
Abstract
Although shrimp are of great economic importance, few full-length shrimp transcriptomes are available. Here, we used Pacific Biosciences single-molecule real-time (SMRT) long-read sequencing technology to generate transcripts from the Pacific white shrimp (Litopenaeus vannamei). We obtained 322,600 full-length non-chimeric reads, from which we generated 51,367 high-quality unique full-length transcripts. We corrected errors in the SMRT sequences by comparison with Illumina-produced short reads. We successfully annotated 81.72% of all unique SMRT transcripts against the NCBI non-redundant database, 58.63% against Swiss-Prot, 45.38% against Gene Ontology, 32.57% against Clusters of Orthologous Groups of proteins (COG), and 47.83% against Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Across all transcripts, we identified 3,958 long non-coding RNAs (lncRNAs) and 80,650 simple sequence repeats (SSRs). Our study provides a rich set of full-length cDNA sequences for L. vannamei, which will greatly facilitate shrimp transcriptome research.
Similar content being viewed by others

Introduction
Whole-transcriptome analysis is of growing importance for animal biology research. However, whole-transcriptome analyses are ineffective without high quality transcript sequences1. Recently, second-generation sequencing (SGS) technologies, such as the Illumina Genome Analyzer, the Roche 454 pyrosequencing platform, and the ABI Solid platform, have facilitated the construction of transcriptome resources for many organisms2,3.
Shrimp are economically- and nutritionally-important crustaceans4. Several transcriptome studies in shrimp have been performed using SGS5, and many expressed sequence tags (ESTs) have been obtained6. However, the construction of transcriptomic sequences using SGS generally requires the assembly of short RNA-seq reads, and without a high-quality genome sequence available as a reference transcriptomic sequences may be misassembled due to reads transcribed from very similar members of multigene families or from highly repetitive regions7. In shrimp, the danger of misassembly may be even greater, as ~80% of the shrimp genome has been estimated to consist of repetitive elements8. Another limitation of SGS is that these technologies generally do not produce full-length transcripts, which are fundamental to studies of structural and functional genomics9,10,11. In addition, gene annotations and transcriptional characterizations of full-length transcripts are more accurate than those of transcript tags assembled from short RNA-sequencing reads7. Finally, alternative splicing, alternative polyadenylation, homologous genes, and superfamily genes are more easily identified based on full-length transcripts12,13,14,15.
Single-molecule real-time (SMRT) sequencing, a third-generation sequencing (TGS) technique recently developed by Pacific Biosciences (PacBio), allows direct sequencing of full-length, single-molecule cDNA sequences with a read length of up to 20 kb9,11,16. Using PacBio SMRT sequencing, intact RNA molecules can be sequenced without the need for fragmentation or post-sequencing assembly9. Thus, full-length transcripts can be constructed using SMRT sequencing.
The Pacific white shrimp (Litopenaeus vannamei) is the most extensively cultured crustacean species in the world, owing to its fast growth and strong disease resistance17,18. In this study, we used SMRT sequencing to construct the L. vannamei transcriptome. This is the first shrimp transcriptome constructed with SMRT.
Results
SMRT sequencing, quality filtering, and error correction
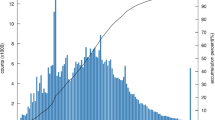
We used RNA extracted from six tissues (hepatopancreas, gills, heart, intestine, muscle, and stomach), collected and pooled from six L. vannamei, to constructed five cDNA libraries, each including cDNA inserts of approximately the same size: <1 kb, 1–2 kb, 2–3 kb, 3–6 kb, and >6 kb. We generated 1,307,853 polymerase reads (30.9 gigabases) across all five libraries. After removing adaptor sequences, low-quality sequences, and short sequences (<50 bp), 12,920,542 sub-reads remained. The mean sequence lengths for five cDNA libraries were 789 bp (<1 kb); 1,438 bp (1–2 kb); 2,304 bp (2–3 kb); 3,766 bp (3–6 kb); and 6,834 bp (>6 kb). We obtained 828,618 ROIs across all five cDNA libraries; the average lengths of the ROIs across the cDNA libraries were 2,018 bp, 2,968 bp, 3,340 bp, 4,235 bp, and 5,913 bp, respectively (Fig. 1). Of the 828,618 ROIs, 322,600 (38.93%) were identified as full-length non-chimeric (FLNC) reads.

ROI read length distribution. Different colors represent different SMRT sequencing libraries with different cDNA insert size ranges.
We performed Illumina library construction and sequencing in parallel to correct the 322,600 FLNC reads. Using Illumina, ~148 million paired-end reads were sequenced, from which ~132 million clean reads were generated after adaptor sequence trimming and low-quality read filtering. We used Proovread19 to correct the FLNC reads based on the Illumina short reads. Proovread indicated that 124,201 FLNC reads (38.50%) contained at least one erroneous inner and/or terminal fragment; these fragments were corrected. We then used iterative clustering for error correction (IEC) to obtain 51,367 unique corrected SMRT transcripts.
To further test the completeness of our transcriptome, we used the Benchmarking Universal Single-Copy Orthologs (BUSCO) pipeline20 to compare our L. vannamei transcriptome to 1,066 conserved arthropod genes. This analysis indicated that 81.0% of the L. vannamei transcriptome (863 genes) encoded complete proteins. Of these genes, 34.3% (366 genes) were complete single-copy BUSCOs, 46.6% (497 genes) were complete duplicated BUSCOs, 3.1% (33 genes) were fragmented BUSCO archetypes, and 16.0% (170 genes) were missing BUSCOs entirely.
Functional annotation of transcripts
Of the 51,367 unique SMRT transcripts, we identified significant matches in the NCBI non-redundant (Nr) protein database for 41,975 (81.72%; E-value ≤ 10−5). Of the species with matches for >1.8% of all L. vannamei transcripts, 15.69% of the hits were from the termite Zootermopsis nevadensis, 9.81% were from L. vannamei, and 8.52% were from the crustacean Daphnia pulex (8.52%; Fig. 2).

Percentage of L. vannamei transcripts with BlastX hits in various species. Transcripts were searched against the NCBI non-redundant protein database, using BlastX with the E-value cutoff set to <10−5. Only species with matches for >1.8% of the L. vannamei transcripts are shown; species matching fewer than 1.8% of all transcripts are classed as ‘Other’.
Our gene ontology (GO) analysis indicated that 9910 of the unique transcripts (42.51%) were enriched in biological processes, 8129 (34.87%) were enriched in molecular functions, and 5272 (22.62%) were enriched in cellular components (Fig. 3). We also identified matches to our unique transcripts in the Swiss-Prot21, Clusters of Orthologous Groups of proteins (COG)22, and Kyoto Encyclopedia of Genes and Genomes (KEGG)23 databases: 30,117 transcripts matched an entry in Swiss-Prot (58.63%), 16,732 transcripts matched an entry in COG (32.57%), and 24,569 transcripts matched an entry in KEGG (47.83%). The functional annotation of all unique transcripts are listed in Supplementary Table 1.

GO classification of the putative functions of the unique transcripts of L. vannamei.
To further identify the protein coding potential of unique transcripts, we predicted ORFs within all unique transcripts. In total, 47,260 unique transcripts were found having the protein coding potential, with an average length of 3,493 bp. The length distribution indicated that most protein-coding unique transcripts were distributed in length from 300 bp to 1,0000 bp, and there were more than 600 transcripts with a length >10,000 bp. (Fig. 4).

Lengths of candidate protein-coding RNAs.
Identification of long non-coding RNAs (lncRNAs)
We used four tools to identity unique transcripts without protein coding potential (i.e., lncRNAs): the Coding Potential Calculator (CPC)24 identified 375 lncRNAs, the Coding-Non-Coding Index (CNCI)25 identified 2,178 lncRNAs, the Coding Potential Assessment Tool (CPAT)26 identified 751 lncRNAs, and Pfam27 identified 4,342 lncRNAs. In total, 5893 unique transcripts were identified as lncRNAs by at least one tool (Fig. 5). After candidate lncRNAs with EMBOSS-predicted ORFs > 100 bp were removed, 3,958 lncRNAs remained. The average length of these lncRNAs was 2,111 bp, with most lncRNAs ranging in length from 300 bp to 4,800 bp (Fig. 6).


Lengths of candidate lncRNAs.
Identification of simple sequence repeats (SSRs)
SSRs are repetitive sequence motifs approximately 1–6 bp long28. We searched for SSRs in the 50,688 unique L. vannamei transcripts longer than 500 bp. We identified 80,650 SSRs across all tested transcripts, with 17,222 (33.98%) unique transcripts containing more than one SSR. Most of the SSRs identified were mono-nucleotide repeats (50.81%), followed by the di-nucleotide repeats (27.55%), tri-nucleotide repeats (18.33%), tetra-nucleotide repeats (2.41%), hexa-nucleotide repeats (0.55%), and penta-nucleotide repeats (0.35%). All SSRs and their primers are listed in Supplementary Table 2.
Comparison with previous L. vannamei transcriptomes
Strikingly, most of the assembled unique transcripts generated by Illumina and 454 sequencing were <1000 bp in length, while the lengths of the SMRT assembled unique transcripts were much more evenly distributed, with a considerable proportion of assembled transcripts ~6000–8000 bp long (Fig. 7). With respect to transcript functional annotations, proportionally more SMRT-sequenced transcripts were annotated than either 454-pyrosequenced transcripts or Illumina-sequenced transcripts (Fig. 8).


Discussion
Full-length cDNA sequences are useful for functional studies of important genes. However, full-length cDNA sequences can often only be generated by rapid amplification of cDNA ends (RACE), which is time consuming, labor intensive, expensive, and inefficient29. To date, very few full-length cDNA sequences have been reported for shrimp. Here, we used PacBio SMRT sequencing to obtain 51,367 high-quality unique full-length transcripts for L. vannamei. This large number of full-length cDNA sequences will greatly facilitate research projects using the shrimp transcriptome.
We compared several previously reported full-length cDNAs from L. vannamei with the corresponding full-length transcripts obtained in this study, including C-type lectin30, prophenoloxidase31, and ferritin32. We found the SMRT transcripts were essentially identical to the RACE cDNAs, with only minor differences at the 5′ and 3′ ends. These differences might have been due to differences in the primer sequences used by SMRT and RACE. Thus, our results suggested that SMRT sequencing is an effective method by which to obtain full-length cDNA sequences from the shrimp transcriptome.
Short-read sequencing (Illumina or 454) has been used to produce transcriptomes of some shrimp species, including L. vannamei17,18,33,34,35,36,37,38,39,40, Fenneropenaeus merguiensis41,42, Macrobrachium rosenbergii43, Triops newberryi44, T. longicaudatus45, Pandalus latirostris46, Fenneropenaeus chinensis47, Palaemon serratus48, and Penaeus monodon49. The average lengths of transcripts obtained in these studies were ~306–1,027 bp. Here, the average length of SMRT-sequenced transcripts was nearly 3 kb, far exceeding those of the previous studies. Our findings thus indicated that long transcripts in shrimp, from both coding and non-coding genes, might be more prevalent than previously estimated33.
Although SMRT sequencing produces longer reads than SGS methods, the SMRT raw data error rate is relatively high50. To correct these errors, it is possible to use the short reads generated by SGS as references51,52. Here, we used Illumina sequences to correct the SMRT reads. As 38.50% of the SMRT FLNC reads contained erroneous fragments (or single-nucleotide bases), our results indicated that error correction processing should be performed before further analysis of SMRT sequences.
LncRNAs are non-coding RNAs that are longer than 200 nucleotides long53,54. LncRNAs evolve rapidly, and are often species-specific in plants or animals55. An accumulating body of evidence has suggested that lncRNAs play essential roles in many important biological processes, such as translation, transcription, differentiation, splicing, immune responses, epigenetic regulation, and cell cycle control54,56,57,58,59. However, no lncRNAs in crustaceans have previously been reported. Here, we identified 3,958 novel lncRNAs in the L. vannamei shrimp transcriptome. These newly identified lncRNAs will be useful for several aspects of shrimp research, including epigenetics, immunology, and phylogenomics.
The SMRT transcriptome obtained here had a longer average transcript length than the transcripts obtained with SGS. Our results suggested that full-length transcripts were more easily annotated than shorter transcripts. Here, 81.72% of unique transcripts were annotated in the Nr database, as compared to 37.80%–73.08% in previously published L. vannamei transcriptomes produced with short-read sequencing17,18,33,34. This suggested that full-length transcripts were annotated more efficiently than the ESTs obtained by assembling short RNA-sequence reads.
Materials and Methods
Animal materials
Specific pathogen-free (SPF) white shrimp (L. vannamei) were obtained from the National and Guangxi Shrimp Genetic Breeding Center (Guangxi Province, China). We removed and pooled the hepatopancreases, gills, hearts, intestines, muscles, and stomachs of six shrimp. Pooled tissues were immediately stored in liquid nitrogen until RNA extraction.
RNA extraction
Total RNA was extracted from the pooled tissues using TRIzol LS Reagent (Invitrogen, USA) following the manufacturer’s instructions, and genomic DNA was removed using DNase I (Invitrogen, USA). RNA purity (OD260/280), concentration, and absorption peak were measured using a NanoDrop 2000 (Thermo Scientific, USA). RNA quality was determined with a Bioanalyser 2100 (Agilent, USA). Only total RNAs with a RIN score >7 were used to construct cDNA libraries for SMRT sequencing.
SMRT library construction, sequencing, and quality control
To construct full-length cDNAs, 10 μg of total RNA was reverse transcribed into cDNA using a SMARTer PCR cDNA Synthesis Kit (Takara, Japan), following the manufacturer’s protocols. Size fractionation and selection were performed using the BluePippin Size Selection System (Sage Science, USA). We prepared five SMRT libraries, each including fragments in one of five size groups: <1 kb, 1–2 kb, 2–3 kb, 3–6 kb, and >6 kb, following the PacBio protocol. Each library was sequenced in three SMRT cells on a PacBio RSII platform using C4 reagents and 3–4 h sequencing movies.
We used PacBio SMRT analysis software v2.3.0 (http://www.pacb.com/products-andservices/analytical-software/smrt-analysis/) to filter out low-quality polymerase reads (read-length <50 bp and read-score <0.75). ROIs were filtered from the sub-reads with the full pass threshold set to ≥0 and the predicted unique accuracy set to ≥0.75. We considered ROIs FLNC reads only if they possessed a 5′-cDNA primer, a 3′-cDNA primer, and a polyA tail preceding the 3′ primer. Then 5′- and 3′-cDNA primers and polyA tail were removed from FLNC according to the Pac-bio recommended procedure (https://github.com/PacificBiosciences/IsoSeq.3).
Illumina library construction and sequencing
The Illumina libraries used to correct the FLNC reads were constructed with the Tru-Seq RNA sample Prep kit (Illumina, USA). Briefly, poly-(A) mRNA was isolated from total RNA using oligo (dT) magnetic beads and then fragmented into 200–700 bp pieces with fragmentation buffer. Double-stranded cDNAs were synthesized using a SuperScript double-stranded cDNA synthesis kit (Invitrogen, USA) with random hexamer primers (Illumina, USA), following the manufacturer’s instructions. Synthesized cDNAs were gen-purified and amplified with PCR. PCR products were sequenced on a single lane of an Illumina HiSeq. 2500 high-throughput sequencer. Raw sequencing reads were quality controlled to remove adaptor sequences, low-quality reads (reads where quality was ≤10% for >50% of all nucleotides), and read with many unknown nucleotides (>10%). Cleaned sequences were used for SMRT error correction.
Quality filtering and error correction of PacBio reads
Nucleotide errors in the FLNC reads were corrected by comparison with the Illumina RNA sequences using Proovread v2.13.13 (https://github.com/BioInf-Wuerzburg/proovread) with parameter coverage set to 507,19. Corrected FLNC reads were clustered into unique (non-redundant) transcripts using the ICE algorithm in the PacBio SMRT analysis software v2.3.0, with quiver polishing set to ≥0.9955,60. We used BUSCO v3.0 (http://busco.ezlab.org/)20 with the BUSCO arthropod dataset (http://busco.ezlab.org/v2/datasets/arthropoda_odb9.tar.gz) to evaluate the completeness of the L. vannamei transcriptome.
Functional annotation of transcripts
We identified functional annotations matching each unique transcript by searching Nr, Swiss-Prot, COG, and KEGG using BlastX with an E-value cut-off of 10−5. Protein function was predicted based on the annotation of the most similar hit across all databases. The unique transcripts identified by BlastX were submitted to blast2GO v4.1 (http://www.blast2go.com)61 to assign GO categories. To identify the protein coding potential of each unique transcript, the ORFs within unique transcripts were predicted using TransDecoder v2.0.1 (https://transdecoder.github.io)62, with default parameters.
Identification of lncRNAs
We identified unique transcripts without protein coding potential as candidate lncRNAs using four tools: CPC v1.0 (http://cpc.cbi.pku.edu.cn/)24, CNCI v2.0 (https://github.com/www-bioinfo-org/CNCI)25, CPAT v1.2 (http://lilab.research.bcm.edu/cpat/index.php)26, and Pfam (http://pfam.xfam.org/)27 with default parameters. We then predicted the ORFs of all candidate lncRNAs selected by at least one tool with EMBOSS getorf v6.1.063; sequences containing ORFs > 100 bp long were discarded.
Identification of SSRs
We used MISA v1.0 (http://pgrc.ipk-gatersleben.de/misa/)64 with default parameters to identify SSRs (mono- to penta-nucleotide repeats) in all corrected unique transcripts longer than 500 bp. SSR primers were designed using primer365 with default parameters.
Comparison with previously published L. vannamei transcriptomes
To evaluate SMRT sequencing performance, we compared the SMRT transcriptome constructed here to two previously published L. vannamei transcriptomes, one obtained using 454 sequencing17 and one obtained using Illumina sequencing18. First, we compared the distributions of transcript lengths among the three transcriptomes. Next, we compared the number of Nr, Swiss-Prot, KEGG, COG and GO hits among the transcriptomes (all functional annotations for each of the three transcriptomes were performed with an E-value cutoff of 10−5).
Data Availability
Raw PacBio sequencing reads are available at NCBI GenBank under the accession SRX3267788, SRX3267789, SRX3267790, SRX3267791, SRX3267792, SRX3267793, SRX3267794, SRX3267795, SRX3267796, SRX3267797, SRX3267798, SRX3267799, SRX3267800, and SRX3267801). Raw Illumina sequencing reads are available at NCBI GenBank under the accession SRX3527198 and SRX3527197. Candidate protein-coding transcripts are available at NCBI GenBank under the accession GGUK00000000. Candidate lncRNA sequences are available at NCBI GenBank under the accession GGUT00000000.
References
Nagalakshmi, U., Waern, K. & Snyder, M. RNA-Seq: a method for comprehensive transcriptome analysis. Current protocols in molecular biology Chapter 4, (Unit4 11), 11–13, https://doi.org/10.1002/0471142727.mb0411s89 (2010).
Metzker, M. L. Sequencing technologies - the next generation. Nature reviews. Genetics 11, 31–46, https://doi.org/10.1038/nrg2626 (2010).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature reviews. Genetics 17, 333–351, https://doi.org/10.1038/nrg.2016.49 (2016).
Castillo-Juarez, H., Campos-Montes, G. R., Caballero-Zamora, A. & Montaldo, H. H. Genetic improvement of Pacific white shrimp [Penaeus (Litopenaeus) vannamei]: perspectives for genomic selection. Frontiers in genetics 6, 93, https://doi.org/10.3389/fgene.2015.00093 (2015).
Santos, C. A., Blanck, D. V. & de Freitas, P. D. RNA-seq as a powerful tool for penaeid shrimp genetic progress. Frontiers in genetics 5, 298, https://doi.org/10.3389/fgene.2014.00298 (2014).
Leu, J. H. et al. A review of the major penaeid shrimp EST studies and the construction of a shrimp transcriptome database based on the ESTs from four penaeid shrimp. Mar Biotechnol (NY) 13, 608–621, https://doi.org/10.1007/s10126-010-9286-y (2011).
Dong, L. et al. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC genomics 16, 1039, https://doi.org/10.1186/s12864-015-2257-y (2015).
Abdelrahman, H. et al. Aquaculture genomics, genetics and breeding in the United States: current status, challenges, and priorities for future research. BMC genomics 18, 191, https://doi.org/10.1186/s12864-017-3557-1 (2017).
Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138, https://doi.org/10.1126/science.1162986 (2009).
Korlach, J. et al. Real-time DNA sequencing from single polymerase molecules. Methods in enzymology 472, 431–455, https://doi.org/10.1016/S0076-6879(10)72001-2 (2010).
Roberts, R. J., Carneiro, M. O. & Schatz, M. C. The advantages of SMRT sequencing. Genome biology 14, 405, https://doi.org/10.1186/gb-2013-14-6-405 (2013).
Conesa, A. et al. A survey of best practices for RNA-seq data analysis. Genome biology 17, 13, https://doi.org/10.1186/s13059-016-0881-8 (2016).
Chen, L., Tovar-Corona, J. M. & Urrutia, A. O. Alternative splicing: a potential source of functional innovation in the eukaryotic genome. International journal of evolutionary biology 2012, 596274, https://doi.org/10.1155/2012/596274 (2012).
Wu, X. et al. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proceedings of the National Academy of Sciences of the United States of America 108, 12533–12538, https://doi.org/10.1073/pnas.1019732108 (2011).
Elkon, R., Ugalde, A. P. & Agami, R. Alternative cleavage and polyadenylation: extent, regulation and function. Nature reviews. Genetics 14, 496–506, https://doi.org/10.1038/nrg3482 (2013).
Au, K. F. et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proceedings of the National Academy of Sciences of the United States of America 110, E4821–4830, https://doi.org/10.1073/pnas.1320101110 (2013).
Chen, X. et al. Transcriptome analysis of Litopenaeus vannamei in response to white spot syndrome virus infection. PloS one 8, e73218, https://doi.org/10.1371/journal.pone.0073218 (2013).
Peng, J. et al. Gonadal transcriptomic analysis and differentially expressed genes in the testis and ovary of the Pacific white shrimp (Litopenaeus vannamei). BMC genomics 16, 1006, https://doi.org/10.1186/s12864-015-2219-4 (2015).
Hackl, T., Hedrich, R., Schultz, J. & Forster, F. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011, https://doi.org/10.1093/bioinformatics/btu392 (2014).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Bairoch, A. & Boeckmann, B. The SWISS-PROT protein sequence data bank. Nucleic acids research 19 Suppl, 2247–2249 (1991).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic acids research 28, 33–36 (2000).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic acids research 27, 29–34 (1999).
Li, A., Zhang, J. & Zhou, Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC bioinformatics 15, 311, https://doi.org/10.1186/1471-2105-15-311 (2014).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25, 3389–3402 (1997).
Yangyang, D., Wu Songfeng, L. J., Yunping, Z., Yaowen, C. & Fuchu, H. Integrated nr Database in Protein Annotation System and Its Localization. Computer Engineering 32, 71–72 (2006).
Sonnhammer, E. L., Eddy, S. R. & Durbin, R. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins 28, 405–420 (1997).
Schlotterer, C. Evolutionary dynamics of microsatellite DNA. Chromosoma 109, 365–371 (2000).
Bower, N. I. & Johnston, I. A. Targeted rapid amplification of cDNA ends (T-RACE)–an improved RACE reaction through degradation of non-target sequences. Nucleic acids research 38, e194, https://doi.org/10.1093/nar/gkq816 (2010).
Ma, T. H., Tiu, S. H., He, J. G. & Chan, S. M. Molecular cloning of a C-type lectin (LvLT) from the shrimp Litopenaeus vannamei: early gene down-regulation after WSSV infection. Fish & shellfish immunology 23, 430–437, https://doi.org/10.1016/j.fsi.2006.12.005 (2007).
Lai, C. Y., Cheng, W. & Kuo, C. M. Molecular cloning and characterisation of prophenoloxidase from haemocytes of the white shrimp, Litopenaeus vannamei. Fish & shellfish immunology 18, 417–430, https://doi.org/10.1016/j.fsi.2004.10.004 (2005).
Hsieh, S. L., Chiu, Y. C. & Kuo, C. M. Molecular cloning and tissue distribution of ferritin in Pacific white shrimp (Litopenaeus vannamei). Fish & shellfish immunology 21, 279–283, https://doi.org/10.1016/j.fsi.2005.12.003 (2006).
Li, C. et al. Analysis of Litopenaeus vannamei transcriptome using the next-generation DNA sequencing technique. PloS one 7, e47442, https://doi.org/10.1371/journal.pone.0047442 (2012).
Zeng, D. et al. Transcriptome analysis of Pacific white shrimp (Litopenaeus vannamei) hepatopancreas in response to Taura syndrome Virus (TSV) experimental infection. PloS one 8, e57515, https://doi.org/10.1371/journal.pone.0057515 (2013).
Xue, S. et al. Sequencing and de novo analysis of the hemocytes transcriptome in Litopenaeus vannamei response to white spot syndrome virus infection. PloS one 8, e76718, https://doi.org/10.1371/journal.pone.0076718 (2013).
Yu, Y. et al. SNP discovery in the transcriptome of white Pacific shrimp Litopenaeus vannamei by next generation sequencing. PloS one 9, e87218, https://doi.org/10.1371/journal.pone.0087218 (2014).
Wei, J. et al. Comparative transcriptomic characterization of the early development in Pacific white shrimp Litopenaeus vannamei. PloS one 9, e106201, https://doi.org/10.1371/journal.pone.0106201 (2014).
Hu, D., Pan, L., Zhao, Q. & Ren, Q. Transcriptomic response to low salinity stress in gills of the Pacific white shrimp, Litopenaeus vannamei. Marine genomics 24(Pt 3), 297–304, https://doi.org/10.1016/j.margen.2015.07.003 (2015).
Johnson, J. G. et al. High CO2 alters the hypoxia response of the Pacific whiteleg shrimp (Litopenaeus vannamei) transcriptome including known and novel hemocyanin isoforms. Physiological genomics 47, 548–558, https://doi.org/10.1152/physiolgenomics.00031.2015 (2015).
Gao, Y. et al. Whole Transcriptome Analysis Provides Insights into Molecular Mechanisms for Molting in Litopenaeus vannamei. PloS one 10, e0144350, https://doi.org/10.1371/journal.pone.0144350 (2015).
Powell, D., Knibb, W., Remilton, C. & Elizur, A. De-novo transcriptome analysis of the banana shrimp (Fenneropenaeus merguiensis) and identification of genes associated with reproduction and development. Marine genomics 22, 71–78, https://doi.org/10.1016/j.margen.2015.04.006 (2015).
Saetan, U., Sangket, U., Deachamag, P. & Chotigeat, W. Ovarian Transcriptome Analysis of Vitellogenic and Non-Vitellogenic Female Banana Shrimp (Fenneropenaeus merguiensis). PloS one 11, e0164724, https://doi.org/10.1371/journal.pone.0164724 (2016).
Rao, R. et al. A transcriptome study on Macrobrachium rosenbergii hepatopancreas experimentally challenged with white spot syndrome virus (WSSV). Journal of invertebrate pathology 136, 10–22, https://doi.org/10.1016/j.jip.2016.01.002 (2016).
Horn, R. L., Ramaraj, T., Devitt, N. P., Schilkey, F. D. & Cowley, D. E. De novo assembly of a tadpole shrimp (Triops newberryi) transcriptome and preliminary differential gene expression analysis. Molecular ecology resources 17, 161–171, https://doi.org/10.1111/1755-0998.12555 (2017).
Seong, J. et al. Transcriptome Analysis of the Tadpole Shrimp (Triops longicaudatus) by Illumina Paired-End Sequencing: Assembly, Annotation, and Marker Discovery. Genes 7 https://doi.org/10.3390/genes7120114 (2016).
Kawahara-Miki, R., Wada, K., Azuma, N. & Chiba, S. Expression profiling without genome sequence information in a non-model species, Pandalid shrimp (Pandalus latirostris), by next-generation sequencing. PloS one 6, e26043, https://doi.org/10.1371/journal.pone.0026043 (2011).
Li, S., Zhang, X., Sun, Z., Li, F. & Xiang, J. Transcriptome analysis on Chinese shrimp Fenneropenaeus chinensis during WSSV acute infection. PloS one 8, e58627, https://doi.org/10.1371/journal.pone.0058627 (2013).
Perina, A., Gonzalez-Tizon, A. M., Meilan, I. F. & Martinez-Lage, A. De novo transcriptome assembly of shrimp Palaemon serratus. Genomics data 11, 89–91, https://doi.org/10.1016/j.gdata.2016.12.009 (2017).
Uengwetwanit, T. et al. Transcriptome-based discovery of pathways and genes related to reproduction of the black tiger shrimp (Penaeus monodon). Marine genomics, https://doi.org/10.1016/j.margen.2017.08.007 (2017).
Au, K. F., Underwood, J. G., Lee, L. & Wong, W. H. Improving PacBio long read accuracy by short read alignment. PloS one 7, e46679, https://doi.org/10.1371/journal.pone.0046679 (2012).
Salmela, L. & Rivals, E. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514, https://doi.org/10.1093/bioinformatics/btu538 (2014).
Koren, S. et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature biotechnology 30, 693–700, https://doi.org/10.1038/nbt.2280 (2012).
Kapranov, P. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488, https://doi.org/10.1126/science.1138341 (2007).
Wapinski, O. & Chang, H. Y. Long noncoding RNAs and human disease. Trends in cell biology 21, 354–361, https://doi.org/10.1016/j.tcb.2011.04.001 (2011).
Wang, B. et al. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nature communications 7, 11708, https://doi.org/10.1038/ncomms11708 (2016).
Chen, X. & Yan, G. Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624, https://doi.org/10.1093/bioinformatics/btt426 (2013).
Bu, D. et al. NONCODEv3.0: integrative annotation of long noncoding RNAs. Nucleic acids research 40, D210–215, https://doi.org/10.1093/nar/gkr1175 (2012).
Mattick, J. S. & Makunin, I. V. Non-coding RNA. Human molecular genetics 15 Spec No 1, R17–29, https://doi.org/10.1093/hmg/ddl046 (2006).
Qureshi, I. A., Mattick, J. S. & Mehler, M. F. Long non-coding RNAs in nervous system function and disease. Brain research 1338, 20–35, https://doi.org/10.1016/j.brainres.2010.03.110 (2010).
Zulkapli, M. M. et al. Iso-Seq analysis of Nepenthes ampullaria, Nepenthes rafflesiana and Nepenthes x hookeriana for hybridisation study in pitcher plants. Genomics data 12, 130–131, https://doi.org/10.1016/j.gdata.2017.05.003 (2017).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676, https://doi.org/10.1093/bioinformatics/bti610 (2005).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512, https://doi.org/10.1038/nprot.2013.084 (2013).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends in genetics: TIG 16, 276–277 (2000).
Beier, S., Thiel, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585, https://doi.org/10.1093/bioinformatics/btx198 (2017).
Untergasser, A. et al. Primer3Plus, an enhanced web interface to Primer3. Nucleic acids research 35, W71–74, https://doi.org/10.1093/nar/gkm306 (2007).
Acknowledgements
This study was supported by grants from the Guangxi scientific research and technology development project (AD16380049, AB16380189, nycytxgxcxtd-03–14, 17-A-04-04, AA17204080-1 and CARS-47) and Eight Osmanthus scholars special funding (BGXZ-NMBDX-04).
Author information
Authors and Affiliations
Contributions
D.Z. and Xiu C. wrote the main manuscript text. Xiao C., Y.Z. and Y.L. designed the experiments. J.P., C.Y., M.P., W.Z. and D.X. carried out the experiments. P.H. and P.W. analyzed the data. All authors approved and read the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, D., Chen, X., Peng, J. et al. Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci Rep 8, 16920 (2018). https://doi.org/10.1038/s41598-018-35066-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35066-3
- Springer Nature Limited
Keywords
This article is cited by
-
Genome-wide QTL and eQTL mapping reveal genes associated with growth rate trait of the Pacific white shrimp (Litopenaeus vannamei)
BMC Genomics (2024)
-
Long non-coding RNAs in the physiology of aquaculture animals: a perspective update
Reviews in Fish Biology and Fisheries (2022)
-
Single-molecule Real-time (SMRT) Sequencing Facilitates Transcriptome Research and Genome Annotation of the Fish Sillago sinica
Marine Biotechnology (2022)
-
Full-length SMRT transcriptome sequencing and microsatellite characterization in Paulownia catalpifolia
Scientific Reports (2021)
-
Single-molecule long-read sequencing of the full-length transcriptome of Rhododendron lapponicum L.
Scientific Reports (2020)