
Abstract
AP endonuclease deficiency causes cell death and embryonic lethality in mammals. However, the physiological roles of AP endonucleases in multicellular organisms remain unclear, especially after embryogenesis. Here, we report novel physiological roles of the AP endonuclease EXO-3 from larval to adult stages in Caenorhabditis elegans, and elucidated the mechanism of the observed phenotypes due to EXO-3 deficiency. The exo-3 mutants exhibited developmental delay, whereas the apn-1 mutants did not. The delay depended on the DNA glycosylase NTH-1 and checkpoint kinase CHK-2. The exo-3 mutants had further developmental delay when treated with AP site-generating agents such as methyl methane sulfonate and sodium bisulfite. The further delay due to sodium bisulfite was dependent on the DNA glycosylase UNG-1. The exo-3 mutants also demonstrated an increase in dut-1 (RNAi)-induced abnormal vulval organogenesis protruding vulva (Pvl), whereas the apn-1 mutants did not. The increase in Pvl was dependent on UNG-1 and CHK-2. Methyl viologen, ndx-1 (RNAi) and ndx-2 (RNAi) enhanced the incidence of Pvl among exo-3 mutants only when combined with dut-1 (RNAi). This further increase in Pvl incidence was independent of NTH-1. These results indicate that EXO-3 prevents developmental delay and Pvl in C. elegans, which are induced via DNA glycosylase-initiated checkpoint activation.
Similar content being viewed by others


Introduction
AP endonucleases are enzymes that function in the repair of DNA damage such as apurinic/apyrimidinic (AP) sites or single-strand breaks with 3′ blocked ends (3′-blocked SSB) in DNA1,2. AP sites are generated by the spontaneous hydrolysis of purine/pyrimidine bases (depurination/depyrimidination) or through the DNA glycosylase activity of monofunctional DNA glycosylases, which cleave N-glycosidic bonds in DNA3,4. Bifunctional DNA glycosylases, which possess both DNA glycosylase activity and AP lyase activity, generate 3′-blocked SSB by cutting N-glycosidic bonds in DNA and subsequently nicking the sugar phosphate backbone at AP sites1. Through the process of DNA replication, AP sites and 3′-blocked SSB can cause double-stranded breaks (DSB), which are considered to be the most deleterious form of DNA lesions and can lead to cell death if not repaired properly5,6. Therefore, the repair of AP sites and 3′-blocked SSB is important to protect cells and organisms against their adverse effects. AP sites and 3′-blocked SSB are processed by different enzymatic activities of AP endonucleases, AP endonuclease activity and 3′-phosphodiesterase activity, respectively1,2,7. Both activities generate SSB with 3′-OH ends, which are required for the subsequent steps of the repair process known as base excision repair (BER), which is carried out by DNA polymerases and DNA ligases3.
The in vivo roles of AP endonucleases in unicellular organisms have been well studied. Insufficient AP endonuclease activity in unicellular organisms, including Escherichia coli (E. coli) and Saccharomyces cerevisiae (S. cerevisiae), increases the spontaneous mutation frequency and heightens the sensitivity to DNA damaging agents such as methyl methane sulfonate (MMS), hydrogen peroxide (H2O2) and gamma rays8,9. On the other hand, the physiological roles of AP endonucleases in multicellular organisms are unclear. The knockout of APEX1, which encodes the main AP endonuclease APEX1 in mice (human APE1 ortholog), and knockdown of zebrafish APEX1 (zfAPEX1) both cause embryonic lethality10,11. In addition to DNA repair activity, APEX1 has redox regulation activity, which is well-conserved among APE1 homologs in mammals. Human APE1 knockdown cells exhibit decreased cellular viability, but this decreased viability is reversed by expression of yeast Apn1 protein, which lacks redox regulation activity12. zfAPEX1 does not have the cysteine residue required for redox regulation activity13. Thus, the embryonic lethality caused by the knockout of mouse APEX1 or knockdown of zfAPEX1 is thought to be due to deficient repair activity, and strongly suggests that the DNA repair activity of AP endonucleases plays an essential role in embryogenesis in multicellular organisms. However, their roles after embryogenesis remain unknown.
Caenorhabditis elegans (C. elegans) is a useful model animal to study the physiological roles of AP endonucleases. Worms deficient in AP endonuclease genes do not exhibit embryonic lethality and can become fertile adults14. Therefore, the physiological effects of the lack of AP endonucleases can be assessed throughout life. In C. elegans, two AP endonucleases, EXO-3 and APN-1, have been identified15. EXO-3 and APN-1 are encoded by the exo-3 and apn-1 genes, respectively. EXO-3 is a homologue of mammalian APE1, but the redox regulatory domain is not conserved13. A series of in vitro experiments revealed that both EXO-3 and APN-1 have AP endonuclease activity and 3′-phosphodiesterase activity16,17. Malfunction of AP endonucleases causes severe phenotypes in C. elegans. Exo-3 (RNAi) worms have a reduced life-span in a cep-1 (C. elegans p53 ortholog)-dependent manner18. Exo-3 mutant worms also exhibit a shortened life-span and reduced self-brood size14. Apn-1 (RNAi) worms have a moderately reduced longevity only when they are exposed to DNA damaging agents19. Apn-1 (RNAi) worms also demonstrate retardation of the division of the P1 blastomere19. Studies conducted thus far have focused on worms before the larval stages or after the adult stages14,18,19. However, the contribution of AP endonucleases during the larval to adult stages remains poorly understood.
To investigate the physiological roles of AP endonucleases from the larval to adult stages in C. elegans, we assessed the impact of AP endonuclease deficiency on development and vulval organogenesis from the larval to adult stages in C. elegans. We report newly identified phenotypes of exo-3(tm4374) mutants: developmental delay and increased dut-1 (RNAi)-induced abnormal vulval organogenesis Pvl. We also present evidence that these phenotypes are induced through a common mechanism where DNA glycosylases initiate DNA damage checkpoint activation.
Results
The expression level of AP endonucleases increases after the L4 stage
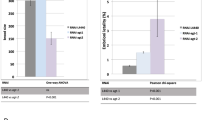
After hatching, C. elegans develop into adults through four larval stages (L1-L4), each punctuated by molting of the cuticle. Adult stages are still subdivided into two stages: the young adult stage and the gravid adult stage. To gain insight into the role of AP endonucleases after embryogenesis, we measured the temporal change in the mRNA expression level of exo-3 or apn-1. At 0, 24 and 48 hours, when most N2 worms are in the egg stage, L1 stage and L4 stage, respectively, no difference in mRNA expression level was found for both exo-3 and apn-1 (Fig. 1a,b). At 60 hours, when most N2 worms are in the young adult stage, the exo-3 and apn-1 expression levels were approximately 13-fold and 2.3-fold higher than those at 0 hours, respectively (Fig. 1a,b). The expression level at 72 hours, when most N2 worms are in the gravid adult stage, was the same at 60 hours (Fig. 1a,b). These results suggest that AP endonucleases are required after embryogenesis, especially after the L4 stage.

Effects of AP endonuclease deficiency on larval development of worms under normal rearing conditions. (a,b) At each time point after birth, N2 worms were harvested, and the total RNA isolated from the worms was subjected to real-time PCR analysis using specific primer sets for exo-3 (a) and apn-1 (b). As an internal control, Y45F10D.4 was used. The values represent the mean ± S.E. (n = 3/each time point). (c) Experimental scheme. (d–f) Representative images of worms at each developmental stage. L4 larvae (d). Young adults (e). Gravid adults (f). Black arrows indicate the vulval position. Developmental stages were assessed based on vulval morphology and brooding of eggs. (g) Proportion of worms at each developmental stage after 3 days of incubation of fertilized eggs. The values indicate the number of worms at each developmental stage/the number of total surviving worms.
The exo-3 mutants exhibit developmental delay
To clarify the contribution of AP endonucleases to worm development from the L4 to adult stages, worms deficient in either or both AP endonucleases (EXO-3 and APN-1) were incubated under normal rearing conditions for 3 days from the fertilized egg stage (Fig. 1c). Developmental stages among the L4, young adult and gravid adult stages were distinguished by the state of vulval morphology and brooding of eggs (Fig. 1d,f). Although all of the N2 worms developed into gravid adults, only 14% of the exo-3 mutants were in the gravid adult stage, 85% were in the young adult stage and 1% was in the larval stage (Fig. 1g), suggesting that EXO-3 deficiency causes the cessation of development or developmental delay. In contrast, all of the apn-1 mutants became gravid adults, and the apn-1;exo-3 mutants were at almost the same developmental stages as the exo-3 mutants (Fig. 1g). Twelve hours later, all the exo-3 and apn-1;exo-3 mutants reached the gravid adult stage (data not shown), indicating that EXO-3 deficiency does not cause cessation of development at the young adult stage, only developmental delay. To precisely investigate how long the delay of the exo-3 mutants was, we measured the reaching time to gravid adult of each worm every two hours and calculated the difference of the average time between N2 (N = 8) and the exo-3 mutants (N = 16). The difference was 6 hours.
The developmental delay in the exo-3 mutants is dependent on the DNA glycosylase NTH-1
It is reasonable to infer that the developmental delay phenotype of exo-3 mutants is caused by the accumulation of AP sites or 3′-blocked SSB in DNA because these are substrates for EXO-315. These structures in DNA can be generated by DNA glycosylases. Of the two DNA glycosylases conserved in C. elegans, UNG-1 generates AP sites through monofunctional DNA glycosylase activity on uracil in DNA, and NTH-1 produces 3′-blocked SSB via bifunctional DNA glycosylase activity on oxidative pyrimidine lesions in DNA. Therefore, we examined the dependency of the delay in the exo-3 mutants on UNG-1 and NTH-1. Three days after developing from eggs, 9% of the ung-1;exo-3 mutants were in the gravid adult stage, 86% were in the young adult stage and 5% were in the larval stage, and these proportions were almost the same as those shown by the exo-3 mutants (11% in the gravid adult stage, 85% in the young adult stage and 4% in the larval stage) (Fig. 2), suggesting that the delay is independent of UNG-1. In contrast, 94% of the nth-1;exo-3 mutants were in the gravid adult stage and 6% were in the young adult stage. The nth-1;ung-1;exo-3 mutants exhibited similar results (Fig. 2), suggesting that the delay is dependent on NTH-1.

Effects of DNA glycosylase deficiency on larval development of exo-3 (tm4374) mutant worms under normal rearing conditions. Proportion of worms at each developmental stage after 3 days of development from eggs. The values indicate the number of worms at each developmental stage/the number of total surviving worms.
The developmental delay of the exo-3 mutants is enhanced by MMS and NaHSO3
To clarify whether AP site-generating agents can cause developmental delay, we conducted a developmental assay using MMS and sodium bisulfite (NaHSO3) (Fig. 3a). MMS is known to indirectly create AP sites20,21 and shown to induce DNA lesions in the genome of C. elegans22. At 3.5 days after the eggs were laid on plates containing 0.94 mM MMS, 97% of N2 were in the gravid adult stage and 3% were in the larval stage, whereas 61% of the exo-3 mutants were in the gravid adult stage, 34% were in the young adult stage and 5% were in the larval stage (Fig. 3b), suggesting that MMS-induced AP sites cause further developmental delay in the exo-3 mutants than in N2. On the other hand, 93% of the apn-1 mutants were in the gravid adult stage, 6% were in the young adult stage and 1% were in the larval stage, which is similar to the results for N2 (Fig. 3b). NaHSO3 damages DNA mainly through deamination of cytosine to uracil23. Four days after developing from the egg stage, all exo-3 mutants not treated with NaHSO3 developed into gravid adults, but 33% of those treated with 10 mM NaHSO3 were in the gravid adult stage, 12% were in the young adult stage and 55% were in the larval stage (Fig. 3c), indicating that NaHSO3 induced developmental delay in the exo-3 mutants. This delay was alleviated in the ung-1;exo-3 mutants, as 79% of those treated with 10 mM NaHSO3 were in the gravid adult stage, 14% were in the young adult stage and 7% were in the larval stage (Fig. 3c), suggesting that the NaHSO3-induced developmental delay was due to UNG-1 activity. Taken together, AP sites may cause developmental delay as well as 3′-blocked SSB generated by NTH-1.

Effects of AP site-generating agents on larval development of worms. (a) Experimental scheme. (b,c) Proportion of worms at each developmental stage 3.5 and 4 days after developing from eggs under 0.94 mM MMS (b) and 10 mM NaHSO3 (c) conditions, respectively. The values indicate the number of worms at each developmental stage/the number of total surviving worms.
The developmental delay of the exo-3 mutants is induced by CHK-2
We next hypothesized that the DNA glycosylase-initiated developmental delay of the exo-3 mutants was due to DNA damage checkpoint activation driven by cleavage products produced by DNA glycosylases. DNA damage checkpoint genes, such as chk-2 and clk-2, have been described in C. elegans24. To test our hypothesis, the developmental assay was conducted under chk-2 (RNAi) or clk-2 (RNAi) conditions (Fig. 4a). Although 14% of the exo-3;control (RNAi) worms were in the gravid adult stage and 84% were in the young adult stage, and exo-3;clk-2 (RNAi) worms demonstrated similar proportions (18% in the gravid adult stage and 82% in the young adult stage), 93% of the exo-3;chk-2 (RNAi) worms were in the gravid adult stage (Fig. 4b). Thus, the knockdown of chk-2 rescued the developmental delay in the exo-3 mutants, suggesting that CHK-2 induces the developmental delay in the exo-3 mutants.

Effects of lack of checkpoint kinases on larval development of exo-3 (tm4374) mutant worms. (a) Experimental scheme for the developmental assay. (b) Proportion of worms at each developmental stage 3 days after development from eggs. The values indicate the number of worms at each developmental stage/the number of total surviving worms.
EXO-3 prevents dut-1 (RNAi)-induced Pvl formation
DUT-1 is a deoxyuridine triphosphate nucleotidohydrolase (dUTPase), which hydrolyzes dUTP into dUMP. Therefore, dut-1 (RNAi) leads to increased dUTP in the nucleotide pool, which is incorporated into DNA through DNA replication instead of dTTP, causing the accumulation of uracil in DNA25,26. It has been reported that dut-1 (RNAi) induces abnormal vulval organogenesis, Pvl, in wild-type N2 worms, and that the dut-1 (RNAi)-induced Pvl is dependent on UNG-127. We speculated that EXO-3 deficiency causes an increase in the incidence of dut-1 (RNAi)-induced Pvl because EXO-3 is needed to repair AP sites after the cleavage of uracil in DNA by UNG-1. To confirm this, dut-1 (RNAi)-induced Pvl formation was observed in AP endonuclease-deficient mutants (Fig. 5a–c). Of the surviving adults 4 days after developing from eggs, 52% were exo-3 mutants with dut-1 (RNAi)-induced Pvl and 13% were N2 worms with Pvl (Fig. 5d), suggesting that EXO-3 deficiency causes an increase in dut-1 (RNAi)-induced Pvl. On the other hand, 15% of the apn-1 mutants had Pvl, which is similar to the proportion of N2 worms with Pvl (Fig. 5d). The apn-1;exo-3 mutants and exo-3 mutants were similar in proportion, with 52% having Pvl (Fig. 5d).

Effects of AP endonuclease deficiency on dut-1 (RNAi)-induced Pvl. (a) Experimental scheme. (b,c) Representative images of vulva of control worms (b) and dut-1 (RNAi)-induced Pvl worms (c). (d) Proportion of Pvl worms 4 days after development from eggs. The values indicate the number of Pvl worms/the number of adult worms.
The increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants is dependent on UNG-1 irrespective of NTH-1
To examine whether the increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants occurred through UNG-1 activity, we investigated the dependency of the phenotype on UNG-1. The percentage of ung-1;exo-3 mutants with Pvl was 2%, which was the same as that of the ung-1 mutants with Pvl (1%) (Fig. 6a), suggesting that the increase in Pvl in the exo-3 mutants is only due to UNG-1 expression.

Effects of DNA glycosylase deficiency and lack of checkpoint kinases on dut-1 (RNAi)-induced Pvl in exo-3 (tm4374) mutant worms. Proportion of Pvl worms 4 days after development from eggs. The values indicate the number of Pvl worms/the number of adult worms. (a) Effects of ung-1 (tm2862) mutation. (b) Effects of chk-2 (RNAi) and clk-2 (RNAi).
Next, we examined whether the cleavage products produced by UNG-1 are needed to induce Pvl, i.e., whether AP sites are transformed into 3′-blocked SSB via the AP lyase activity of NTH-1. The percent of exo-3 mutants with Pvl was comparable to that of nth-1;exo-3 mutants (Fig. 7b), suggesting that NTH-1 is not necessary for dut-1 (RNAi)-induced Pvl.

Effects of oxidative DNA damaging agents on dut-1 (RNAi)-induced Pvl in exo-3 (tm4374) and exo-3 (tm4374);nth-1 (ok724) mutant worms by using the double knockdown technique. (a) Experimental scheme. (b) Proportion of Pvl worms 4 days after development from eggs. The values indicate the number of Pvl worms/the number of adult worms. The values represent the mean ± SD (n = 3). *P < 0.05; one-way ANOVA with Tukey’s test for multiple comparisons.
The increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants is driven by CHK-2
We hypothesized that the UNG-1-dependent increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants can occur via DNA checkpoint activation driven by cleavage products produced by UNG-1 in addition to developmental delay. It has also been reported that dut-1 (RNAi)-induced Pvl is caused by the checkpoint kinase CLK-227. Therefore, we examined whether the increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants was dependent on CHK-2 and CLK-2. Although 49% of exo-3;control (RNAi) worms had dut-1 (RNAi)-induced Pvl, only 10% of the exo-3;chk-2 (RNAi) worms had it (Fig. 6b). On the other hand, the proportion of exo-3;clk-2 (RNAi) worms with Pvl was almost the same (44%) as that of the exo-3;control (RNAi) worms. Therefore, the knockdown of chk-2 rescued the increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants, as observed for developmental delay. These results suggest that the increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants occurs via CHK-2 expression.
Oxidative DNA damaging agents enhanced the proportion of Pvl in the exo-3 mutants only when combined with dut-1 (RNAi)
To investigate other DNA damaging agents that cause an increase in Pvl in the exo-3 mutants, we evaluated whether Pvl formation is enhanced by oxidative DNA damaging agents such as ndx-1 (RNAi), ndx-2 (RNAi) and methyl viologen (MV). NDX-1 and NDX-2 hydrolyze 8-oxo-dGDP into 8-oxo-dGMP24,28. Accordingly, ndx-1 (RNAi) and ndx-2 (RNAi) lead to an increase in 8-oxo-dGDP in the nucleotide pool. The presence of 8-oxo-dGDP reduces the 8-oxo-dGTPase activity of NDX-4, causing 8-oxo-dGTP to accumulate in the pool24. 8-oxo-dGTP is incorporated to DNA during DNA replication, resulting in the accumulation of 8-oxoG in DNA24,28. MV generates superoxide radicals, which subsequently cause oxidative lesions in DNA29. However, single treatment with ndx-1 (RNAi), ndx-2 (RNAi) or MV had no effect on the incidence of Pvl in the exo-3 mutants. (data not shown). Next, we examined whether, when combined with the dut-1 (RNAi) treatment, each oxidative DNA damaging agent further enhanced the increase in Pvl in the exo-3 mutants (Fig. 7a). Each treatment tested enhanced the increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants by approximately 10% (Fig. 7b), suggesting that oxidative lesions in DNA can also cause an increase in Pvl.
In in vitro experiments, NTH-1 was found to possess weak DNA glycosylase activity toward 8-oxoG in DNA, in addition to its much higher activity toward oxidative pyrimidine lesions30. Thus, we suspect that the higher increase in dut-1 (RNAi)-induced Pvl by the additional oxidative agents depends on the activity of NTH-1. Although we confirmed the dependency of the phenotype on NTH-1, NTH-1 deficiency did not alter the proportion of worms exhibiting Pvl (Fig. 7b).
Discussion
In this study, we investigated the in vivo contribution of AP endonucleases from the larval to adult stages in C. elegans by evaluating development and vulval organogenesis in AP endonuclease gene mutants, and clarified that AP endonuclease EXO-3 deficiency causes developmental delay and an increased incidence of dut-1 (RNAi)-induced Pvl via DNA glycosylase-initiated checkpoint activation (Fig. 8).

A model for the mechanisms of developmental delay and the increase in Pvl in the exo-3 mutant worms. Cleavage products produced by either NTH-1 or UNG-1 can cause checkpoint activation, leading to developmental delay when worms lack EXO-3, but not when worms lack APN-1. Moreover, UNG-1-inducing checkpoint activation results in an increase in dut-1 (RNAi)-induced Pvl when worms lack EXO-3, but not when they lack APN-1.
The exo-3 mutants demonstrated developmental delay, whereas the apn-1 mutants did not (Fig. 1e), suggesting that EXO-3 has a more important role than APN-1 during development from the larval to adult stages.
The delay in the exo-3 mutants was completely dependent on NTH-1 (Fig. 2), suggesting that it is caused by 3′-blocked SSB generated by NTH-1. We therefore hypothesized that AP sites cannot cause developmental delay in the exo-3 mutants. However, MMS and NaHSO3 induced further developmental delay in the exo-3 mutants (Fig. 3b,c), and we found that the delay by NaHSO3 was dependent on UNG-1 (Fig. 3c), suggesting that AP sites also caused developmental delay in the exo-3 mutants, although the further delay may be caused by a mixture of both AP sites and cleaved AP sites with 3′-blocked SSB by NTH-1.
The developmental delay in the exo-3 mutants was also due to CHK-2 (Fig. 4b). CHK-2 is an ortholog of mammalian Chk2, which is activated in response to several DNA damaging agents that cause DSB31, but there is no direct evidence that C. elegans CHK-2 is involved in a checkpoint mechanism driven by SSB. As 3′-blocked SSB can generate DSB during DNA replication6, as can AP sites32, it is possible that the resulting DSB activated the CHK-2 response, thereby leading to developmental delay. It was recently reported that ATM, which positively regulates Chk2 activity in response to DSB-generating agents33,34, is also activated by SSBs in human cells35. Thus, it is possible that 3′-blocked SSB directly activate CHK-2 via an ATM-1/CHK-2 pathway.
The exo-3 mutants exhibited an increased incidence of dut-1 (RNAi)-induced Pvl (Fig. 5d), but the apn-1 mutants did not, suggesting that EXO-3 has a more important role than APN-1 in vulval organogenesis and development. The increase in Pvl in the exo-3 mutants was completely dependent on UNG-1 (Fig. 6a). This suggests that Pvl is caused by UNG-1-generating AP sites. The increase in Pvl occurred irrespective of NTH-1 (Fig. 7b), suggesting that the transformation of AP sites into 3′-blocked SSB by NTH-1 is not needed to induce the increase in Pvl.
Dengg et al. previously reported that dut-1 (RNAi)-induced Pvl occurs via the checkpoint kinase CLK-227. CLK-2 may be activated by replication fork collapse-mediated DSB because they found that dut-1(RNAi) enhances the accumulation of RPA-1, ATL-1 (ATR ortholog in C. elegans) and RAD-51 in mitotic germ cells based on UNG-1 activity27. In this study, we tried to clarify whether the increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants was dependent on CLK-2, but knockdown of CLK-2 had no effect on the increased incidence of Pvl (Fig. 6b). This discrepancy may result from the methods used to compromise CLK-2 function. Dengg et al. used clk-2 mutant worms, whereas we used clk-2 (RNAi) worms, as reported previously24. Instead of CLK-2, another checkpoint kinase, CHK-2, was found to be a causal factor of the increase in Pvl (Fig. 6b). It is possible that a mechanism similar to CLK-2 activation causes Pvl formation through CHK-2 in the exo-3 mutants.
The increase in dut-1 (RNAi)-induced Pvl in the exo-3 mutants was further enhanced by oxidative DNA damaging agents such as ndx-1 (RNAi), ndx-2 (RNAi) and MV (Fig. 7b). The damaging agents may have only caused Pvl in the exo-3 mutants when combined with dut-1 (RNAi) because of a threshold of DNA lesions needed for Pvl. Combining each oxidative damaging agent with dut-1 (RNAi) results in more DNA lesions containing 8-oxoG than single dut-1 (RNAi) treatment. However, a direct homolog of MutM or OGG1 cannot be detected in C. elegans. As a candidate protein to incise 8-oxoG in DNA in vivo in C. elegans, we examined NTH-1. However, the effects of ndx-1 and ndx-2 knockdown on Pvl were independent of NTH-1 (Fig. 7b). A novel DNA glycosylase that can incise 8-oxoG may be responsible for this phenotype, but further studies are needed.
Pvl is formed by prevention of ras/notch/wnt signaling pathway36,37,38. We demonstrated that the checkpoint activation caused by UNG-1 results in the induction of Pvl, while the downstream pathway to induce Pvl still remains unclear. Thus, it is possible that checkpoint activation affects Pvl induction through modulation of other pathways such as ras/notch/wnt signaling pathways. However, there is no evidence of the link between checkpoint activation and such signaling pathways.
It is reasonable that checkpoint activation causes developmental delay, while it seems paradoxical that the activation also causes Pvl induction. The reason for the point is that developmental delay induced by checkpoint activation is predicted to be a mechanism for preventing mutagenesis, which provides worms beneficial effects. In contrast, Pvl caused by the activation seems to have no valuable effects. However, it is probable that the induction of Pvl, resulting in egg-laying-defective (Egl) worms27, is a mechanism for preventing the accumulation of mutations in the next generation.
This study demonstrated that EXO-3 prevents DNA glycosylase-initiated checkpoint activation in order for worms to grow at a normal speed and with normal vulva. Although there have been many studies reported a correlation between BER and biological phenomena, such as carcinogenesis and aging, few studies demonstrating causation have been conducted39,40. Due to the availability of C. elegans mutants and the characteristics of C. elegans mutants lacking AP endonucleases enabling them to mature past embryonic stages and produce the next generation, we were able to examine causal relationships between BER and many biological phenomena throughout life. Further studies on AP endonucleases using C. elegans will provide more detailed information about the in vivo roles of AP endonucleases in multicellular organisms.
Methods
C. elegans strains and culture conditions
The wild-type strain (Bristol N2) and RB877 [nth-1(ok724) III]30 were obtained from the Caenorhabditis Genetics Center, University of Minnesota. TM4374 [exo-3(tm4374) I]41, TM6691 [apn-1(tm6691) II]14 and TM2862 [ung-1(tm2862) III]42 were supplied by the National Bioresource Project for the Nematode (Tokyo Women’s Medical College). All mutants were backcrossed with N2 worms at least 3 times to remove background mutations, and the homozygous mutant progeny were used in the following experiments. The apn-1;exo-3, nth-1;exo-3, ung-1;exo-3 and nth-1;ung-1;exo-3 mutants were generated by crossing each strain. Deletion in the exo-3, apn-1, nth-1 and ung-1 genes was confirmed by PCR utilizing the same 2 primer pairs from a previous study14. In general, worms were cultured at 20 °C on NGM agar plates containing 0.3% (w/v) NaCl, 0.25% (w/v) polypeptone, 0.005% (w/v) cholesterol, 1 mM MgSO4, 1 mM CaCl2 and 25 mM potassium phosphate, at pH 6.0 with 0.17% (w/v) agar and a lawn of Escherichia coli (E. coli) OP50.
Synchronization of worms
To obtain synchronized eggs, we made gravid adult worms lay eggs for approximately 2 hours on the same plates used for subsequent assays. Synchronized eggs were transferred to new plates for each assay.
Bacteria-mediated RNA interference
For knockdown experiments, we used a well-established RNA interference (RNAi) method43,44. C. elegans dut-1, ndx-1, ndx-2, chk-2 (Y60A3A.12) and clk-2 (C07H6.6) complementary DNA (cDNA) clones were amplified by PCR from a cDNA library using the same 2 primer pairs from a previous study24,28. The amplified PCR products were subcloned into the plasmid L4440 for bacteria-mediated feeding RNAi (RNAi plasmid). Double RNAi experiments were performed by mixing an equal amount of overnight cultures of E. coli HT115 (DE3) that had been transformed with the respective RNAi plasmids, and then plating the mixture on NGM plates containing 0.1 mM IPTG and 100 μg/ml ampicillin (RNAi plates)24. As a negative control for RNAi, the transformant harboring L4440 was used.
Measurement of developmental speed
To assay the effects of AP endonuclease deficiency on larval development, synchronized eggs were placed on normal NGM plates, NGM plates containing 0.94 mM MMS or 10 mM NaHSO3, or RNAi plates. After incubation at 20 °C for 3, 3.5 (MMS) or 4 (NaHSO3) days, developmental stages of surviving worms were assessed based on vulval morphology and brooding of eggs to distinguish young adult worms from gravid adult worms. The proportion of worms at each developmental stage among the total number of surviving worms was calculated.
Measurement of the proportion of Pvl worms
To assay the effects of AP endonuclease deficiency on organogenesis, synchronized eggs were placed on dut-1 (RNAi) plates (plus additional RNAi and 0.1 mM methyl viologen (MV), if necessary). After incubation at 20 °C for 4 days, the numbers of total adult worms and Pvl worms were counted, and the proportion of Pvl worms among adult worms was calculated. Significance was determined using one-way ANOVA with Tukey’s test for multiple comparisons.
Microscopy
Observation and imaging of C. elegans were performed using an OLYMPUS SZX16 microscope (OLYMPUS, Japan).
Real-time PCR analysis
Worms (N2 at 0, 24, 48, 60 or 72 hr) were collected and lysed using TriPure isolation reagent (Roche, Basel, Switzerland). Then, total RNA was extracted from the above supernatant with NucleoSpin RNA (Takara Bio, Shiga, Japan), as recommended by the manufacturer. First-strand cDNA was synthesized from total RNA using oligo-dT primer and ReverTra Ace (Toyobo, Osaka, Japan). Real-Time PCR was performed using Light-Cycler 96 (Roche) with THUNDERBIRD SYBER qPCR Mix (Toyobo, Osaka, Japan). The thermal cycler conditions were as follows: an initial denaturation step at 95 °C for 60 sec, followed by 45 three-step PCR cycles of 95 °C for 15 sec, 60 °C for 30 sec and 72 °C for 45 sec. Gene amplification specificity was confirmed by melting curve analysis. The following are the primer sequences: APN1, 5′-GCTATCAGGAAATTGAAGCA-3′ and 5′-TCCAGTTTAGAGGTTTCTTC-3; EXO3, 5′-CGGAGATGGAGGAGACGTTTA-3′ and 5′-TCTGGGTCACCGATTCCTTTG-3′; Y45F10D.4, 5′-CGAGAACCCGCGAAATGTCGGA-3′ 5′-GCCTCATCTTCCCTGGCAACCG-3′. The two-tailed Student’s t-test was used for statistical analysis.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Levin, J. D. & Demple, B. Analysis of class II (hydrolytic) and class I (β-lyase) apurinic/apyrimidinic endonucleases with a synthetic DNA substrate. Nucleic Acids Res. 18, 5069–5075 (1990).
Winters, T. A., Henner, W. D., Russell, P. S., McCullough, A. & Jorgensen, T. J. Removal of 3′-phosphoglycolate from DNA strand-break damage in an oligonucleotide substrate by recombinant human apurinic/apyrimidinic endonuclease 1. Nucleic Acids Res. 22, 1866–1873 (1994).
Krokan, H. E., Standal, R. & Slupphaug, G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 325, 1–16 (1997).
Loeb, L. A. & Preston, B. D. Mutagenesis by apurinic apyrimidinic sites. Ann. Rev. Genet 20, 201–30 (1986).
Roos, W. P. & Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 12, 440–450 (2006).
Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. 98, 8241–8246 (2001).
Izumi, T. et al. Mammalian DNA base excision repair proteins: Their interactions and role in repair of oxidative DNA damage. Toxicology 193, 43–65 (2003).
Cunningham, R. P., Saporito, S. M., Spitzer, S. G. & Weiss, B. Endonuclease IV (nfo) mutant of Escherichia coli. J. Bacteriol. 168, 1120–1127 (1986).
Ramotar, D., Popoff, S. C., Gralla, E. B. & Demple, B. Cellular Role of Yeast Apnl Apurinic Endonuclease/3′-Diesterase: Repair of Oxidative and Alkylation DNA Damage and Control of Spontaneous Mutation. Mol. Cell. Biol. 11, 4537–4544 (1991).
Xanthoudakis, S., Smeyne, R. J., Wallace, J. D. & Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci USA 93, 8919–8923 (1996).
Wang, Y., Shupenko, C. C., Melo, L. F. & Strauss, P. R. DNA repair protein involved in heart and blood development. Mol. Cell. Biol. 26, 9083–9093 (2006).
Fung, H. & Demple, B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell 17, 463–470 (2005).
Georgiadis, M. M. et al. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 643, 54–63 (2008).
Kato, Y., Moriwaki, T., Funakoshi, M. & Zhang-Akiyama, Q. M. Caenorhabditis elegans EXO-3 contributes to longevity and reproduction: Differential roles in somatic cells and germ cells. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 772, 46–54 (2015).
Shatilla, A., Leduc, A., Yang, X. & Ramotar, D. Identification of two apurinic/apyrimidinic endonucleases from Caenorhabditis elegans by cross-species complementation. DNA Repair (Amst). 4, 655–670 (2005).
Shatilla, A., Ishchenko, A. A., Saparbaev, M. & Ramotar, D. Characterization of Caenorhabditis elegans exonuclease-3 and evidence that a Mg2+-dependent variant exhibits a distinct mode of action on damaged DNA. Biochemistry 44, 12835–12848 (2005).
Yang, X. et al. Functional characterization of the Caenorhabditis elegans DNA repair enzyme APN-1. DNA Repair (Amst). 11, 811–822 (2012).
Schlotterer, A. et al. Apurinic/apyrimidinic endonuclease 1, p53, and thioredoxin are linked in control of aging in C. elegans. Aging Cell 9, 420–432 (2010).
Zakaria, C. et al. Caenorhabditis elegans APN-1 plays a vital role in maintaining genome stability. DNA Repair (Amst). 9, 169–176 (2010).
Drabløs, F. et al. Alkylation damage in DNA and RNA - Repair mechanisms and medical significance. DNA Repair (Amst). 3, 1389–1407 (2004).
Shrivastav, N., Li, D. & Essigmann, J. M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 31, 59–70 (2009).
Hunter, S. E. et al. In vivo repair of alkylating and oxidative DNA damage in the mitochondrial and nuclear genomes of wild-type and glycosylase-deficient Caenorhabditis elegans. DNA Repair (Amst). 11, 857–863 (2012).
Burgers, P. M. & Klein, M. B. Selection by genetic transformation of a Saccharomyces cerevisiae mutant defective for the nuclear uracil-DNA glycosylase. J.Bacteriol. 166, 905–913 (1986).
Sanada, Y. & Zhang-Akiyama, Q. M. An increase of oxidised nucleotides activates DNA damage checkpoint pathway that regulates post-embryonic development in Caenorhabditis elegans. Mutagenesis 29, 107–114 (2014).
Lari, S. U., Chen, C. Y., Vertéssy, B. G., Morré, J. & Bennett, S. E. Quantative determination of uracil residues in Escherichia coli DNA: Contribution of ung. dug, and dut genes to uracil avoidance. DNA Repair (Amst). 5, 1407–1420 (2006).
Duncan, B. K. & Weiss, B. Specific mutator effects of ung (uracil-DNA glycosylase) mutations in Escherichia coli. J. Bacteriol. 151, 750–755 (1982).
Dengg, M. et al. Abrogation of the CLK-2 checkpoint leads to tolerance to base-excision repair intermediates. EMBO Rep. 7, 1046–1051 (2006).
Sanada, U. et al. NDX-1 protein hydrolyzes 8-oxo-7, 8-dihydrodeoxyguanosine-5′- diphosphate to sanitize oxidized nucleotides and prevent oxidative stress in Caenorhabditis elegans. J. Biochem. 150, 649–657 (2011).
Yonei, S., Noda, A., Tachibana, A. & Akasaka, S. Mutagenic and cytotoxic effects of oxygen free radicals generated by methylviologen (paraquat) on Escherichia coli with different DNA-repair capacities. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 163, 15–22 (1986).
Morinaga, H. et al. Purification and characterization of Caenorhabditis elegans NTH, a homolog of human endonuclease III: Essential role of N-terminal region. DNA Repair (Amst). 8, 844–851 (2009).
Ahn, J., Urist, M. & Prives, C. The Chk2 protein kinase. DNA Repair (Amst). 3, 1039–1047 (2004).
Guillet, M. & Boiteux, S. Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. EMBO J. 21, 2833–2841 (2002).
Matsuoka, S., Huang, M. & Elledge, S. J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science (80-.). 282, 1893–1897 (1998).
Zhao, H., Traganos, F. & Darzynkiewicz, Z. Phosphorylation of p53 on Ser15 during cell cycle caused by Topo I and Topo II inhibitors in relation to ATM and Chk2 activation. Cell Cycle 7, 3048–3055 (2008).
Khoronenkova, S. V. & Dianov, G. L. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc. Natl. Acad. Sci. 112, 3997–4002 (2015).
Eisenmann, D. M. & Kim, S. K. Protruding vulva mutants identify novel loci and wnt signalling factors that function during Caenorhabditis elegans vulva development. Genetics 156, 1097–1116 (2000).
Korcsmáros, T. et al. Signalogs: Orthology-based identification of novel signaling pathway components in three metazoans. PLoS One 6, (2011).
Flex, E. et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum. Mol. Genet. 23, 4315–4327 (2014).
Krokan, H. E. & Bjøra’s, M. Base Excision Repair. Cold Spring Harb Perspect Biol. 5, (2013).
Wallace, S. S. Base excision repair: A critical player in many games. DNA Repair (Amst). 19, 14–26 (2014).
SenGupta, T. et al. Base excision repair AP endonucleases and mismatch repair act together to induce checkpoint-mediated autophagy. Nat. Commun. 4, (2013).
Nakamura, N. et al. Cloning and characterization of uracil-DNA glycosylase and the biological consequences of the loss of its function in the nematode Caenorhabditis elegans. Mutagenesis 23, 407–413 (2008).
Timmons, L. & Fire, A. Specific interference by ingested dsRNA. Nature 395, 854 (1998).
Kamath, R. S. & Ahringer, J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–321 (2003).
Acknowledgements
We thank Dr. Elizabeth Nakajima for critically reading the manuscript. We also thank Dr. Hiroko Udaka for the helpful guidance on qPCR experiments. We appreciated the National Bioresource Project (NBRP) for the Experimental Animal “Nematode C. elegans” of Japan (Mitani laboratory). Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40OD010440). This work was financially supported in part by Grants-in-Aid for Scientific Research (16K00545) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to Q.-M. Zhang-Akiyama).
Author information
Authors and Affiliations
Contributions
M.M. and Q.Z. designed the project. M.M. designed experiments. M.M., Y.H., M.F. and A.T. performed research. All authors contributed to the analysis of the results. M.M. drafted the manuscript, and all authors refined the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miyaji, M., Hayashi, Y., Funakoshi, M. et al. AP endonuclease EXO-3 deficiency causes developmental delay and abnormal vulval organogenesis, Pvl, through DNA glycosylase-initiated checkpoint activation in Caenorhabditis elegans. Sci Rep 8, 16736 (2018). https://doi.org/10.1038/s41598-018-35063-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35063-6
- Springer Nature Limited