaSki, SiHa and HeLa cervical carcinoma cell lines. J. Gen. Virol. 80(Pt 7), 1725–1733, https://doi.org/10.1099/0022-1317-80-7-1725 (1999).
Hudelist, G. et al. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: different levels of viral integration are correlated with lesion grade. Gynecol. Oncol. 92, 873–880, https://doi.org/10.1016/j.ygyno.2003.11.035 (2004).
Liu, Y. et al. Genome-wide profiling of the human papillomavirus DNA integration in cervical intraepithelial neoplasia and normal cervical epithelium by HPV capture technology. Sci. Rep. 6, 35427, https://doi.org/10.1038/srep35427 (2016).
Li, H. et al. Preferential sites for the integration and disruption of human papillomavirus 16 in cervical lesions. J. Clin. Virol. 56, 342–347, https://doi.org/10.1016/j.jcv.2012.12.014 (2013).
Schirmer, M., D’Amore, R., Ijaz, U. Z., Hall, N. & Quince, C. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics 17, 125, https://doi.org/10.1186/s12859-016-0976-y (2016).
Warren, C. J. et al. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 89, 688–702, https://doi.org/10.1128/JVI.02383-14 (2015).
Kukimoto, I. et al. Hypermutation in the E2 gene of human papillomavirus type 16 in cervical intraepithelial neoplasia. J. Med. Virol. 87, 1754–1760, https://doi.org/10.1002/jmv.24215 (2015).
Chen, J. & Furano, A. V. Breaking bad: The mutagenic effect of DNA repair. DNA Repair (Amst) 32, 43–51, https://doi.org/10.1016/j.dnarep.2015.04.012 (2015).
Lamble, S. et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13, 104, https://doi.org/10.1186/1472-6750-13-104 (2013).
Soderlund-Strand, A., Carlson, J. & Dillner, J. Modified general primer PCR system for sensitive detection of multiple types of oncogenic human papillomavirus. J. Clin. Microbiol. 47, 541–546, https://doi.org/10.1128/JCM.02007-08 (2009).
Schmitt, M. et al. Bead-based multiplex genotyping of human papillomaviruses. J. Clin. Microbiol. 44, 504–512, https://doi.org/10.1128/JCM.44.2.504-512.2006 (2006).
Beaudenon, S. et al. A novel type of human papillomavirus associated with genital neoplasias. Nature 321, 246–249, https://doi.org/10.1038/321246a0 (1986).
Van Doorslaer, K. et al. The Papillomavirus Episteme: a central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 41, D571–578, https://doi.org/10.1093/nar/gks984 (2013).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539, https://doi.org/10.1038/msb.2011.75 (2011).
Untergasser, A. et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 40, e115, https://doi.org/10.1093/nar/gks596 (2012).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120, https://doi.org/10.1128/AEM.01043-13 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, https://doi.org/10.14806/ej.17.1.200 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Kielbasa, S. M., Wan, R., Sato, K., Horton, P. & Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493, https://doi.org/10.1101/gr.113985.110 (2011).
Acknowledgements
We thank Mona Hansen and Hanne Kristiansen-Haugland for DNA sample extraction and HPV genotyping, and Tobias Neidel for primer design for HPV31, 33 and 45. This work was funded by a grant from South-Eastern Norway Regional Health Authority (project number 2016020).
Author information
Authors and Affiliations
Contributions
S.L. designed primers, performed the experiments, analysed the results and drafted the manuscript text. S.U.U. contributed to the data analysis. M.L. and P.E. performed the pilot experiments and P.E. designed the initial TaME-seq assay concept. R.M. contributed to the primer design process and designed primers. I.K.C. and O.H.A. contributed to study design and result interpretation. T.B.R. contributed to the study design, data analysis and result interpretation. All authors contributed to writing, reading and approving the manuscript.
Corresponding author
Ethics declarations
Competing Interests
S.L., M.L., P.E., R.M., I.K.C., O.H.A. and T.B.R. and their corresponding institutions have filed a patent application at the technology transfer company Inven2, Oslo, Norway on the protocol described here.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lagström, S., Umu, S.U., Lepistö, M. et al. TaME-seq: An efficient sequencing approach for characterisation of HPV genomic variability and chromosomal integration. Sci Rep 9, 524 (2019). https://doi.org/10.1038/s41598-018-36669-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36669-6
- Springer Nature Limited
This article is cited by
-
TaME-seq2: tagmentation-assisted multiplex PCR enrichment sequencing for viral genomic profiling
Virology Journal (2023)
-
Human papillomavirus integration transforms chromatin to drive oncogenesis
Genome Biology (2023)
-
A novel tailed primer nucleic acid test for detection of HPV 16, 18 and 45 DNA at the point of care
Scientific Reports (2023)
-
HPV genotyping by L1 amplicon sequencing of archived invasive cervical cancer samples: a pilot study
Infectious Agents and Cancer (2022)
-
PVAmpliconFinder: a workflow for the identification of human papillomaviruses from high-throughput amplicon sequencing
BMC Bioinformatics (2020)