
Abstract
Staphylococcus aureus pathogenicity islands (SaPIs) are a type of mobile genetic element that play a significant role in the pathogenesis and virulence of this microorganism. SaPIs are integrated in the chromosome under the control of the master repressor Stl, but they can be horizontally transferred at a high frequency due to certain bacteriophages. Thus, a phage protein can bind to the SaPI Stl and induce the SaPI cycle, spreading the SaPI virulence factors to other bacterial populations. We report the dissemination mechanism of SaPIs mediated by endogenous prophages in S. aureus clinical strains. We reveal the induction of SaPIs by a co-resident prophage in seven clinically relevant strains, and we further study this mechanism in MW2, a community-acquired methicillin-resistant S. aureus strain that contains two bacteriophages (ɸSa2mw and ɸSa3mw) and one SaPI (SaPImw2) encoding for three enterotoxins (sec, sel and ear). ɸSa2mw was identified as responsible for SaPImw2 induction, and the specific phage derepressor protein DUF3113 was determined. The Stl-DUF3113 protein interaction was demonstrated, along with the existence of variants of this protein in S. aureus phages with different abilities to induce SaPI. Both Stl and DUF3113 are present in other Staphylococcus species, which indicates that this is a generalised mechanism.
Similar content being viewed by others

Introduction
Staphylococcus aureus is a widespread pathogen that colonises the skin and mucosa of healthy adults. S. aureus is found in both hospital and community environments, causing a variety of infectious diseases from minor skin infections to serious conditions such as osteomyelitis or endocarditis1,2. The high pathogenicity of this microorganism is due to the broad spectrum of virulence factors present in its genome. Remarkably, many of these virulence factors such as toxins, adhesins or invasins are part of its mobile genetic elements (MGE)3,4.
The acquisition of most MGEs is produced by horizontal transfer, Staphylococcus aureus pathogenicity islands (SaPIs) and the bacteriophages are two closely related MGEs that have been identified in S. aureus and whose mobilisation mechanisms have been studied4,5. SaPIs contain virulence genes that can be disseminated amongst bacteria populations by means of certain phages6,7. SaPIs act as molecular parasites, using different mechanisms to interfere with the normal biology of phages, to encapsidate in phage particles and to promote their own spread8,9,10,11,12.
SaPIs, as bacteriophages, can be packaged using two different mechanisms: pac and cos13. They can be packaged in full-sized procapsids by pac phages or packaged in DNA units delimited by cos sites in cos phages. Additionally, pac SaPIs can interfere with pac phages via several proteins such as TerS or CpmAB to reduce the capsid size and exclusively pack the island genome14,15,16. Recently, SaPIs that do not encode TerS or CpmAB homologues have been identified, they are cos SaPIs and they can use the packaging mechanisms of pac and cos phages17. These cos SaPIs encode for Ccm, which is responsible for small capsid formation and interferes with the packaging mechanism of cos phages18.
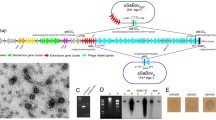
In general, SaPIs are repressed and integrated in the host chromosome. The normal repressed state of SaPIs is due to the master regulator Stl19. Stl acts as a repressor, binding to a region between two divergent promoters and inhibiting the transcription of SaPI genes20,21. The SaPI life cycle starts by the infection of a helper phage or by activation of an endogenous helper prophage by the SOS response. This response can be caused by oxidative stress, exposure to UV-irradiation or antibiotic treatment22,23,24. Once the prophage is activated or the cell is infected by the phage, a bacteriophage protein binds to Stl, triggering the release of the Stl repressor from the SaPI DNA. Disruption of the Stl-SaPI DNA complex allows the expression of SaPI proteins and the initiation of the excision-replication-packaging (ERP) cycle of the SaPI25 (Fig. 1).

SaPI cycle after prophage induction. (1) SOS response after mitomycin C addition. (2) The prophage is induced and start the gene transcription, while the SaPI resides passively in the genome under the control of the global repressor Stl. (3) The interaction between the Stl and a specific phage derepressor protein induces the SaPI cycle. (4) The SaPI gene transcription starts. (5) SaPI is excised from the chromosome in covalently closed circular (CCC) state and (6) starts the replication. (7) The phage synthetises the procapsids and tails. (8) SaPI L indicates linear monomers released from the phage heads if the prophage is able to mobilise the SaPI.
The interaction between Stl and the phage derepressor protein is the first step of the SaPI cycle. In the different SaPIs, Stl sequences are not conserved. On the other hand, derepressor proteins are specific for each interaction. Therefore, bacteriophages have different capacities to induce SaPIs according to whether they present derepressor proteins which are able to bind to specific Stls. All phage derepressor proteins studied so far are known as moonlighting proteins because they have more than one function, apart from their role in SaPI induction, a second function is supposed to be beneficial for the phage. The functions of four bacteriophage proteins have already been described. Dimeric and trimeric dUTPases of various phages are derepressor proteins of SaPIbov1 and SaPIbov5. Sri protein, which binds to DnaI and inhibits host cell replication, is the derepressor protein of SaPI1. SaPIbov2 was shown to be induced by ORF15 of the 80α phage, but its function still remains unknown25,26,27,28 and the single-strand annealing protein (SSAP) family, involved in homologous recombination, induces SaPI229. These four phage derepressors are able to induce only a few SaPIs. Furthermore, it has been shown that Stl repressors may have different domains to interact with unrelated phage proteins but perform the same conserved function29,30. Therefore, there are still many uncertainties about SaPI induction, and the identification of new derepressor proteins is key to understanding this induction mechanism and the subsequent dissemination to other bacteria.
SaPI induction by helper phages has been studied in phi11, phi12 and 80α lysogens, but there has been no demonstration of SaPI induction by endogenous phages in clinical strains. In this work, we demonstrate the ability of certain quiescent bacteriophages to induce resident SaPIs. We studied the SaPI induction process in 14 S. aureus clinical strains and focused on SaPImw2 induction in the MW2 strain. MW2 is a community-acquired methicillin-resistant S. aureus (MRSA) strain that presents 19 unique toxins31,32, some of them encoded in the SaPImw2 pathogenicity island. We demonstrated that the ɸSa2mw phage is able to induce SaPImw2, and we identified the phage derepression protein responsible for the induction and horizontal transference of the SaPImw2 genome. Finally, derepressor homologous proteins were identified in various phages. All of the proteins showed different abilities to interact with Stl and, consequently, different capacity to induce SaPImw2.
Results
SaPI induction by endogenous phages in clinical strains
SaPI induction by prophages has been extensively studied in laboratory strains, mainly in lysogens from different phages in the non-lysogenic strains RN4220 and RN450. To confirm whether this mechanism occurs naturally, a total of 14 S. aureus clinical strains were treated with mitomycin C to activate the SOS response and induce endogenous phages to check whether these phages are able to activate the resident SaPIs (Fig. 1). The clinical S. aureus strains MRSA252, E-MRSA16, Mu50, TW20, MN8, USA300, COL, N315, MW2, A, B, C, D and E33, were selected because of their broad characterisation and because their genomes have already been sequenced. All of these strains contain SaPIs and bacteriophages integrated in the chromosome (Table 1).
SaPI induction was tested by Southern blot, as indicated in the methods section. The blots showed that 7 strains contain prophages able to induce the resident SaPIs (Fig. 2). In MW2, C and USA300 strains the hybridization signal of the specific probe for each SaPI was observed in the bulk DNA as well as in a band that corresponds to SaPI linear monomers (L) released from the small phage capsids, as it was observed in previous works, in presence of a phage able to induce and encapsidate the SaPI25,34. Instead, a second band in A, E, N315 and COL strains, at the bottom of the gel (Fig. 2), is the covalently closed circular molecules (CCC) which correspond to the SaPI induced but not packaged25,34,35.

SaPI induction of S. aureus clinical strains by endogenous phages. In total, 14 strains were treated with mitomycin C (2 µg/mL) to activate the SOS response and induce the resident prophages. Samples were isolated at 0 and 90 minutes, separated in agarose gel and blotted with the SaPI integrase-specific probe. The upper band corresponds to chromosomal bulk DNA including SaPI, and the lower bands correspond to covalently closed circular SaPI (CCC) excised from the genome or SaPI monomers released from the phage capsid (L) if the helper phage mediated induction and packaging.
To explain the absence of an L SaPI band in A, E, N315 and COL strains, corresponding with SaPIs that have been packaged, the SaPI and prophage sequences of the clinical strains were analysed to identify whether they are cos or pac types (Table 1). pac SaPIs contain a group of genes related to phage packaging and interference, known as Operon I, and a small terminase that recognises the unique pac site. In contrast, cos SaPIs present a highly conserved phage cos site and a different packaging and interference module (Operon 1 like), and lacks the small terminase. The differences in migration and the assignment in two types (CCC or L) of the lower band among the analysed strains are consistent with the presence of cos and pac SaPIs and phages. MW2, C and USA300 encode cos SaPI and cos phages with similar cos sites, so the SaPIs can be induced and mobilised by their endogenous phages. A, E, N315 and COL contain pac SaPIs and cos phages, so there might be induction but not packaging in these strains.
After induction, SaPI is excised from the chromosome and circularised. To clearly confirm that SaPI induction has occurred, SaPI excision from the chromosome was confirmed by PCR using specific primers recognising the flanking region of each SaPI integrase type (Supplementary Fig S1). After treatment with mitomycin C, all of the strains that produced a band that migrated at a position suggesting SaPI induction by Southern blot analysis could be confirmed as the excised product by PCR. Additionally, SaPI circularisation was tested by PCR using a pair of primers set divergently at both termini of each SaPI. Only the strains positive for SaPI induction were tested, and all showed a band indicating SaPI circularisation (Supplementary Fig. S2).
These results confirm that the SaPIs present in the strains A, E, C, MW2, USA300, COL and N315 are able to excise from the chromosome and circularisation after induction.
Analysis of the global repressor stl sequences of the 7 induced SaPIs revealed that the stl gene is highly divergent among different SaPIs19 (Supplementary Fig. S3). Some SaPIs contain identical stls such as USA300 and COL or MW2 and C. The SaPI from strain A contains an stl, which has already been described, and the derepressor was identified as a Sri protein25. A Redβ protein was recently identified as the derepressor of SaPI-N31529. In contrast, the other Stl repressor have not yet been studied, and the phage proteins responsible for interaction with them remain unknown (Table 1).
In this study, we paid special attention to the SaPI mobilisation mechanism of these clinical strains. We focused on identifying the phage protein responsible for induction of the SaPIs present in the C and MW2 strains, which encode for the same stl. MW2 is a community-acquired methicillin-resistant S. aureus strain that encodes multiple virulence factors, some of them in SaPImw2. Consequently, it is important to understand how this SaPI is mobilised and how its virulence factors are spread.
ɸSa2mw from MW2 induces SaPImw2 replication and transfer
To follow SaPImw2 replication and mobilisation, a chloramphenicol (Cm) marker was introduced in the SaPI genome, which resulted in strain GTM750 (Supplementary Table S1). The chloramphenicol acetyl transferase (cat) gene was inserted, replacing enterotoxin type C (sec4), an accessory gene of the pathogenicity island. SaPImw2 belongs to a SaPI subfamily in which the classical SaPI operon I is not present and contains a conserved phage cos site sequence (cggcgggggc)17,18,35. As SaPImw2 is a cos SaPI, in the replacement, the SaPI genome size must not be affected, because the SaPI size genome is a prerequisite for high-frequency SaPI transfer by cos phages18.
ɸSa2mw and ɸSa3mw phages were identified in the human clinical strain MW2 genome. ɸSa2mw is a cos phage that presents the same cos site sequence as that of SaPImw2. For this reason, we hypothesised that ɸSa2mw could be responsible for SaPImw2 mobilisation. To examine this idea, we generated ɸSa2mw lysogen in the non-lysogenic strain RN4220, which yielded strain GTM752. In addition, SaPImw2 entC::cat was transduced into the GTM752 strain, which generated GTM753.
SaPImw2 induction of GTM753 was tested by Southern blot (Fig. 3A), and we demonstrated that ɸSa2mw has the ability to mobilise SaPImw2 entC::cat to a recipient strain RN4220 from GTM753 (Table 2). On the other hand, SaPIs interfere with the reproduction of their helper phage, blocking plaque formation and reducing plaque number36,37. The phage titre was reduced by more than one order of magnitude in presence of SaPImw2 entC::cat (GTM753) with regard to RN4220 ɸSa2mw (GTM752), which confirmed the interference. The relationship between SaPI and the bacteriophage is a parasitism relationship, in which SaPI benefits but the phage is impaired (Table 2). All these results indicate that ɸSa2mw is responsible for the SaPImw2 induction and packaging in small capsids.

(A) Southern blot of φSa2mw, φ12, φ11 and 80α lysogens in SaPImw2-positive strains (blotted with SaPI integrase type III probe) (B). Diagram of the derivative pCN41 (pGTM2), which contains the regulatory region of SaPImw2 fused to blazTT. (C) The lysogen of phages φSa2mw, φ12, φ11 and 80α containing pGTM2 were assayed for β-lactamase activity after 2 h of induction with mitomycin C (2 µM). Data are result of three independent experiments.
Previous studies have demonstrated that the interaction of a phage protein with the Stl repressor interrupts the binding of Stl to the intergenic region of the SaPI regulation module, which causes initiation of the SaPI cycle25. To demonstrate this mechanism for MW2, we constructed the plasmid pGTM2 (Supplementary Table S2), a derivative pCN41 with the reporter gene β-lactamase fused with the intergenic regulatory region of SaPImw2, including the stl gene (Fig. 3B). pGTM2 was introduced into the lysogenic strain GTM752 of ɸSa2mw, which created GTM754. The strain was then induced with mitomycin C, and the expression of the β-lactamase reporter was measured as indicated in the methods section. As is shown in Fig. 3C, expression of the β-lactamase reporter gene appeared when phage ɸSa2mw was induced, so we concluded that ɸSa2mw activates transcription by specifically disrupting the Stl–DNA complex.
DUF3113 is responsible for derepression of SaPImw2
To identify which protein of ɸSa2mw could be responsible for the interaction with Stl and, therefore, for SaPImw2 induction, a total of 7 regions of ɸSa2mw were cloned into the plasmid pGTM3 (pCU1_Pcad) (Fig. 4A). Due to the previously described inducers are encoded in the replication module, the 7 selected regions correspond to this region. The cloning resulted in plasmids pGTM4 to pGTM10, which were introduced into GTM755 (RN4220 pGTM2) and induced with CdCl2 (2 µM). As shown in Fig. 4B, pGTM8 was responsible for the activity of β-lactamase. The region of pGTM8 is composed of 9 open reading frames (ORFs), from MW1424 to MW1416. All of these ORFs were cloned individually into pGTM3 (pCU1_Pcad), generating 9 new plasmids (from pGTM11 to pGTM19), and were transformed into GTM755 (RN4220 pGTM2). In this case, pGTM11 showed β-lactamase activity and corresponded to ORF MW1424. This ORF encodes for a hypothetical protein, DUF3113 (UniProtKB code A0A0H3JWD1_STAAW), which belongs to the DUF3113 superfamily.

(A) Diagram of the ɸSa2mw genome with the cloned regions marked. (B) β-lactamase activity assay of the derivative pGTM3 (pCU1_Pcad) with different regions of φSa2mw in GTM754 (RN4220 φSa2mw pGTM2) strains. (C) Southern blot of GTM805 containing SaPImw2 and complemented with a plasmid overexpressing DUF313ɸSa2mw. One millilitre of the culture was collected after 2 h of induction with 2 μM of CdCl2. The lower band corresponds to CCC because there was no helper phage present. Southern blot was performed with the wild-type strains (GTM750 and GTM753) and the DUF3113 mutant deletion in MW2 (GTM756) and GTM753 (GTM757). Both Southern blots were hybridised with a SaPI integrase type III probe.
Previous studies have indicated that SaPIs protect bacteria from exogenous phage-mediated infection, as long as this exogenous phage is able to induce the SaPI cycle37. Furthermore, it has also been noted that phages with mutations in the derepressor gene are able to form plaques in strains that contain an inducible SaPI29,36,37. We demonstrate in this study that SaPImw2 blocks plaque formation by ɸSa2mw. However, after several attempts to infect a SaPImw2-positive strain (GTM751) with ɸSa2mw lysate, two plaques were obtained. The sequence of the DUF3113 derepressor gene from the phage genomes obtained from the plaques showed mutations in the ribosome-binding site and one mutation in the start codon (atg to atc; M1I), which suggests that this gene is responsible for SaPImw2 induction (Supplementary Fig. S4).
Overexpression of DUF3113 by cloning into a vector pCN51, producing pGTM20, and transforming into the RN4220 SaPImw2 entC::cat, generating GTM805 strain, confirmed that DUF3113 is the unique protein responsible for SaPImw2 induction (Fig. 4C). Moreover, an inframe deletion using allelic exchange with the pMAD vector in this gene was carried out in GTM750 (MW2 SaPImw2 entC::cat) and in GTM753 (RN4220 ɸSa2mw SaPImw2 entC::cat), which generated GTM756 and GTM757, respectively. To confirm our previous results, SaPImw2 mobilisation was tested by Southern blot. No SaPI specific band was detected when DUF3113 was not present (Fig. 4C).
Finally, to confirm that DUF3113 is responsible for SaPImw2 mobilisation, SaPI and phage titres were studied by induction with mitomycin C of the ɸSa2mw lysogen wt and DUF3113 mutant strain and in the absence and presence of SaPImw2 entC::cat (Table 2). Phage titres indicate that the phage mutant seems to be as viable as the wild type. Therefore, DUF3113 gene is not essential for the phage in laboratory conditions. On the other hand, the mutant phage was not able to transduce SaPImw2entC::cat, but when DUF3113 was overexpressed (pGTM20) in the mutant strain (GTM759), the SaPI titer was fully restored.
DUF3113 interacts with SaPImw2 Stl
SaPI induction occurs through direct protein interaction between the phage derepressor protein and the SaPI repressor Stl25,38. To test the in vitro interaction between DUF3113ɸSa2mw and StlSaPImw2, a size-exclusion chromatography assay was performed, and all the relevant peak fractions were analysed by SDS-PAGE. His6-tagged StlSaPImw2 and His6-tagged DUF3113ɸSa2mw were expressed (plasmids pGTM22 and pGTM23) and purified. Figure 5A shows three chromatograms of His6-StlSaPImw2, His6-DUF3113ɸSa2mw and the complex His6-StlSaPImw2-His6-DUF3113ɸSa2mw. The His6-StlSaPImw2 chromatogram (blue) showed two peaks (PK116 and PK165), and the His6-DUF3113ɸSa2mw chromatogram (green) showed one peak (PK187). However, the complex His6-StlSaPImw2 - His6-DUF3113ɸSa2mw (orange) eluted in 3 peaks: PK116, PK135, PK187. PK116 and PK187 corresponded to specific elution volumes from free His6-StlSaPImw2 and His6-DUF3113ɸSa2mw, respectively. However, the new peak, PK135, was associated with the protein complex, as was confirmed by SDS-PAGE.

(A) Size-exclusion chromatography assays. Blue: Free His6-StlSaPImw2 chromatogram (PK116 and PK165). Green: His6-DUF3113ɸSa2mw chromatogram (PK187). Orange: Chromatogram corresponding to the interaction between His6-StlSaPImw2 and His6-DUF3113ɸSa2mw. As confirmed by SDS-PAGE (top insert), PK116 belonged to His6-StlSaPImw2, PK187 to His6-DUF3113ɸSa2mw and PK135 to the complex between the two proteins. (B) Size exclusion chromatography of the pull-down assays. Orange: Chromatogram corresponding to the pull-down of His6-StlSaPImw2 with untagged DUF3113ɸSa2mw, in which one peak was observed for PK135, corresponding to the interaction of both proteins. Blue: Chromatogram of the pull-down of His6-StlSaPImw2 with untagged DUF3113ɸSa2c. Two peaks, PK116 and PK165, corresponding to free His6-StlSaPImw2 were observed. All peaks were analysed by SDS-PAGE. Inj1: injection of His6-StlSaPImw2 (32,5 KDa) and His6-DUF3113ɸSa2mw (10.6 KDa); Inj2: injection of His6-StlSaPImw2; Inj3: injection of His6-DUF3113ɸSa2mw; Inj4: injection corresponding to the pull-down of His6-StlSaPImw2 - untagged DUF3113ɸSa2mw (7.1 KDa); and Inj5: injection corresponding to the of pull-down His6-StlSaPImw2-untagged DUF3113ɸSa2c (7.1 KDa). A degradation product from protein His6-StlSaPImw2 was observed in PK165 (asterisk).
In addition, to confirm the Stl-DUF3113 interaction, pull-down assays were performed. For this, His6-tagged StlSaPImw2 and untagged DUF3113ɸSa2mw were expressed (plasmids pGTM22 and pGTM24), and His6-tagged StlSaPImw2 was bound to a metal affinity resin and incubated with untagged DUF3113ɸSa2mw. The co-elution of both proteins was confirmed by SDS-PAGE (Supplementary Fig. S5) and by size-exclusion chromatography (Fig. 5B). Again, a peak at 135 mL was detected, corresponding to the interaction of both proteins, as confirmed by SDS-PAGE. The negative control, untagged DUF3113ɸSa2mw, was not retained by the resin (data not shown). Furthermore, to check the specificity of StlSaPImw2-DUF3113ɸSa2mw, the Stl of SaPIbov1, which is not induced by DUF3113ɸSa2mw, was used as a control (His6-StlSaPIbov1). This protein construct was also expressed and incubated with untagged DUF3113ɸSa2mw; however, in this case, no interaction was detected (Supplementary Fig S5). Finally, the interaction between His6-StlSaPImw2 and DUF3113ɸSa2c, which does not derepress SaPImw2 (see below), was evaluated and, as expected, they did not co-elute (Fig. S5). The lack of interaction between these two proteins was also confirmed by size-exclusion chromatography (Fig. 5B). Here, only two peaks (PK116 and PK165) were detected, corresponding to His6-StlSaPImw2.
All these results indicate that DUF3113ɸSa2mw binds directly and specifically to StlSaPImw2.
Diverse DUF3113 phage proteins display different capacities to induce SaPImw2
Because there are significant differences in the bacteriophage capacities to induce SaPIs8, the ability of different phages such as 80α, ɸ11 and ɸ12 to induce SaPImw2 was tested by Southern blot (Fig. 3A). ɸ11 cannot induce SaPImw2, whereas 80α and ɸ12 are able to induce SaPImw2. ɸ12 is a cos phage like ɸSa2mw and contains the same cos sequence. In the same way as that shown for ɸSa2mw, SaPImw2 induction by ɸ12 showed a lower band corresponding to a SaPI monomer packaged in small capsids. Most likely, this was due to the presence of the ccm gene in SaPImw2, which is responsible for conducting packaging in small capsids. On the other hand, Ccm protein is responsible for cos phage interference18. In contrast, 80α is a pac phage, and there was no lower band after induction, probably because SaPImw2 does not encode for CpmA and CpmB, which are responsible for the formation of smaller capsids than those normally generated by the pac phages.
The transducing titres of SaPImw2 entC::cat (SaPI titres) showed that 80α and ɸ12 mobilised this SaPI successfully. As expected, ɸ11 was unable to transfer SaPImw2 (Table 2). To study the capacity of the phages to bind to StlSaPImw2 and increase β-lactamase expression, the plasmid pGTM2 was introduced in 80α, ɸ11 and ɸ12 lysogens (Fig. 3C). All tested phages showed different abilities to derepress the regulatory region and induce SaPImw2.
An in silico approach was carried out to look for DUF3113ɸSa2mw homologues in these phages and check whether the allelic variants of DUF3113 were also able to induce SaPImw2. The phage ɸ12 contains a protein with 100% identity with DUF3113ɸSa2mw. Phages 80α and ɸ11 encode DUF3113ɸSa2mw homologues in ORF25 and ORF19, respectively, both with 61% amino acid identity. As mentioned earlier, strain C, with a resident SaPI also mobilised by an endogenous bacteriophage (Fig. 2), carries two prophages in its genome. These phages have been designated ɸ2c and ɸ6c in this study. Both phages also present DUF3113ɸSa2mw homologues with 98% and 58% amino acid identity, respectively (Fig. 6A).

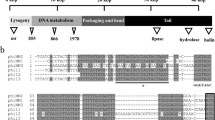
(A) Sequence alignment of DUF3113 homologues in S. aureus phages using PRALINE45. The scoring scheme assigns 0 as the least-conserved alignment position, with values up to 10 (asterisk) for the most-conserved alignment position (B). Southern blot of SaPImw2 excision after induction of 3xFLAG-tagged homologues to the DUF3113 protein from different phages cloned into pCN51 after 2 h of CdCl2 treatment. (C) Western blot of 3xFLAG-tagged DUF3113 homologues cloned into pGTM3 (pCU1_ Pcad) with the same RBS (D). Sequence alignment of DUF3113 homologues of different staphylococcal species.
To determine whether the different DUF3113 proteins are able to induce SaPImw2, DUF3113 genes from phages 80α, ɸ11, ɸ2c and ɸ6c were tagged with 3xflag, cloned into the plasmid pCN51 and introduced into the RN4220 SaPImw2entC::cat (GTM751) strain. SaPImw2 induction was tested by Southern blot after treatment with CdCl2 (Fig. 6B). DUF3113 from phages 80α and ɸ6c induced SaPImw2, while the homologous protein from ɸ11 and ɸ2c could not induce it.
As in the previous experiment using lysogens, the β-lactamase expression was measured after cloning DUF3113 homologues genes from different phages into pGTM3 (pCU1_Pcad) tagged with 3xflag and then transformed into GTM755 (RN4220 pGTM2). The β-lactamase activity varied between the tested strains, which supported the Southern blot results (Fig. 6B). However, after Western blotting was performed, differences in the levels of protein expression were observed. These differences could be due to either the expression level of the derepressor phage proteins or to differences in the amino acid sequences of these proteins. To this end, we cloned the homologous proteins tagged with 3xflag with the same ribosome binding site (RBS of DUF3113) into pCN51 and pCN41 plasmids, which were then transformed into GTM751 and GTM755 (RN4220 pGTM2), respectively. Southern blot showed the same differences in SaPImw2 induction as those seen in Fig. 6B when the proteins were equally expressed (Fig. 6C). Different allelic variants differ in their capability to derepress SaPImw2 due to differences in the ability to bind to StlSaPImw2.
To determine whether DUF3113 is also present in another Staphylococcus spp, an in silico study was performed. We found DUF3113 protein in phages from Staphylococcus argenteus, Staphylococcus warneri, Staphylococcus haemolyticus, Staphylococcus schweitzeri and Staphylococcus simiae (Fig. 6D). In addition, we discovered an S. argenteus clinical strain, BN75, which contains a SaPI that encodes for a Stl similar to StlSaPImw2 (99% of identity) and a phage with a DUF3113 protein similar to DUF3113ɸSa2mw (61% identity). In the same way, we located a SaPI that encodes for a Stl and a phage with a DUF3113 in the S. aureus strain TCH70 showing 100% identity to present in MW2. These results demonstrate that the mechanism described is general in Staphylococcus spp.
Discussion
SaPIs encode for virulence factors such as superantigens, epidermolytic toxins, biofilm formation genes, and iron transport or antibiotic-resistance genes (penicillin, fosfomycin and fusidic acid). Therefore, these MGE have a high clinical relevance34. For this reason, it is very important to know how these SaPIs are mobilised and, consequently, how they spread their virulence factors among bacteria of the same species and even among bacteria of different genera. In this paper, we characterised the mobilisation mechanism of SaPImw2 carrying genes for enterotoxin sec, sel and ear. The last of the listed genes is linked to ampicillin resistance.
The Stl sequences of the SaPIs are not conserved among different families, and, therefore, each SaPI family is induced by different phage proteins25. There are many SaPIs with unknown inducers. It has recently been demonstrated that the Stl sequences of certain SaPIs have acquired multiple domains to bind to different phage inductor proteins29. On the other hand, SaPIs and bacteriophages are in continuous co-evolution. Subsequently, much remains to be understood about the SaPI induction mechanism.
Prophages and SaPIs are abundant in the S. aureus genome. Most strains contain more than one prophage and at least one SaPI39. Based on the fact that SaPI interference mechanisms are counterselective for the phages and that the phages capable of relieving Stl repression are not able to form plaques on a SaPI-containing strain25, endogenous phages able to induce co-resident SaPIs are considered unlikely to be found. However, this study showed that SaPI induction by endogenous phages naturally occurs in human clinical S. aureus isolates, as we demonstrated for the MW2, USA300, COL, N315, A, C and E strains.
There are some possible explanations for this finding. SaPIs have different interference mechanisms that negatively affect the phage packaging machinery. On the one hand, pac SaPIs encode for the genes cpmA, cpmB, ppi, ptiA, ptiB and ptiM, which are responsible for interference with pac phage packaging. On the other hand, cos SaPIs, which encode the ccm gene, cause severe interference in cos phage packaging18. However, there are no described interference mechanisms between pac SaPIs and cos phages. Therefore, in strains COL, N315, A and E, in which the SaPIs are pac and their co-resident phages are cos, bacteriophages may have been acquired by horizontal transfer regardless of the SaPI presence, since they were not affected by interference mechanisms.
In contrast, MW2, as described, contains the cos SaPI SaPImw2, which is induced and packaged by the cos phage ɸSa2mw. In this case, we think that SaPImw2 may have been acquired after the phage.
Finally, strain C has two cos prophages and one cos SaPI. We describe that the cos SaPIc is induced by cos ɸ6c, but probably, this phage is not responsible for packaging because the cos site and its flanking region, which is important for mobilisation and packaging, are completely different between SaPIc and ɸ6c. However, ɸ2c does present a cos site and its flanking region with 89% identity with SaPIc, which suggests that this phage is probably able to mobilise this SaPI. Therefore, the assumption is that ɸ6c is the responsible for the induction, while ɸ2c is mainly involved in packaging. Phage major capsid protein (CP) is the target for Ccm SaPI protein interference. These two proteins present in the C-terminal have high structural similarity18. In our case, Ccm SaPIc presents a high structural similarity with CPɸ2c and no similarity with CPɸ6c. Therefore, it is probable that SaPIc may only interfere with ɸ2c (Supplementary Fig. S7). In this case, we suggest two possibilities: the SaPI was the last transferred or, more interestingly, the ɸ6c may be the last acquired, since it is not affected by interference mechanisms.
DUF3113, identified as the SaPImw2 derepressor, is a small, non-essential protein and is sufficient for SaPImw2 derepression. All derepressor phage proteins have a dual role: a primary function related to the phage biology and a secondary function related to SaPI mobilisation. The best studied example is the dUTPase protein, which prevents the incorporation of uracil into DNA and is able to mobilise SaPIbov125,28,38,40,41. DUF3113 is a protein with an unknown function for the phage. In our study, deletion of the gene did not change phage replication (data not shown) and did not decrease the phage titre, which indicates that in laboratory conditions, DUF3113 is not an essential protein.
In other phages, DUF3113-homologous proteins were chosen to try to understand the interaction mechanism with the Stl repressor and subsequent SaPI induction. DUF3113ɸ11 and DUF3113ɸ2c variants did not derepress SaPImw2. Instead, DUF311380α and DUF3113ɸ6c variants were more effective at inducing SaPImw2 than DUF3113ɸSa2mw. The higher capacity to induce SaPImw2 corresponds with greater affinity for the Stl repressor. Overexpression of DUF311380α and DUF3113ɸ6c variants in a SaPImw2-positive strain affected bacterial growth because SaPIs overexpression has a high cost for the bacteria. On the other hand, overexpression of DUF3113ɸSa2mw produced normal bacterial growth of the SaPI-positive strains, even though there was SaPImw2 induction. Possibly, the lower capacity to induce the SaPI cycle caused SaPI and the bacteriophage to be in equilibrium within MW2.
As previously mentioned, strain C presents two prophages that seem to be hijacked by SaPIc, which is mobilised in a coordinated way. ɸ6c induces the SaPI, and ɸ2c is involved in packaging. Phage collaboration can also be suspected in the MW2 strain. The SaPI titre from MW2 (ɸSa2mw and ɸSa3mw) was three orders of magnitude higher than that of ɸSa2mw lysogen, which indicates that although ɸSa2mw is responsible for SaPImw2 induction, ɸSa3mw increases SaPImw2 mobilisation. In this case, both phages are cos phages, but the cos site sequence of ɸSa2mw is identical to that of SaPImw2, so this phage could both induce and encapsidate SaPImw2. However, lysis of the MW2 culture after mitomycin C treatment was higher than that in the ɸSa2mw lysogen. This suggests that ɸSa3mw is only involved in lysis, which improves the mobilisation rate.
In conclusion, our study demonstrates the existence of SaPI mobilisation in clinical S. aureus strains. Additionally, the study shows signs of a new SaPIs strategy using the machinery of more than one phage in a coordinated way to increase SaPI transduction. A new derepressor protein has been characterised, but more extensive studies are necessary to identify other derepressor proteins and to better understand the mechanism of SaPI mobilisation and the consequent transfer of virulence factors.
Methods
Bacterial growth
The bacterial strains used in this study are listed in Supplementary Table S1. S. aureus strains were cultured in tryptic soy broth (TSB) or agar (TSA) plates supplemented with erythromycin (10 μg /ml, Sigma-Aldrich) or chloramphenicol (20 μg /ml, Sigma-Aldrich) as needed. Escherichia coli were grown in Luria-Bertani broth (LB) supplemented with ampicillin (100 μg /ml, Sigma-Aldrich) or kanamycin (50 μg /ml, Sigma-Aldrich) as appropriate. Both were grown at 37 °C.
Phage isolation and transduction
S. aureus strains were grown up to OD540: 0.2–0.3 and were then treated with mitomycin C (2 μg /ml) to activate the SOS response and induce the resident prophages. Bacterial cultures were incubated at 32 °C and 80 rpm for 4 h or until lysis occurred. The cultures were filtered (0.22 µm filters), and the obtained phage lysates were diluted and spotted on the susceptible prophage-free RN4220 strain42. Phage from the centre of an individual plaque were extracted. PCR of the phage integrases was performed to check the lysogeny. GTM752 was obtained using this method.
To test the phage-induced SaPI mobilisation (SaPI o transducing titre), different dilutions of the filtered lysates (100 µl) were used to infect 1 mL of the acceptor strain RN4220 up to OD540 = 1.4 at 37 °C for 15 min and plated in TSA containing the corresponding antibiotic. After 24 h at 37 °C, the colonies were counted.
DNA Methods
DNA manipulations were performed using standard procedures. The primers used in this study are listed in Supplementary Table S3.
Southern blot was performed by standard procedures43. For sample preparation, S. aureus strains were grown to OD540: 0.2–0.3 and were then induced with mitomycin C (2 μg/ml). The cultures were incubated at 32 °C and 80 rpm. Samples were taken 90 minutes after phage induction. Then, 1 ml of the sample was centrifuged, and the pellet was resuspended in 50 µL lysis buffer (47.5 µl of TES-sucrose: 10 mM Tris, 1 mM EDTA, 100 mM NaCl, 0.5 M sucrose and 2.5 µl of lysostaphin (5 mg/ml, Sigma-Aldrich)). After 30 min at 37 °C, 47.5 µl of 2% SDS and 2.5 µl of proteinase K (20 mg/ml) were added, and the mixture was incubated at 55 °C for 30 min. Next, 11 µL of loading buffer dye 10X was added, and the mixture was shaken for 20 min. The samples were cold (N2)/heat (65 °C) treated three times.
The standard SDS minilysates were resolved on a 0.7% agarose gel at 20 V overnight, transferred to a nylon membrane (Roche) via capillary action and blotted with specific Digoxigenin-labelled probes and anti-DIG antibodies. Specific primers (Supplementary Table S3) for SaPI integrase genes were used to generate DIG-labelled probes. Detection was carried out according to the Immun StarTM AP substrate pack (BIO-RAD).
For Western blot, the strains were treated with CdCl2 (2 μM) and harvested (1 ml) after 2 h. The pellets were resuspended in 200 μl lysis buffer (50 mM Tris-HCl, 20 mM MgCl2, 30% raffinose and 1 μl of lysostaphin) and incubated 1 h at 37 °C. SB buffer with β-mercaptoethanol was added, and the samples were heated 10 min at 95 °C. Samples were electrophoresed on a 15% SDS-PAGE gel, transferred to a PVDF membrane and incubated with 5% non-fat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) for 60 min. The membrane was washed with TBST, incubated with anti-FLAG antibodies (Sigma-Aldrich) and revealed according to the protocol supplied by the manufacturer.
To delete the DUF3113 gene and introduce the Cm marker in SaPImw2, we used the plasmid pMAD44, as previously described25. pGTM1 was constructed by 3-piece and pGTM21 by 2-piece overlap assembly PCR. The primer pairs used are listed in Supplementary Table S3. The whole fragments were subsequently cloned into the pMAD vector, and the plasmids were transformed by electroporation into S. aureus RN4220 and transduced to MW2 and different lysogens. Allelic replacement was carried out by a two-step procedure. First, the pMAD plasmid was integrated into the chromosome by homologous recombination under non-permissive conditions (44 °C), and in the second step, the plasmid was avoided under a permissive temperature (30 °C). Mutation was confirmed by DNA sequencing.
Plasmid construction
The plasmids expressing allelic variants of DUF3113 proteins were prepared by cloning PCR products obtained with the oligonucleotide primers listed in Supplementary Table S3. Proteins were expressed in S. aureus under inducing conditions from the cadmium-inducible promoter (Pcad) in the expression vector pCN51, as previously described25.
β-lactamase activity assay
The plasmid pCN41 was used to clone the intergenic regulatory region of SaPImw2 fused to the β-lactamase reporter gene contained in this plasmid, which generated pGTM2. A cadmium-inducible promoter was introduced into the pCU1 plasmid and used to overexpress allelic variants of the DUF3113 protein. Plasmids were electroporated into RN4220 pGTM2, and the transformed strains were grown up to OD540: 0.2 and induced with CaCl2 (1 µM). Lysogens of different phages were also transformed with pGTM2 and induced with mitomycin C. Samples were taken after 2 h for a β-lactamase activity assay using nitrocefin as a substrate. Changes of the OD at 490 nm were registered during 20 min (every 30 s) in the Synergy H1 microplate reader. β-lactamase activity units were calculated using the formula (ΔVmax(OD490)*1000/min)/OD650.
Protein expression and purification
Stl and DUF3113 genes were amplified by PCR and cloned into the pET28a vector. The primers were designed to generate an N-terminal 6xHis-tag fusion Stl protein and a 6xHis-tag fusion DUF3113 protein. The plasmids were transformed into E. coli BL21(DE3). E. coli cells containing the recombinant plasmids were propagated in 500 ml of LB broth (50 µg/ml kanamycin). Cultures were grown until the OD650 reached 0.6–0.8, then isopropyl-β-D-thiogalactopyranoside (IPTG, Thermo Scientific) was added to reach a concentration of 1 mM. After induction, the cultures were grown overnight at 16 °C and 200 rpm. Cells were harvested by centrifugation (5000 g for 10 min at 4 °C) and resuspended in phosphate buffer (20 mM Na-phosphate pH 7,4, 300 mM NaCl, 10% glycerol and 0,1 mM PMSF). The cells were lysed by sonication (Amplitude 70% 3 min: 3 s ON and 10 s OFF) and centrifuged (11000 g for 10 min). Supernatants were applied to a metal affinity resin (TALON, Clontech) and incubated for one hour at RT. The hexahistidine-tagged proteins were eluted with 100–500 mM imidazole gradient buffers. Elution fractions were analysed by SDS/PAGE and by size-exclusion chromatography.
For pull-down assays, the primers were designed to generate N-terminal 6xHis-tag fusion Stl proteins and untagged DUF3113 proteins. The 6xHis-tag Stls were incubated for 1 h with metal affinity resin and for 1 h with untagged DUF3113 proteins. Pull-down elution was performed with 250 mM imidazole buffer. Elution fractions were analysed by SDS/PAGE and by size-exclusion chromatography.
Size-exclusion chromatography
A SuperdexTM 75 26/60 column running on a ÄKTATM pure25 system (GE Healthcare Life Sciences) was used to study the interaction of the selected proteins. The column was equilibrated in 20 mM phosphate buffer containing 300 mM NaCl at pH 7.4, and the elution was performed at 2.6 ml/min at 25 °C. The protein concentration was 17 μM for 6xHis-tag StlSaPImw2 and 40 μM for 6xHis-tag DUF3113ɸSa2mw. The injection volume was 7.5 mL. Calibration of the column was performed with aldolase (158 KDa), covalbumina (75 KDa), BSA (66 KDa), ovalbumina (44 KDa), dimer ribonuclease (27.4 KDa), dimer ubiquitin (17 KDa), ribonuclease (13.5 KDa) and ubiquitin (8.5 KDa) (Supplementary Fig S6). Peak protein fractions were analysed by SDS-PAGE.
References
Riewerts Eriksen, N. H., Espersen, F., Thamdrup Rosdahl, V. & Jensen, K. Carriage of Staphylococcus aureus among 104 healthy persons during a 19-month period. Epidemiol. Infect. 115, 51–60 (1995).
Wertheim, H. F. et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5, 751–762 (2005).
Lindsay, J. A. & Holden, M. T. G. Staphylococcus aureus: Superbug, super genome? Trends in Microbiology 12, 378–385 (2004).
Ruzin, A., Lindsay, J. & Novick, R. P. Molecular genetics of SaPI1–a mobile pathogenicity island in Staphylococcus aureus. Mol. Microbiol. 41, 365–77 (2001).
Malachowa, N. & Deleo, F. R. Mobile genetic elements of Staphylococcus aureus. Cellular and Molecular Life Sciences 67, 3057–3071 (2010).
Tallent, S. M., Langston, T. B., Moran, R. G. & Christie, G. E. Transducing particles of Staphylococcus aureus pathogenicity island SaPI1 are comprised of helper phage-encoded proteins. J. Bacteriol. 189, 7520–7524 (2007).
Tormo, M. Á. et al. Staphylococcus aureus pathogenicity island DNA is packaged in particles composed of phage proteins. J. Bacteriol. 190, 2434–2440 (2008).
Maiques, E. et al. Role of staphylococcal phage and SaPI integrase in intra- and interspecies SaPI transfer. J. Bacteriol. 189, 5608–5616 (2007).
Chen, J. & Novick, R. P. Phage-Mediated Intergeneric Transfer of Toxin Genes. Science (80-.). 323, 139–141 (2009).
Ram, G., Chen, J., Ross, H. F. & Novick, R. P. Precisely modulated pathogenicity island interference with late phage gene transcription. Proc. Natl. Acad. Sci. 111, 14536–14541 (2014).
Ram, G., Chen, J., Ross, H. F. & Novick, R. P. An insight into staphylococcal pathogenicity island-mediated interference with phage late gene transcription. Bacteriophage 5, e1028608 (2015).
Penadés, J. R. & Christie, G. E. The Phage-Inducible Chromosomal Islands: A Family of Highly Evolved Molecular Parasites. Annu. Rev. Virol. 2, 181–201 (2015).
Feiss, M. & Rao, V. B. The bacteriophage DNA packaging machine. Adv. Exp. Med. Biol. 726, 489–509 (2012).
Ubeda, C. et al. Specificity of staphylococcal phage and SaPI DNA packaging as revealed by integrase and terminase mutations. Mol. Microbiol. 72, 98–108 (2009).
Damle, P. K. et al. The roles of SaPI1 proteinsgp7 (CpmA) and gp6 (CpmB) in capsid size determination and helper phage interference. Virology 432, 277–282 (2012).
Ram, G. et al. Staphylococcal pathogenicity island interference with helper phage reproduction is a paradigm of molecular parasitism. Proc. Natl. Acad. Sci. 109, 16300–16305 (2012).
Quiles-Puchalt, N. et al. Staphylococcal pathogenicity island DNA packaging system involving cos-site packaging and phage-encoded HNH endonucleases. Proc. Natl. Acad. Sci. 111, 6016–6021 (2014).
Carpena, N., Manning, K. A., Dokland, T., Marina, A. & Penade, R. Convergent evolution of pathogenicity islands in helper cos phage interference. https://doi.org/10.1098/rstb.2015.0505 (2016).
Úbeda, C. et al. SaPI mutations affecting replication and transfer and enabling autonomous replication in the absence of helper phage. Mol. Microbiol. https://doi.org/10.1111/j.1365-2958.2007.06027.x (2008).
Papp-Kádár, V., Szabó, J. E., Nyíri, K. & Vertessy, B. G. In Vitro analysis of predicted DNA-binding sites for the Stl repressor of the Staphylococcus aureus SaPIBov1 pathogenicity island. PLoS One 11, (2016).
Surányi, É. V. et al. Exploiting a phage-bacterium interaction system as a molecular switch to decipher macromolecular interactions in the living cell. Viruses 10, (2018).
Witkin, E. M. et al. Ultraviolet mutagenesis and inducible DNA repair in Escherichia coli. Bacteriol. Rev. 40, 869–907 (1976).
Maiques, E. et al. β-lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J. Bacteriol. 188, 2726–2729 (2006).
Smith, C. L. & Oishi, M. Early events and mechanisms in the induction of bacterial SOS functions: analysis of the phage repressor inactivation process in vivo. Proc Natl Acad Sci USA 75, 1657–1661 (1978).
Tormo-Más, M. Á. et al. Moonlighting bacteriophage proteins derepress staphylococcal pathogenicity islands. Nature 465, 779–782 (2010).
Dearborn, A. D. & Dokland, T. Mobilization of pathogenicity islands by Staphylococcus aureus strain Newman bacteriophages. Bacteriophage 2, 70–78 (2012).
Hill, R. L. L. et al. Derepression of SaPIbov1 Is Independent of φNM1 Type 2 dUTPase Activity and Is Inhibited by dUTP and dUMP. J. Mol. Biol. 429, 1570–1580 (2017).
Leveles, I. et al. Structure and enzymatic mechanism of a moonlighting dUTPase. Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 2298–2308 (2013).
Bowring, J. et al. Pirating conserved phage mechanisms promotes promiscuous staphylococcal pathogenicity island transfer. Elife 6, e26487 (2017).
Nyíri, K., Kohegyi, B., Micsonai, A., Kardos, J. & Vertessy, B. G. Evidence-based structural model of the staphylococcal repressor protein: Separation of functions into different domains. PLoS One 10, (2015).
Salgado-Pabon, W. et al. Staphylococcus aureus beta-toxin production is common in strains with the beta-toxin gene inactivated by bacteriophage. J. Infect. Dis. 210, 784–792 (2014).
Baba, T. et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359, 1819–1827 (2002).
Viana, D. et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat. Genet. 47, 361–6 (2015).
Novick, R. P., Christie, G. E. & Penadés, J. R. The phage-related chromosomal islands of Gram-positive bacteria. Nature Reviews Microbiology 8, 541–551 (2010).
Chen, J. et al. Intra- and inter-generic transfer of pathogenicity island-encoded virulence genes by cos phages. ISME J. 9, 1260–1263 (2015).
Lindsay, J. A., Ruzin, A., Ross, H. F., Kurepina, N. & Novick, R. P. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 29, 527–543 (1998).
Frígols, B. et al. Virus Satellites Drive Viral Evolution and Ecology. PLoS Genet. 11, (2015).
Tormo-Más, M. ángeles et al. Phage dUTPases Control Transfer of Virulence Genes by a Proto-Oncogenic G Protein-like Mechanism. Mol. Cell 49, 947–958 (2013).
Novick, R. P. & Ram, G. The Floating (Pathogenicity) Island: A Genomic Dessert. Trends in Genetics 32, 114–126 (2016).
Vértessy, B. G. & Tóth, J. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 42, 97–106 (2009).
Alite, C. et al. Dissecting the link between the enzymatic activity and the SaPI inducing capacity of the phage 80α dUTPase. Sci. Rep. 7, (2017).
Kreiswirth, B. N. et al. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–12 (2011).
Russell, D. W. & Sambrook, J. Molecular cloning: A laboratory manual. Cold Spring Harbour (2001).
Arnaud, M., Chastanet, A. & Débarbouillé, M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891 (2004).
Simossis, V. A. & Heringa, J. PRALINE: A multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 33, (2005).
Acknowledgements
We thank José Rafael Penadés Casanova for helpful comments on the manuscript. This work was supported by grants RYC-2012-12246, SAF2017-82251-R, SAF2014-56986-R and Consolider-Ingenio CSD2009-00006 to M.A.T.-M., SAF2017-89229-R to A.P.-L., and BES-2015-073651 to M.C-A. from the Ministerio de Economia y Competitividad (Spain).
Author information
Authors and Affiliations
Contributions
Designed the study: M.A.T.-M. Performed the experiments: M.C-A., K.G.-M., M.Z., L.O and P.B-Q. Analysed the data: M.A.T.-M., M.C-A., A.P.L and L.O. Supervised the research: M.A.T.-M., A.P.L. and J.P. Wrote the paper: M.A.T.-M. and M.C-A. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cervera-Alamar, M., Guzmán-Markevitch, K., Žiemytė, M. et al. Mobilisation Mechanism of Pathogenicity Islands by Endogenous Phages in Staphylococcus aureus clinical strains. Sci Rep 8, 16742 (2018). https://doi.org/10.1038/s41598-018-34918-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34918-2
- Springer Nature Limited
Keywords
This article is cited by
-
Molecular characterization and antibiotic resistance of Staphylococcus aureus strains isolated from patients with diarrhea in Korea between the years 2007 and 2022
Food Science and Biotechnology (2024)
-
Isolation and characterization of novel Staphylococcus aureus bacteriophage Hesat from dairy origin
Applied Microbiology and Biotechnology (2024)
-
Multi-drug resistant bacteria isolates from lymphatic filariasis patients in the Ahanta West District, Ghana
BMC Microbiology (2022)