
Abstract
Lithium rich layered oxide xLi2MnO3∙(1−x)LiMO2 (M = Mn, Co, Ni, etc.) materials are promising cathode materials for next generation lithium ion batteries. However, the understanding of their electrochemical kinetic behaviors is limited. In this work, the phase separation behaviors and electrochemical kinetics of 0.5Li2MnO3∙0.5LiCoO2 materials with various Li2MnO3 domain sizes were studied. Despite having similar morphological, crystal and local atomic structures, materials with various Li2MnO3 domain sizes exhibited different phase separation behavior resulting in disparate lithium ion transport kinetics. For the first few cycles, the 0.5Li2MnO3∙0.5LiCoO2 material with a small Li2MnO3 domain size had higher lithium ion diffusion coefficients due to shorter diffusion path lengths. However, after extended cycles, the 0.5Li2MnO3∙0.5LiCoO2 material with larger Li2MnO3 domain size showed higher lithium ion diffusion coefficients, since the larger Li2MnO3 domain size could retard structural transitions. This leads to fewer structural rearrangements, reduced structural disorders and defects, which allows better lithium ion mobility in the material.
Similar content being viewed by others

Introduction
Much research has been done to develop alternative cathode materials for lithium ion batteries. Commercial LiCoO2 materials have a limited practical capacity of around 140 mAh.g−1, poor thermal stability, are expensive and relatively toxic. Moreover, LiCoO2 is structurally unstable, resulting in a large capacity decay as the cycle numbers increase1,2,3. Lithium rich layered oxide, xLi2MnO3∙(1−x)LiMO2 (M = Mn, Co, Ni, among others) cathode materials have attracted much attention as potential next generation cathode materials for lithium ion batteries. This is due to their high specific capacity, ≥250 mAh.g−1, with an operating voltage range of 2.0–4.8 V, leading to a high energy density that is ≥900 Wh.kg−1 2,4,5,6. This cathode material family consists of two compounds, Li2MnO3 and LiMO2. It is generally accepted that Li2MnO3 acts as a stabilizer enabling LiMO2 to maintain its overall structural stability when used as a cathode material2,4,6,7. The large capacity in the first charge process was ≥4.4 V due to activation of Li2MnO3. The charge compensation mechanisms occurring in this Li extraction process in the high voltage region may involve irreversible oxygen release from the lattice4,8,9, reversible oxygen redox (O−2/O2n−, n = 1, 2 or 3)10,11,12, or a combination of these two mechanisms13,14. Thackeray et al.4 reported that this material class is a composite material. These cathode materials often present domains of Li2MnO3 and LiMO2 components with a high degree of structural integration in the nanoscale regime. This can be observed using high-resolution transmission electron microscopy (HRTEM). These cathode materials often reveal a phase transition from a layered Li2MnO3 component to a spinel-like structure during cycling2,4,6,15,16,17. This phase transition is believed to be a cause of the large capacity decay and dramatic voltage drop leading to a high energy density loss as cycle numbers increase. Moreover, the low electrical conductivity of the Li2MnO3 component leads to poor rate capability18. This indicates that the electrochemical performance of lithium rich layered oxide cathode materials is largely determined by the Li2MnO3 component. Furthermore, structural properties of these cathode materials, including cation ordering and phase separation, are significantly dependent on the synthesis methods used19,20,21,22.
Generally, electrochemical performance of lithium ion batteries is directly related to lithium ion transport between their cathode and anode materials. Lithium ion transport affects reversible capacity, cycling stability, and rate capability of the electrode materials. The electrochemical kinetics of lithium rich layered oxide cathode materials have been studied by a few research groups using an electrochemical impedance spectroscopy (EIS) and a galvanostatic intermittent titration technique (GITT)23,24,25,26,27. The GITT measurement was performed to examine the lithium ion diffusion coefficients of lithium rich layered oxide composite cathode materials, including 0.5Li2MnO3·0.5LiNi0.5Mn0.5O223 and 0.5Li2MnO3·0.5LiMn0.42Ni0.42Co0.16O224. The results revealed that the calculated lithium ion diffusion coefficients of these cathode materials were very small, ranging from 10−14 cm2.s−1 to 10−18 cm2.s−1 and were primarily dependent upon Li2MnO3 activation24,27,28. The values are much lower than those of layered cathode materials such as LiCoO2 (10−7∼10−11 cm2.s−1)29 and LiMn1/3Ni1/3Co1/3O2 (10−9∼10−10 cm2.s−1)30.
The 0.5Li2MnO3∙0.5LiCoO2 material is one type of lithium rich layered oxide cathode that has been widely studied and is considered a potential candidate for next generation high energy density cathode materials for lithium ion batteries31,32,33,34,35. However, lithium ion transport behavior of this cathode material has not been investigated. Therefore, it is essential to study the parameters that affect the electrochemical kinetics of 0.5Li2MnO3∙0.5LiCoO2. So, herein, we report these electrochemical kinetics in terms of lithium ion diffusion in 0.5Li2MnO3∙0.5LiCoO2 with various Li2MnO3 domain sizes during cycling. The galvanostatic intermittent titration technique (GITT) was used for this purpose. The Li2MnO3 domain size has a large impact on the degree of Li2MnO3 activation as well as the Li-ion transport behavior of composite-based cathode materials. This work provides a better understanding of the important parameters that influence electrochemical kinetic behaviors of lithium rich layered oxide materials for next generation cathodes for lithium ion batteries.
Results and Discussion
Morphology characterization
High resolution transmission electron microscopy (HRTEM) was used to verify that the 0.5Li2MnO3∙0.5LiCoO2 materials in this study had significantly different Li2MnO3 domain sizes. Transmission electron microscopy (TEM) was used to show that both samples have similar average particle sizes of approximately 230 nm with similar particle size distributions. Their particle size distributions were quite broad, ranging from 100 nm up to 400 nm, as demonstrated in Fig. 1(a,c). The Li2MnO3 domain with a space group C2/m and the LiCoO2 domain with a space group \(R\bar{{\rm{3}}}m\) were clearly observed in individual particles of both materials as demonstrated in Fig. 1(b,d). The presence of Li2MnO3 and LiCoO2 domains can confirm that both materials formed a composite system, consistent with previous reports36,37,38. Since the Li2MnO3 domains appear to be irregular in shape, an image processing technique employing ImageJ software, was used to determine the 2D area of individual domains. The detailed processing procedures are briefly presented as follows. We first selected images showing clear domains. We then selected a domain of interest and carefully traced it. Then, the area inside this trace was determined using the measuring function of the ImageJ software. The process was repeated 5 times/domain of interested. The 2D areas obtained from several 200 nm particle domains were then averaged. Standard deviations of the 2D area representing LMO domain were determined. The standard deviations were found to be 18% and 12% for L-LMO and S-LCO samples, respectively. Figure 1(b) shows that the L-LMO material consists of larger average Li2MnO3 domain sizes of around 2309 ± 413 nm2, while Fig. 1(d) reveals that the S-LMO material consists of smaller average Li2MnO3 domain sizes of about 266 ± 31 nm2.

TEM and HRTEM images of the 0.5Li2MnO3∙0.5LiCoO2 materials with larger Li2MnO3 domain sizes (L-LMO, a,b) and the 0.5Li2MnO3∙0.5LiCoO2 material with smaller Li2MnO3 domain sizes (S-LMO, c,d).
The different Li2MnO3 domain sizes of the 0.5Li2MnO3·0.5LiCoO2 composite cathodes may be an essential parameter that significantly affects lithium ion diffusion behaviors of these cathode materials. A good understanding of the relationship between these structural characteristics and electrochemical kinetics is key to developing this cathode material class for practical applications. The impact of the Li2MnO3 domain size on the electrochemical kinetics of the 0.5Li2MnO3·0.5LiCoO2 material class is the central focus of this study.
Crystal structure characterization
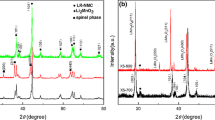
X-ray diffraction (XRD) was performed to analyze the crystal structure of the S-LMO and L-LMO materials and is illustrated in Fig. 2. The diffraction peaks obtained from the materials could be indexed to both Li2MnO3 (monoclinic, space group C2/m) and LiCoO2 (rhombohedral, space group \(R\bar{{\rm{3}}}m\)) phases, possessing a typically layered α-NaFeO2 structure. Weak diffraction peaks in the 2θ range of 20°-25° correspond to the Li and Mn ion ordering in the transition metal layers of the Li2MnO3 component39,40,41,42,43 could be observed in both materials. Rietveld refinements were performed using two structural models (those of Li2MnO3 and LiCoO2) as illustrated in Fig. S1. The results showed that these materials had quite similar lattice constants, as demonstrated in Table S1. This suggests that the crystal structures of the Li2MnO3 and LiCoO2 phases in the two materials are very similar.

X-ray diffraction profiles of the 0.5Li2MnO3∙0.5LiCoO2 materials with a larger Li2MnO3 domain (L-LMO, bottom) and a smaller Li2MnO3 domain size (S-LMO, top).

XANES spectra and k2-weighted Fourier-transformed EXAFS signals at the Mn (a,b) and Co (c,d) K-edges of the 0.5Li2MnO3∙0.5LiCoO2 materials.
The XRD peaks of the S-LMO material presented well merged XRD patterns of the Li2MnO3 and LiCoO2 phases. This indicates a good degree of mixing between these components as well as a peak broadening effect resulting from the nano-sized crystalline domains. The XRD peaks of the L-LMO material revealed clear separation of the XRD peaks at 2θ values above 35°. This result indicates a larger degree of separation of the Li2MnO3 and LiCoO2 phases44 as well as crystals with larger domain sizes crystals than the S-LMO material. Generally, lithium rich layered oxide materials often form a composite structure in terms of a phase separation between their Li2MnO3 and LiCoO2 components4,45,46. This result indicates that the crystal structures of the Li2MnO3 and LiCoO2 phases in the two materials are very similar and that their phase separation behaviors are significantly different.
Local atomic structure characterization
X-ray absorption spectroscopy (XAS) was used to examine the local atomic structure of the 0.5Li2MnO3∙0.5LiCoO2 materials with various Li2MnO3 domain sizes. The XANES spectra at Mn and Co K-edges are presented in Fig. 3(a,c), respectively. There are three main features in the absorption spectra, including the pre-edge (feature A) and the small shoulder (feature B), and the absorption edge (feature C). The pre-edge (feature A) occurs due to the transition of electrons from a 1s state to an unoccupied 3d state. The weak shoulder (feature B) results from the transition of electrons from a 1s state to an unoccupied 4p state with a shakedown process, followed by a ligand to metal charge transfer. The main absorption edge (feature C) corresponds to the transition of electrons from a 1s state to an unoccupied 4p state without the shakedown process. The position of the main absorption edge directly relates to the oxidation states of the Mn and Co species. For Mn and Co K-edges, both materials presented main absorption edges in the same position, which can be confirmed from the first derivatives of their XAS spectra, as illustrated in Fig. S2, suggesting that the Mn and Co atoms in both materials possessed the same valance states. This indicated that the local environments of the Mn and Co atoms in both materials exhibited very small disordered octahedral sites47,48. Moreover, the XANES profiles correspond to those in previous works that used XAS to investigate the same cathode material type. They revealed that Mn and Co atoms had average valance states of 4+ and 3+, respectively22,45,49. As presented in Fig. 3(a,c), the normalized absorption spectra at the Mn and Co edges of both materials overlapped with few differences between them. The results suggest that Mn and Co atoms in the S-LMO and L-LMO materials are in similar environments with quite similar local structures.
Figure 3(b,d) illustrate the Fourier-transformed EXAFS signals at the Mn and Co K-edges, respectively. There are two main peaks. The first peak corresponds to the Mn and Co adsorbing atoms that occupy octahedral sites, surrounded by six oxygen atoms (Mn-O for the Mn K-edge and Co-O for the Co K-edge). The second peaks are due to the interactions between Mn and Co adsorbing atoms and transition metal (TM) atoms in the transition metal layers (Mn-TM for Mn K-edge and Co-TM for Co K-edge). These peaks appear at similar positions (RMn−O = 1.60 Å, RMn−TM = 2.49 Å, RCo−O = 1.49–1.51 Å, and RCo−TM = 2.36 Å). However, the amplitudes of the Mn-TM and Co-TM peaks are noticeably different. The Mn-TM peak intensity of the S-LMO material is higher than that of the L-LMO material. In contrast, the Co-TM peak intensity of the L-LMO material is higher than that of the S-LMO material. This indicates that in this coordination shell, the Mn and Co have different types and numbers of neighboring species. It is notable that there is an equal amount of the Li2MnO3 and LiCoO2 components in both the materials. However, in the S-LMO material, the Li2MnO3 domain size is small, so it is more distributed. In the L-LMO material, the Li2MnO3 domain size is large, so the individual Li2MnO3 and LiCoO2 domains are more locally isolated. For the Mn-TM peak, its lower peak intensity is an indication of weaker X-ray scattering. This phenomenon results because the Mn atoms in the S-LMO material with a smaller Li2MnO3 domain size are surrounded by a larger number of Co atoms resulting in better scattering than Mn alone. There are 3 Li atoms and 3 Mn atoms in this coordination shell, and Li has a very low X-ray scattering power. This leads to weaker Mn-Mn/Li peak in the EXAFS spectra for the material with large Li2MnO3 domains. In the case of the Co K-edge, the Co-TM peak of the S-LMO material was lower than that of the L-LMO material. This is because the Co atoms in the 0.5Li2MnO3∙0.5LiCoO2 material with smaller Li2MnO3 domain size interacted with a larger number of Mn atoms, producing less scattering than Co. The XAS experimental results revealed that the 0.5Li2MnO3∙0.5LiCoO2 materials have the similar overall local structures with subtle differences in EXAFS spectra due to the different Li2MnO3 and LiCoO2 domain sizes, indicating different phase separation behaviors.
Electrochemical characterizations
A galvanostatic cycling technique was performed to examine the effect of various Li2MnO3 domain sizes on electrochemical properties of the 0.5Li2MnO3∙0.5LiCoO2 materials. The results are given in Fig. 4. The first charging voltage profiles (black curves) of both materials revealed two distinct plateaus at around 3.9 V and 4.5 V, as shown in Fig. 4(a,b). The first voltage plateau at 3.9 V corresponds to the oxidation of Co3+ to Co4+ in the LiCoO2 component. The second voltage plateau at 4.5 V corresponds to extraction of lithium ions from the structure of the Li2MnO3 component. Various charge theories have been introduced to describe the charge compensation mechanisms at this higher voltage plateau, including irreversible oxygen loss from the lattice. This contributes to large irreversible capacity loss4,8,9, and reversible oxygen redox, which induces reversible capacity10,11,12. However, Chen and Islam50 postulated that lithium ion extraction from the Li2MnO3 structure involves oxidation of oxygen, but the resulting oxygen holes (O−) are not thermodynamically stable. This leads to formation of an oxygen dimer, resulting in oxygen release from the lattice. The charge compensation mechanisms of the cathode materials in this study, occurring at 4.5 V, were dominated by an irreversible oxygen release from the Li2MnO3 structure revealing a high initial irreversible capacity loss. The charge compensation involving the oxygen release from the lattice always produces Li2O and layered MnO2 phases. The formation of the Li2O phase induces an increase in the initial charge capacity and a large irreversible capacity in the first cycle4,36. Moreover, the sloping discharge curves from 3.8 to 2.0 V resulted from intercalation of lithium ions into the layered MnO2 component51. The electrochemical reactions of these composite-based cathode materials can be described as follows. While lithium ions are de-intercalated from the LiCoO2 component at voltages of 3.9 V to 4.5 V, depleted lithium layers are formed in the LiCoO2 structure. Lithium ions in octahedral sites on lithium and manganese layers of the Li2MnO3 component diffuse into the depleted lithium to compensate for lithium ions in the LiCoO2 structure4,52,53. This compensation allows the overall structure of the cathode materials to remain stable during cycling. Nevertheless, during the discharge processes, lithium ions intercalate into the MnO2 component to form a layered LiMnO2 phase until the Li2MnO3 component is completely consumed. It is well established that the layered LiMnO2 phase often transforms into a spinel-like phase upon cycling, causing a large capacity and voltage drop as the cycle numbers increase. Controlling the activation of the Li2MnO3 component is an essential key to improving the cycling stability of composite-based cathode materials. Thus, strategies to retard this phase transformation from a layered Li2MnO3 phase to spinel-liked phases were introduced in several previous studies. These strategies include using suitable testing conditions (appropriate cut off voltage and current density)3 and controlling the Li2MnO3 domain size54. Ghanty et al. revealed that a large Li2MnO3 domain size can reduce the spinel phase transformation because a Li2MnO3 component with a large domain size is quite difficult to activate and produce a layered LiMnO2 phase. This leads to less spinel phase formation upon continuous cycling54.

Voltage profiles (a,b) and differential capacity (c) of the 0.5Li2MnO3∙0.5LiCoO2 materials with larger Li2MnO3 domain (L-LMO) and smaller Li2MnO3 domain sizes (S-LMO).
Differential capacity plots obtained from differentiation of capacity as a function of its voltage profile for both materials at various cycle numbers at selected voltages ranging from 4.4 to 4.6 V are presented in Fig. 4(c). This was done to show more detail about the influence of the Li2MnO3 domain size on activation of the Li2MnO3 component during cycling. The oxidation peak between 4.5 V and 4.6 V corresponded to lithium and oxygen extraction from the Li2MnO3 component55. For the S-LMO material, the oxidation peak could not be clearly observed after the second cycle due to complete activation of the Li2MnO3 component after the second cycle resulting from its small domain size. In contrast, for the L-LMO material, an oxidation peak was clearly observed in subsequent cycles. In this material, the Li2MnO3 component was not completely activated in the first few cycles because the large Li2MnO3 domain size made lithium and oxygen extraction from the Li2MnO3 component was quite difficult. The presence of the oxidation peak suggested that Li2MnO3 activation still took place and the peak confirms the presence of a Li2MnO3 component during subsequent cycles. The remaining Li2MnO3 component after extended cycles stabilized the overall cathode structure during repeated cycling, resulting in higher cycling stability.
Lithium ion diffusion coefficients reflect the degree of lithium ion transport inside electrode materials. This is a crucial kinetic parameter for ion insertion/extraction in cathode materials. An understanding of lithium ion diffusion behaviors in electrode materials is very important to improve the electrochemical performance of lithium rich layered oxide cathode materials. A galvanostatic intermittent titration technique (GITT) was performed to study lithium ion diffusion behaviors in the 0.5Li2MnO3∙0.5LiCoO2 materials. It is well established that the lithium ion diffusion coefficient \(({D}_{{{Li}}^{+}})\) of lithium ions can be determined by Fick’s 2nd Law of Diffusion, which can be written as56,57:
where mB and MB denote the molecular weight and mass of the active material, respectively. VM is the molar volume of the prepared material, obtained from crystallographic data. S is the individual particle surface area of the electrode, and L is the thickness of the electrode. Given that the cell voltage profile (E) as a function of τ1/2 is quite linear (as shown in Fig. S3), Equation 1 can be further simplified24,58:
Indeed, the electrochemical reactions of lithium rich layered oxide cathode materials during cycling are very complex. They include lithium ion diffusion, oxygen loss, and a transition from a layered structure to a spinel form. Previously, Zhu and Wang59 used GITT to study the lithium ion diffusion coefficient in a LiFePO4 cathode material that exhibited a phase transformation upon cycling. Lithium ion diffusion coefficients of each phase (α and β phases) were separately determined to achieve a clear understanding of lithium ion diffusion behaviors in this material. However, the current study aims to investigate the overall lithium ion diffusion coefficients of 0.5Li2MnO3·0.5LiCoO2 cathode materials, which can be considered pseudo- or apparent diffusion coefficients25,60. Full details of this analysis for the lithium rich layered oxide cathode materials will be presented elsewhere (SK in preparation).
The voltage relaxation profiles using GITT and the calculated lithium ion diffusion coefficients during the charging and discharging processes of 0.5Li2MnO3·0.5LiCoO2 materials are shown in Fig. 5. During charging, two distinct diffusion coefficients can be observed. The first is believed to occur due to lithium ion extraction from the LiCoO2 structure and the second corresponds to lithium ion extraction from the Li2MnO3 structure accompanied by a structural transformation. During the initial charging process (voltages below 4.4 V), where lithium ions are extracted from the LiCoO2 component, the lithium ion diffusions coefficients are independent of the cell voltage showing constant values of around 10−14 cm2.s−1. At higher voltages (above 4.4 V), the lithium ion diffusion coefficients decreased rapidly reaching around 10−19 cm2.s−1 when the electrodes were charged to 4.8 V, corresponding to the lithium and oxygen extraction from the Li2MnO3 component. As can be seen, the lithium ion diffusion coefficients of the Li2MnO3 component are lower than those from the LiCoO2 component due to the low ionic conductivity of the Li2MnO3 component. Additionally, the structural transition from a layered Li2MnO3 component to a spinel form can induce large structural disorders and defects, which negatively impact lithium ion diffusivity. During discharge, the two distinct lithium ion diffusion coefficients were observed. They result from lithium ion insertion into the Li1−xCoO2 structure at higher voltages and activation of MnO2 (during the 1st and subsequent cycles) and the transformed spinel phases (during the 2nd and subsequent cycles) at lower voltages, respectively. The lithium ion diffusion coefficients resulting from the insertion of lithium ions into the Li1−xCoO2 structure showed higher values than those into the activated MnO2 and the spinel structures. This occurs due to the large structural disorders and defects formed during the spinel phase transition.

GITT profiles and calculated lithium ion diffusion coefficients during the 1st, 2nd and 20th cycles of the 0.5Li2MnO3∙0.5LiCoO2 materials with larger Li2MnO3 domain (L-LMO, a,b) and smaller Li2MnO3 domain sizes (S-LMO, c,d).
Figure 6 presents the calculated lithium ion diffusion coefficients as a function of cell voltage of the S-LMO and L-LMO materials. This figure compares the effect of the Li2MnO3 domain size on lithium ion diffusion coefficients of the 0.5Li2MnO3·0.5LiCoO2 materials. It is notable that the impact of the Li2MnO3 domain size on the electrochemical kinetics of the 0.5Li2MnO3∙0.5LiCoO2 materials can be clearly observed, especially in the voltage range of 4.4 to 4.8 V. The S-LMO material shows slightly higher lithium ion diffusion coefficients in this voltage range than the L-LMO material during the first two cycles. This result is not surprising because the smaller Li2MnO3 domain size provides shorter lithium ion diffusion paths, hence higher lithium ion diffusion coefficients. However, the difference is not dramatic because the average particle size of these two materials is quite similar, as can observed in Fig. 1. For extended cycles, the L-LMO material appears to have better lithium ion mobility, with lithium ion diffusion coefficients in this voltage range almost two orders of magnitude higher than the S-LMO material. These phenomena can be explained based on structural disorders and defects resulting during phase transformation of the Li2MnO3 component.

The effects of Li2MnO3 domain size on lithium ion diffusion coefficients as a function of cell voltage during charging (a) and discharging (b) of the 0.5Li2MnO3∙0.5LiCoO2 materials.
Figure 7 shows high resolution transmission electron microscopy (HRTEM) images of the 0.5Li2MnO3∙0.5LiCoO2 materials after cycling for 30 cycles. Three main crystalline phases can be indexed in both materials. They are the Li2MnO3, LiCoO2, and spinel-like (LiMn2O4) domains. Additionally, the HRTEM image of the S-LMO material shows a large area with no lattice fringes, indicating significant formation of disorder phase(s) after extended cycling. The S-LMO material also appears to have greater spinel-like and disorder regions than the L-LMO material, as illustrated in Fig. 7(b,d). These defects act as barriers for lithium ion transport in the electrode materials. For the S-LMO material, the phase transition after Li2MnO3 phase activation induced a larger structural rearrangement, causing abundant lattice disorders and defects24,27,28. Moreover, in the absence the Li2MnO3 component after extended cycles, the overall structure of the cathode could not be stabilized, leading to degradation. These phenomena causes low lithium ion diffusion coefficients and poor cycling stability. The cycling stability and rate capability of the 0.5Li2MnO3∙0.5LiCoO2 materials with various Li2MnO3 domain sizes are presented in Fig. S4. The L-LMO material with larger Li2MnO3 domains can retard the formation of these structural imperfections during cycling better than the S-LMO material. This is reflected in higher lithium ion diffusion coefficients after 20 cycles. The GITT measurements and HRTEM images reveal the relationship between the Li2MnO3 component activation and lithium ion coefficients of a composite-based cathode material upon cycling. The results show that the 0.5Li2MnO3∙0.5LiCoO2 material with a large Li2MnO3 domain size can potentially retard spinel phase evolution during cycling with improved structural stability and electrochemical properties.

TEM and HRTEM images of the 0.5Li2MnO3∙0.5LiCoO2 materials with a larger Li2MnO3 domain size (L-LMO, a,b) and the 0.5Li2MnO3∙0.5LiCoO2 material with a smaller Li2MnO3 domain size (S-LMO, c,d) after cycling for 30 cycles.
Conclusions
Composite-based layered 0.5Li2MnO3∙0.5LiCoO2 cathode materials with various Li2MnO3 domain sizes were characterized and showed quite similar morphological, crystal and local atomic structures. However, the materials exhibited different phase separation behaviors. Lithium ion diffusion coefficients depend significantly on activation of the Li2MnO3 component, resulting in various cycling stabilities and levels of rate performance. The 0.5Li2MnO3∙0.5LiCoO2 material with larger Li2MnO3 domains shows a higher cycling stability and rate capability than the material with smaller Li2MnO3 domains. The Li2MnO3 domain size affects phase transition behaviors of the Li2MnO3 component after activation. For the first few cycles, the 0.5Li2MnO3∙0.5LiCoO2 material with a small Li2MnO3 domain size revealed higher lithium ion diffusion coefficients due to shorter lithium ion diffusion path lengths. However, after an extended number of cycles, the 0.5Li2MnO3∙0.5LiCoO2 material with the large Li2MnO3 domain size provided higher lithium ion diffusion coefficients, since the larger Li2MnO3 domain size can retard structural transitions. This leads to fewer structural rearrangements, reduced structural disorders and defects, which allow better lithium ion mobility in the material. The current work shows that controlling the Li2MnO3 component activation through an appropriate Li2MnO3 domain size is an effective strategy for improving both cycling stability and rate capability of lithium rich layered oxide cathode materials.
Materials and Methods
Cathode materials preparation
A sol-gel method was used to prepare Li2MnO3, LiCoO2, and 0.5Li2MnO3·0.5LiCoO2 materials. Raw materials included CH3COOLi·2H2O (Aldrich), Mn(CH3COO)2·4H2O (Aldrich), Co(CH3COO)2·4H2O (Aldrich) and ascorbic acid (with the molar ratio of metal ions to ascorbic acid of 2:1). To prepare each material, required amounts of the precursors were dissolved in ethyl alcohol and mixed with an aqueous solution of ascorbic acid under continuous stirring at a constant temperature of 80 °C until a viscous gel was formed. After that, the obtained gel was first pre-heated at 300 °C for 3 h to eliminate organic constituents, re-heated for crystallization at 800 °C in air for 10 h, and followed by furnace cooling to room temperature. Fine crystalline powders of Li2MnO3, LiCoO2, and 0.5Li2MnO3·0.5LiCoO2 (S-LMO) materials were obtained.
Another 0.5Li2MnO3·0.5LiCoO2 material was synthesized by a ball-milling route. Stoichiometric amounts of the Li2MnO3 and LiCoO2 materials prepared in the previous step. The mixed powders and zirconia balls (weight ratio of ball to powder was 20:1) were placed in 90 mL of ethanol in a sealed Teflon bottle. Then, the mixed powders were ball milled using a rotational speed of 180 rpm for 72 h. The obtained mixture was evaporated overnight at 80 °C in a vacuum drying box, followed by final firing at 800 °C in air for 10 h, and followed by furnace cooling to room temperature. Fine crystalline powder of 0.5Li2MnO3·0.5LiCoO2 material with larger Li2MnO3 domain sizes (L-LMO) was obtained.
Structure and morphology characterization
X-ray diffraction (XRD) (Empyrean, PANalytical) was employed to study the crystal structure of the materials using Cu-Kα radiation operated at 45 kV and 40 mA. The data were recorded with a step size of 0.003° and a step time of 30 s/step over a 2θ range from 15° to 70° using Ni as a filter. X-ray absorption spectroscopy (XAS) (BL. 2.2, SLRI, Thailand) was performed to investigate the local atomic structure of the prepared materials. Morphology of the 0.5Li2MnO3·0.5LiCoO2 materials and the presence of Li2MnO3 and LiCoO2 domains were observed using transmission electron microscopy (TEM) (JEOL, JEM-2100 Plus).
Electrochemical measurements
To prepare the electrodes, 78 wt% of synthesized materials, 11 wt% of Super P carbon black (Alfa Aesar) as a conductive additive, and 11 wt% of polyvinylidene fluoride as a binder (PVDF, Arkema) dissolved in N-methyl-2-pyrollidone (NMP, Aldrich) solvent were mixed using a horizontal shaker for 2 h to form a slurry. The obtained slurry was coated on an Al foil by a doctor blade technique and dried overnight in vacuum at 80 °C. Swagelok type cells were fabricated in an Ar- filled glovebox using Li metal foil as an anode (Alfa Aesar), 1 M LiPF6 dissolved in ethyl carbonate (EC), dimethyl carbonates (DMC), and diethyl carbonate (DEC) (4:3:3 in volume) (MTI) as an electrolyte, and Celgard 2400 as a separator. Cycling stability of the prepared electrodes was studied using a galvanostatic charge/discharge test (BST8-MA, MTI) with a voltage window of 2.0–4.6 V and current rate of C/5 at 30 °C. A galvanostatic intermittent titration technique (GITT) (WBCS-300, WonATech) was performed to study lithium ion diffusion behaviors in the 0.5Li2MnO3·0.5LiCoO2 cathode materials, using a voltage window of 2.0–4.8 V.
References
Li, G., Zhou, S., Wang, P. & Zhao, J. Halogen-doping in LiCoO2 cathode materials for Li-ionBatteries: insights from ab initio calculations. RSC advances 5, 107326–107332, https://doi.org/10.1039/C5RA21258H (2015).
Thackeray, M. M., Johnson, C. S., Vaughey, J. T., Li, N. & Hackney, S. A. Advances in manganese-oxide ‘composite’ electrodes for lithium-ion batteries. Journal of Materials Chemistry 15, 2257–2267, https://doi.org/10.1039/B417616M (2005).
Yu, H. et al. High-energy ‘composite’ layered manganese-rich cathode materials via controlling Li2MnO3 phase activation for lithium-ion batteries. Physical Chemistry Chemical Physics 14, 6584–6595, https://doi.org/10.1039/C2CP40745K (2012).
Thackeray, M. M. et al. Li2MnO3-stabilized LiMO2 (M = Mn, Ni, Co) electrodes for lithium-ion batteries. Journal of Materials Chemistry 17, 3112–3125, https://doi.org/10.1039/B702425H (2007).
Yan, J., Liu, X. & Li, B. Recent progress in Li-rich layered oxides as cathode materials for Li-ion batteries. RSC Advances 4, 63268–63284, https://doi.org/10.1039/C4RA12454E (2014).
Ates, M. N. et al. Mitigation of Layered to Spinel Conversion of a Li-Rich Layered Metal Oxide Cathode Material for Li-Ion Batteries. Journal of The Electrochemical Society 161, A290–A301, https://doi.org/10.1149/2.040403jes (2014).
Nayak, P. K. et al. Al Doping for Mitigating the Capacity Fading and Voltage Decay of Layered Li and Mn-Rich Cathodes for Li-Ion Batteries. Advanced Energy Materials 6, 1502398–n/a, https://doi.org/10.1002/aenm.201502398 (2016).
Lu, Z., MacNeil, D. D. & Dahn, J. R. Layered Cathode Materials Li [ Ni x Li(1/3−2x/3)Mn(2/3−x/3)]O2 for Lithium-IonBatteries. Electrochemical and Solid-State Letters 4, A191–A194, https://doi.org/10.1149/1.1407994 (2001).
Armstrong, A. R. et al. Demonstrating Oxygen Loss and Associated Structural Reorganization in the Lithium Battery Cathode Li[Ni0.2Li0.2Mn0.6]O2. Journal of the American Chemical Society 128, 8694–8698, https://doi.org/10.1021/ja062027 + (2006).
Sathiya, M. et al. Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nature Materials 12, 827, https://doi.org/10.1038/nmat3699 (2013).
Xu, J. et al. Elucidating anionic oxygen activity in lithium-rich layered oxides. Nature Communications 9, 947, https://doi.org/10.1038/s41467-018-03403-9 (2018).
Tang, M. et al. Operando EPR for Simultaneous Monitoring of Anionic and Cationic Redox Processes in Li-Rich Metal Oxide Cathodes. The Journal of Physical Chemistry Letters 8, 4009–4016, https://doi.org/10.1021/acs.jpclett.7b01425 (2017).
Koga, H. et al. Different oxygen redox participation for bulk and surface: A possible global explanation for the cycling mechanism of Li1.20Mn0.54Co0.13Ni0.13O2. Journal of Power Sources 236, 250–258, https://doi.org/10.1016/j.jpowsour.2013.02.075 (2013).
Genevois, C. et al. Insight into the atomic structure of cycled lithium-rich layered oxide Li1.20Mn0.54Co0.13Ni0.13O2 using HAADF STEM and electron Nanodiffraction. J. Phys. Chem. C 119, 75–83, https://doi.org/10.1021/jp509388j (2015).
Song, B. et al. Mitigated phase transition during first cycle of a Li-rich layered cathode studied by in operando synchrotron X-ray powder diffraction. Physical Chemistry Chemical Physics 18, 4745–4752, https://doi.org/10.1039/C5CP04801J (2016).
Amalraj, S. F. et al. Phase Transitions in Li2MnO3 Electrodes at Various States-of-Charge. Electrochimica Acta 123, 395–404, https://doi.org/10.1016/j.electacta.2014.01.051 (2014).
Francis Amalraj, S. et al. Study of the electrochemical behavior of the “inactive” Li2MnO3. Electrochimica Acta 78, 32–39, https://doi.org/10.1016/j.electacta.2012.05.144 (2012).
Wang, Z. Q., Wu, M. S., Xu, B. & Ouyang, C. Y. Improving the electrical conductivity and structural stability of the Li2MnO3 cathode via P doping. Journal of Alloys and Compounds 658, 818–823, https://doi.org/10.1016/j.jallcom.2015.11.013 (2016).
Doan, T. N. L., Yoo, K., Hoang, T. K. A. & Chen, P. Recent Developments in Synthesis of xLi2MnO3 · (1−x)LiMO2 (M = Ni, Co, Mn) Cathode Powders for High-Energy Lithium RechargeableBatteries. Frontiers in Energy Research 2, https://doi.org/10.3389/fenrg.2014.00036 (2014).
Zheng, J. et al. Mitigating Voltage Fade in Cathode Materials by Improving the Atomic Level Uniformity of Elemental Distribution. Nano Letters 14, 2628–2635, https://doi.org/10.1021/nl500486y (2014).
Lu, Z., Chen, Z. & Dahn, J. R. Lack of Cation Clustering in Li[NixLi1/3−2x/3Mn2/3−x/3]O2 (0 < x ≤ 1/2) and Li[CrxLi(1−x)/3Mn(2−2x)/3]O2 (0 < x < 1). Chemistry of Materials 15, 3214–3220, https://doi.org/10.1021/cm030194s (2003).
Bareño, J. et al. Long-Range and Local Structure in the Layered Oxide Li1.2Co0.4Mn0.4O2. Chemistry of Materials 23, 2039–2050, https://doi.org/10.1021/cm200250a (2011).
Zheng, J. et al. Electrochemical Kinetics and Performance of Layered Composite Cathode Material Li[Li0.2Ni0.2Mn0.6]O2. Journal of The Electrochemical Society 160, A2212–A2219, https://doi.org/10.1149/2.090311jes (2013).
Yu, H. et al. Electrochemical kinetics of the 0.5Li2MnO3[middle dot]0.5LiMn0.42Ni0.42Co0.16O2 ‘composite’ layered cathode material for lithium-ion batteries. RSC Advances 2, 8797–8807, https://doi.org/10.1039/C2RA20772A (2012).
Li, Z. et al. Electrochemical Kinetics of the Li[Li0.23Co0.3Mn0.47]O2 Cathode Material Studied by GITT and EIS. The Journal of Physical Chemistry C 114, 22751–22757, https://doi.org/10.1021/jp1088788 (2010).
Shi, S. J. et al. Combustion synthesis and electrochemical performance of Li[Li0.2Mn0.54Ni0.13Co0.13]O2 with improved rate capability. Journal of Power Sources 228, 14–23, https://doi.org/10.1016/j.jpowsour.2012.11.091 (2013).
Shi, S. J. et al. Synthesis and electrochemical performance of Li1.131Mn0.504Ni0.243Co0.122O2 cathode materials for lithium ion batteries via freeze drying. Journal of Power Sources 221, 300–307, https://doi.org/10.1016/j.jpowsour.2012.08.031 (2013).
Shin, Y., Ding, H. & A. Persson, K. Revealing the Intrinsic Li Mobility in the Li2MnO3 Lithium-Excess Material. Chemistry of Materials 28, 2081–2088, https://doi.org/10.1021/acs.chemmater.5b04862 (2016).
Park, M., Zhang, X., Chung, M., Less, G. B. & Sastry, A. M. A review of conduction phenomena in Li-ion batteries. Journal of Power Sources 195, 7904–7929, https://doi.org/10.1016/j.jpowsour.2010.06.060 (2010).
Shaju, K. M., Subba Rao, G. V. & Chowdari, B. V. R. Influence of Li-Ion Kinetics in the Cathodic Performance of Layered Li( Ni1/3Co1/3Mn1/3)O2. Journal of The Electrochemical Society 151, A1324–A1332, https://doi.org/10.1149/1.1775218 (2004).
Zhang, S. et al. Tuning Li2MO3 phase abundance and suppressing migration of transition metal ions to improve the overall performance of Li- and Mn-rich layered oxide cathode. Journal of Power Sources 380, 1–11, https://doi.org/10.1016/j.jpowsour.2018.01.045 (2018).
Ku, K. et al. Suppression of Voltage Decay through Manganese Deactivation and Nickel Redox Buffering in High-Energy Layered Lithium-Rich Electrodes. Advanced Energy Materials 8, 1800606, https://doi.org/10.1002/aenm.201800606 (2018).
Shang, H. et al. Suppressing Voltage Decay of a Lithium-Rich Cathode Material by Surface Enrichment with Atomic Ruthenium. ACS Applied Materials & Interfaces 10, 21349–21355, https://doi.org/10.1021/acsami.8b06271 (2018).
Zhao, T. et al. Three-dimensional Li1.2Ni0.2Mn0.6O2 cathode materials synthesized by a novel hydrothermal method for lithium-ion batteries. Journal of Alloys and Compounds 757, 16–23, https://doi.org/10.1016/j.jallcom.2018.05.058 (2018).
Luo, M. et al. Effects of doping Al on the structure and electrochemical performances of Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode materials. Ionics 24, 967–976, https://doi.org/10.1007/s11581-017-2269-5 (2018).
Kim, S. et al. Synthesis of layered–layered xLi2MnO3·(1−x)LiMO2 (M = Mn, Ni, Co) nanocomposite electrodes materials by mechanochemical process. Journal of Power Sources 220, 422–429, https://doi.org/10.1016/j.jpowsour.2012.07.135 (2012).
Kang, S.-H., Johnson, C. S., Vaughey, J. T., Amine, K. & Thackeray, M. M. The Effects of Acid Treatment on the Electrochemical Properties of 0.5Li2MnO3∙0.5LiNi0.44Co0.25Mn0.31O2 Electrodes in Lithium Cells. Journal of The Electrochemical Society 153, A1186–A1192, https://doi.org/10.1149/1.2194764 (2006).
Kang, S. H. et al. Interpreting the structural and electrochemical complexity of 0.5Li2MnO3·0.5LiMO2 electrodes for lithium batteries (M = Mn0.5−xNi0.5−xCo2x, 0 ≤ x ≤ 0.5). Journal of Materials Chemistry 17, 2069–2077, https://doi.org/10.1039/B618715C (2007).
Bréger, J. et al. High-resolution X-ray diffraction, DIFFaX, NMR and first principles study of disorder in the Li2MnO3–Li[Ni1/2Mn1/2]O2 solid solution. Journal of Solid State Chemistry 178, 2575–2585, https://doi.org/10.1016/j.jssc.2005.05.027 (2005).
Amalraj, F. et al. Synthesis of Integrated Cathode Materials xLi2MnO3⋅ (1−x)LiMn1/3Ni1/3Co1/3O2 (x = 0.3, 0.5, 0.7) and Studies of Their Electrochemical Behavior. Journal of The Electrochemical Society 157, A1121–A1130, https://doi.org/10.1149/1.3463782 (2010).
Buzlukov, A. et al. Li-Rich Mn/Ni Layered Oxide as Electrode Material for Lithium Batteries: A 7Li MAS NMR Study Revealing Segregation into (Nanoscale) Domains with Highly Different Electrochemical Behaviors. The Journal of Physical Chemistry C 120, 19049–19063, https://doi.org/10.1021/acs.jpcc.6b07532 (2016).
West, W. C., Soler, J. & Ratnakumar, B. V. Preparation of high quality layered-layered composite Li2MnO3–LiMO2 (M = Ni, Mn, Co) Li-ion cathodes by a ball milling–annealing process. Journal of Power Sources 204, 200–204, https://doi.org/10.1016/j.jpowsour.2012.01.011 (2012).
Inoue, T., Yamada, Y. & Ariyoshi, K. Synthesis and Characterization of Lithium Cobalt Manganese Oxides (LiCoO2-Li2MnO3) As Positive Electrode Material for Lithium-IonBatteries. Meeting Abstracts MA2016-02, 427 (2016).
McCalla, E., Lowartz, C. M., Brown, C. R. & Dahn, J. R. Formation of Layered–Layered Composites in the Li–Co–Mn Oxide Pseudoternary System during Slow Cooling. Chemistry of Materials 25, 912–918, https://doi.org/10.1021/cm304002b (2013).
Long, B. R. et al. Effect of Cooling Rates on Phase Separation in 0.5Li2MnO3·0.5LiCoO2 Electrode Materials for Li-Ion Batteries. Chemistry of Materials 26, 3565–3572, https://doi.org/10.1021/cm501229t (2014).
Lengyel, M. et al. Trace level doping of lithium-rich cathode materials. Journal of Materials Chemistry A 4, 3538–3545, https://doi.org/10.1039/C5TA07764H (2016).
Ahn, D. et al. Effects of cobalt-intercalation and polyaniline coating on electrochemical performance of layered manganese oxides. Journal of Materials Chemistry 21, 5282–5289, https://doi.org/10.1039/C0JM03548C (2011).
Hye Kim, S. & Kim, C. Improving the rate performance of LiCoO2 by Zr doping. Journal of Electroceramics 23, 254–257, https://doi.org/10.1007/s10832-008-9414-5 (2008).
Croy, J. R., Balasubramanian, M., Kim, D., Kang, S.-H. & Thackeray, M. M. Designing High-Capacity, Lithium-Ion Cathodes Using X-ray Absorption Spectroscopy. Chemistry of Materials 23, 5415–5424, https://doi.org/10.1021/cm2026703 (2011).
Chen, H. & Islam, M. S. Lithium Extraction Mechanism in Li-Rich Li2MnO3 Involving Oxygen Hole Formation and Dimerization. Chemistry of Materials 28, 6656–6663, https://doi.org/10.1021/acs.chemmater.6b02870 (2016).
Nayak, P. et al. Al Doping for Mitigating the Capacity Fading and Voltage Decay of Layered Li and Mn-Rich Cathodes for Li-Ion Batteries (2016).
Grey, C., Yoon, W.-S., Reed, J. & Ceder, G. Electrochemical Activity of Li in the Transition-Metal Sites of O3 Li[Li(1−2x)/3Mn(2−x)/3NixO2. Electrochemical and Solid-State Letters 7, A290–A293, https://doi.org/10.1149/1.1783113 (2004).
Kang, K. & Ceder, G. Factors that affect Li mobility in layered lithium transition metal oxides. Physical Review B 74, 094105 (2006).
Ghanty, C., Basu, R. N. & Majumder, S. B. Electrochemical performances of 0.9Li2MnO3–0.1Li(Mn0.375Ni0.375Co0.25)O2 cathodes: Role of the cycling induced layered to spinel phase transformation. Solid State Ionics 256, 19–28, https://doi.org/10.1016/j.ssi.2013.12.032 (2014).
Ghanty, C., Basu, R. N. & Majumder, S. B. Performance of Wet Chemical Synthesized xLi2MnO3-(1−x)Li(Mn0.375Ni0.375Co0.25)O2 (0.0 ≤ x ≤ 1.0) Integrated Cathode for Lithium Rechargeable Battery. Journal of The Electrochemical Society 159, A1125–A1134, https://doi.org/10.1149/2.081207jes (2012).
Weppner, W. & Huggins, R. A. Determination of the Kinetic Parameters of Mixed‐Conducting Electrodes and Application to the System Li3Sb. Journal of The Electrochemical Society 124, 1569–1578, https://doi.org/10.1149/1.2133112 (1977).
Shaju, K. M., Subba Rao, G. V. & Chowdari, B. V. R. Electrochemical Kinetic Studies of Li-Ion in O2-Structured Li2/3(Ni1/3Mn2/3)O2 and Li(2/3) +x(Ni1/3Mn2/3)O2 by EIS and GITT. Journal of The Electrochemical Society 150, A1–A13, https://doi.org/10.1149/1.1521754 (2003).
Shaju, K. M., Subba Rao, G. V. & Chowdari, B. V. R. EIS and GITT studies on oxide cathodes, O2-Li(2/3)+x(Co0.15Mn0.85)O2 (x = 0 and 1/3). Electrochimica Acta 48, 2691–2703, https://doi.org/10.1016/S0013-4686(03)00317-7 (2003).
Zhu, Y. & Wang, C. Galvanostatic Intermittent Titration Technique for Phase-Transformation Electrodes. The Journal of Physical Chemistry C 114, 2830–2841, https://doi.org/10.1021/jp9113333 (2010).
Dahbi, M., Urbonaite, S. & Gustafsson, T. Combustion synthesis and electrochemical performance of Li2FeSiO4/C cathode material for lithium-ion batteries. Journal of Power Sources 205, 456–462, https://doi.org/10.1016/j.jpowsour.2012.01.079 (2012).
Acknowledgements
This work has been partially supported by the Institute of Nanomaterials Research and Innovation for Energy (IN-RIE), Khon Kaen University and the National Nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of Research Network NANOTEC (RNN). S.K. wishes to acknowledge the support of the Thailand Graduate Institute of Science and Technology (TGIST) (Grant No. TG-33-12-56-007D).
Author information
Authors and Affiliations
Contributions
P.L. and N.M. designed this research. S.K. and W.L. carried out the experiments. S.K., N.M., H.K., W-S.Y., S.M. and W.L. analyzed data. P.L., N.M. and S.K. wrote the manuscript. All authors contributed to the scientific discussion and manuscript revisions.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaewmala, S., Limphirat, W., Yordsri, V. et al. Structural and Electrochemical Kinetic Properties of 0.5Li2MnO3∙0.5LiCoO2 Cathode Materials with Different Li2MnO3 Domain Sizes. Sci Rep 9, 427 (2019). https://doi.org/10.1038/s41598-018-36593-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36593-9
- Springer Nature Limited