
Abstract
The highly conserved Rap1 GTPases perform essential functions during neuronal development. They are required for the polarity of neuronal progenitors and neurons as well as for neuronal migration in the embryonic brain. Neuronal polarization and axon formation depend on the precise temporal and spatial regulation of Rap1 activity by guanine nucleotide exchange factors (GEFs) and GTPases-activating proteins (GAPs). Several Rap1 GEFs have been identified that direct the formation of axons during cortical and hippocampal development in vivo and in cultured neurons. However little is known about the GAPs that limit the activity of Rap1 GTPases during neuronal development. Here we investigate the function of Sema3A and Plexin-A1 as a regulator of Rap1 GTPases during the polarization of hippocampal neurons. Sema3A was shown to suppress axon formation when neurons are cultured on a patterned substrate. Plexin-A1 functions as the signal-transducing subunit of receptors for Sema3A and displays GAP activity for Rap1 GTPases. We show that Sema3A and Plexin-A1 suppress the formation of supernumerary axons in cultured neurons, which depends on Rap1 GTPases.
Similar content being viewed by others


Introduction
Small GTPases of the Ras superfamily perform essential functions throughout neuronal development and in mature neurons1. The highly conserved Rap1 GTPases encoded by the Rap1a and Rap1b genes in mammals are required for the polarity of neuronal progenitors and neurons as well as for neuronal migration in the embryonic brain1,2,3,4,5. In the developing brain, newborn neurons that initially have a multipolar morphology become polarized by forming an axon and a leading process1,6,7,8,9. In culture, dissociated neurons from the embryonic hippocampus or cortex undergo a similar differentiation but polarize without the need for a patterned exogenous signal7,10. After attaching to the culture substrate neurons first extend several undifferentiated neurites (stage 2 of neuronal polarization) before one of them becomes an axon and extends rapidly. The inactivation of Rap1 GTPases impairs the formation of axons during cortical and hippocampal development in vivo and in cultured neurons1,11. Neuronal polarization and axon formation depend on the precise temporal and spatial regulation of Rap1 activity by GEFs and GAPs. Rapgef1 (also called C3G), Rapgef2 and Rapgef6 have been identified as the Rap1 GEFs that are required for the development of the neocortex and hippocampus12,13,14,15,16. However little is known about the GAPs that limit the activity of Rap1 GTPases during neuronal development14.
The plexins are integral membrane proteins with an intracellular domain that shows sequence similarity to GAPs with dual specificity for Ras and Rap GTPases17,18,19,20,21. The mutation of conserved arginine residues in this GAP domain is sufficient to abolish their activity in vitro and in vivo20,21,22,23. Plexins are receptors for the semaphorins, a large family of secreted and membrane-bound proteins that act as axon guidance signals but also perform important functions in other tissues24,25,26. The nine plexins in mammals can be subdivided into four subfamilies (PlexinA to -D). In vitro assays first showed that Plexin-B1 acts as a GAP for R- and M-Ras but not H-Ras27,28,29,30,31. Subsequently, a structural analysis of the GAP domain combined with biochemical assays demonstrated that Plexin-A1 and -C1 specifically regulate Rap1 and Rap2 GTPases20,21,32,33,34. The phenotype of plexin mutants confirmed that GAP activity is essential for their function in vivo and provided evidence for a regulation of Ras and Rap1 GTPases22,23,35.
The A-type plexins act as the signal-transducing subunit of receptors for the secreted Sema3A in a complex with Neuropilin-1 as the ligand binding subunit24,36,37,38,39,40. Semaphorins perform important functions not only during axon guidance but also in the regulation of neuronal migration and the formation of axons, dendrites and synapses25,36,41,42,43. Sema3A directs the orientation of axons and apical dendrites in the developing cortex44,45,46,47. It can also regulate the establishment of neuronal polarity by promoting the formation of dendrites and suppressing the extension of axons when neurons are cultured on a patterned substrate with stripes of immobilized Sema3A48,49. In cultures of sensory neurons from embryonic dorsal root ganglia, Sema3A accelerates the establishment of neuron polarity50. It remains to be investigated if plexins regulate the activity of Rap1 GTPases during neuronal polarization.
Here we show that Sema3A and Plexin-A1 suppress the formation of supernumerary axons in cultured neurons. Hippocampal but not cortical neurons from Sema3A knockout embryos form multiple axons in culture. While inactivation of Plexin-A1 results in the formation of supernumerary axons constitutively active Plexin-A1 inhibits axon formation. These effects can be rescued by the knockdown of Rap1B and the expression of active Rap1B, respectively, indicating that Plexin-A1 suppresses axon formation by regulating Rap1 GTPases.
Results
Hippocampal neurons from Sema3a knockout mice form multiple axons
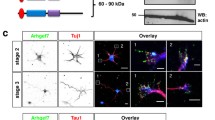
To determine if the establishment of neuronal polarity is affected in the absence of Sema3A neurons were isolated from the hippocampus and cortex of E17 Sema3a−/− and Sema3a+/− embryos and analyzed by staining with an anti-MAP2 and the Tau-1 antibody as markers for dendrites and axons, respectively (Fig. 1a,c). The majority (60 ± 5%) of the Sema3−/− neurons extended several long neurites that were positive for both axonal and dendritic markers at day 3 days in culture (3 days in vitro, d.i.v.) compared to only 9 ± 4% in cultures from Sema3a+/− embryos as control (Fig. 1c). To evaluate axon formation with a second axonal marker, cultures were analyzed at 4, 5 and 6 d.i.v. by staining with the Tau-1 or an anti-Ankyrin G (AnkG) antibody as a marker for the axonal initial segment (AIS) (Fig. 1b,c). A large proportion of the Sema3a−/− neurons formed multiple Tau-1 positive axons (44 ± 1%) and multiple AIS (43 ± 1%) at 4 d.i.v. while only few were observed in Sema3a+/− controls (axons: 11 ± 2%; AIS: 8 ± 1%). The number of Sema3a−/− neurons with multiple Tau-1-positve axons increased to 63 ± 3% (AnkG: 58 ± 1%) at 5 d.i.v. and 71 ± 1% (AnkG: 73 ± 1%) at 6 d.i.v. (Fig. 1c). Thus, Sema3A-deficient neurons from the embryonic hippocampus showed defects in neuronal polarization. Neurons extended multiple neurites that initially display mixed axonal and dendritic characteristics at 3 d.i.v. but become supernumerary axons at 4 d.i.v. When cultures of cortical neurons from Sema3a+/− and Sema3a−/− embryos were analyzed no significant defect in neuronal polarization was observed at any of the analyzed time points (Fig. 1a,c). These results show that the knockout of Sema3a induces the formation of supernumerary axons by hippocampal but not cortical neurons.

Hippocampal neurons from Sema3a−/− embryos extend supernumerary axons in culture. (a–c) Cultures of hippocampal or cortical neurons from E17 Sema3a+/− or Sema3a−/− embryos were analyzed at 3, 4, 5 and 6 d.i.v. by staining with an anti-MAP2 (green, dendrites) and the Tau-1 (a, red, axons) or an anti-AnkG antibody (b, red, AIS marked by arrow heads). Representative images of hippocampal neurons at the indicated times in culture are shown. The scale bar is 25 μm. (c) The percentage of unpolarized hippocampal neurons without an axon or AIS (0, red), polarized neurons with a single axon or AIS (1, blue), neurons with multiple axons or AIS (>1, green) and neurons with neurites that are positive for both axonal and dendritic markers (in, yellow) is shown (Student’s t-test and two-way ANOVA; n = 3 independent experiments with >150 neurons per genotype; values are means ± s.e.m., **p < 0.01, ***p < 0.001 compared to control as indicated).
The knockdown of Sema3A induces cell-autonomous defects
Secreted Sema3A tightly binds to the cell surface of the producing cells and can act as an autocrine signal51. To investigate this possibility we used an miRNA expression vector to knock down Sema3A in cultured neurons. Since only a small number of neurons were transfected, a knockdown reduces the concentration of Sema3A in the medium only minimally compared to cultures from knockout embryos. The efficiency of the Sema3A knockdown construct was confirmed by Western blot after co-expression with tagged Sema3A in HEK 293 T cells (Fig. 2a) and by immunofluorescence staining of cultured hippocampal neurons (Suppl. Fig. S1a). Hippocampal neurons were transfected with the Sema3A knockdown vector and analyzed at 3 d.i.v. (Fig. 2b,c). Suppression of Sema3a induced the formation of multiple Tau-1 positive axons in 45 ± 1% of the neurons compared to 8 ± 2% in controls. By contrast, non-transfected neurons in the same culture where not affected by the knockdown indicating a cell-autonomous function of Sema3A in cultured neurons (Suppl. Fig. S1b). The phenotype of the Sema3A knockdown could be rescued by the expression of an RNAi-resistant Sema3A construct (Sema3A-res; Fig. 2) confirming the specificity of the knockdown. Only 15 ± 2% of the neurons that were co-transfected with the RNAi vector and the expression vector for Sema3A-res extended multiple axons and 78 ± 3% formed a single axon. These results suggest that Sema3A acts as an autocrine signal to suppress the formation of supernumerary axons.

Knockdown of Sema3A induces the formation of supernumerary axons. (a) HEK 293 T cells were transfected with vectors for FLAG-Sema3A (S3A) or RNAi-resistant FLAG-Sema3A-res (S3Ares) and an shRNA directed against Sema3A (S3A RNAi) or pcDNA6.2-GW/EmGFP-miR (Ctrl). The expression of Sema3A and GFP was analyzed by Western blot (WB) using an anti-FLAG antibody. The molecular weight is indicated in kDa. (b) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green), an shRNA against Sema3A (Sema3A RNAi) or pcDNA6.2-GW/EmGFP-miR (control), and a vector for RNAi-resistant FLAG-Sema3A-res (Sema3A-res) or pBK-CMV (control) as indicated. Neurons were analyzed at 3 d.i.v. by staining with an anti-MAP2 (red, dendrites) and the Tau-1 antibody (blue, axon). Representative images of transfected neurons are shown. The scale bar is 25 μm. (c) The percentage of unpolarized neurons without an axon (0, red), polarized neurons with a single axon (1, blue) and neurons with multiple axons (>1, green) is shown (Student’s t-test and two-way ANOVA; n = 3, independent experiments with >150 neurons per experiment; values are means ± s.e.m, ***p < 0.001 compared to control as indicated; n.s., not significant).
Plexin-A but not Plexin-B activity is required for neuronal polarity
Sema3A acts through receptors that contain an A-type plexin as the signal transducing subunit17,26,52,53. To investigate whether plexins are involved in the regulation of neuronal polarization, we used a Plexin-A1 construct (Plexin-A1Δcyt) with a deletion of the intracellular domain that has a dominant-negative effect by forming non-functional receptor complexes19,39,52. Neurons from the hippocampus of E18 rat embryos were transfected with vectors for Plexin-A1Δcyt and analyzed at 3 d.i.v. by staining with an anti-MAP2 and the Tau-1 antibody (Fig. 3a). Expression of Plexin-A1Δcyt increased the number of neurons with multiple axons from 7 ± 1% in controls to 61 ± 2% (Fig. 3b). Deletion of the semaphorin domain releases Plexin-A1 from its auto-inhibited state and results in a constitutively active receptor (Plexin-A1Δsema)54. Expression of Plexin-A1Δsema had the opposite effect of dominant-negative Plexin-A1. The majority of neurons were negative for Tau-1 staining and the percentage of unpolarized neurons was increased to 69 ± 3% (control: 9 ± 1%; Fig. 3b). These results indicate that interfering with the function of A-type plexins disrupts the establishment of neuronal polarity.

A-type plexins suppress the formation of axons. (a,b) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green, control), dominant-negative Plexin-B1Δcyt (PlxnB1-DN), Plexin-A1Δcyt (PlxnA1-DN) or constitutively active PlexinA1Δsema (PlxnA1-CA) and analyzed at 3 d.i.v. by staining with an anti-MAP2 (red, dendrites) and the Tau-1 antibody (blue, axon). Representative images of transfected neurons are shown. The scale bar is 25 μm. (b) The percentage of unpolarized neurons without an axon (0, red), polarized neurons with a single axon (1, blue) and neurons with multiple axons (>1, green) is shown (Student’s t-test and two-way ANOVA; n = 3, independent experiments with >150 neurons per experiment for; values are means ± s.e.m, ***p < 0.001 compared to control; n.s. not significant).
To test a possible involvement of B-type plexins in axon formation, we used dominant-negative Plexin-B1Δcyt. After expression of Plexin-B1Δcyt, hippocampal neurons polarize normally and extend a single Tau-1 positive axon (Fig. 3a). No significant difference was detectable between controls (neurons with a single axon: 85 ± 4%) and the expression of Plexin-B1Δcyt (82 ± 25%; Fig. 3b). Thus, inhibition of Plexin-B1 has no effect on axon formation.
Suppression of Plexin-A1 induces multiple axons
The dominant-negative Plexin-A1Δcyt construct may affect all receptors that include a member of the Plexin-A subfamily. To identify which A-type plexin regulates neuronal polarity, we used established RNAi vectors for PlexinA1 - A441. Rat hippocampal neurons were transfected with these RNAi vectors and axon formation was analyzed at 3 d.i.v. (Fig. 4a,b). After knockdown of Plexin-A1, the number of neurons with multiple axons increased from 7 ± 2% in controls to 51 ± 4% (Fig. 4b). In addition, 20 ± 4% of the neurons formed neurites that were positive for both axonal and dendritic markers. By contrast, after knockdown of Plexin-A2, -A3 or -A4 no significant increase in the extension of supernumerary axons was observed (neurons with a single axon: Plexin-A2: 77 ± 4%, Plexin-A3: 74 ± 3%; Plexin-A4: 72 ± 2%; control: 77 ± 5%). Staining with an anti-Plexin-A1 antibody confirmed that the knockdown efficiently suppressed the expression of Plexin-A1 that is present in all neurites of polarized hippocampal neurons (Fig. 4c). The formation of supernumerary axons after knockdown of Plexin-A1 could be reversed by co-expressing murine Plexin-A1 that contains mismatches in the RNAi target site compared to the rat sequence confirming the specificity of the Plexin-A1 knockdown in rat neurons (Suppl. Fig. S2). These results show that Plexin-A1 is required during neuronal polarization to prevent the formation of supernumerary axons.

Knockdown of Plexin-A1 induces the formation of supernumerary axons. (a,b) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green) and shRNAs against Plexin-A1, -A2. -A3, or -A4 or pSUPER (control) and analyzed at 3 d.i.v. by staining with an anti-MAP2 (red, dendrites) and the Tau-1 antibody (blue, axon). Representative images of transfected neurons are shown. The scale bar is 25 μm. (b) The percentage of unpolarized neurons without an axon (0, red), polarized neurons with a single axon (1, blue), neurons with multiple axons (>1, green) and neurons with neurites that are positive for both axonal and dendritic markers (in, yellow) is shown (Student’s t-test and two-way ANOVA; n = 3, independent experiments with >150 neurons per experiment; values are means ± s.e.m, **p < 0.01 compared to control). (c) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green) and an shRNA against Plexin-A1 (RNAi) or pSUPER (control) and analyzed at 3 d.i.v. by staining with an anti-Plexin-A1 (red) and the Tau-1 antibody (blue, axon). Representative images of transfected neurons are shown. The scale bar is 25 μm.
Plexin-A1 acts upstream of Rap1 GTPases during neuronal polarization
Structural and biochemical analyses showed that Plexin-A1 regulates Rap1 GTPases that are required for the formation of axons1,11,20,21. The inactivation of the Rap1 GAP Plexin-A1 may, therefore, result in the extension of supernumerary axons due to increased Rap1 activity. To investigate this possibility, we tested if the formation of multiple axons after expression of dominant-negative Plexin-A1Δcyt can be blocked by a knockdown of Rap1B. Hippocampal neurons were co-transfected with vectors for Plexin-A1Δcyt and an shRNA directed against Rap1B11 and analyzed at 3 d.i.v. (Fig. 5a). After knockdown of Rap1B, only 19 ± 4% of the transfected neurons extended an axon compared to 84 ± 3% in controls (Fig. 5b) as shown before11. The induction of supernumerary axons by Plexin-A1Δcyt was prevented by the knockdown of Rap1B (unpolarized neurons: 26 ± 5%, neurons with a single axon: 66 ± 3%, neurons with multiple axons: 8 ± 4%) indicating that it depends on Rap1B. The specificity of the Rap1B knockdown was verified by co-expression of an Rap1B construct with mismatches in the RNAi target site that rescued the loss of axons after Rap1B knockdown (Suppl. Fig. S3).

Knockdown of Rap1B counteracts the induction of supernumerary axons by dominant-negative Plexin-A1. (a) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green, control), an shRNA against Rap1B or pSHAG-1 (control) and Plexin-A1Δcyt (PlxnA1-DN) or pBK-CMV (control). The establishment of neuronal polarity was analyzed at 3 d.i.v. by staining with an anti-MAP2 (red, dendrites) and the Tau-1 antibody (blue, axons). Representative images of transfected neurons are shown. The scale bar is 25 μm. (b) The percentage of unpolarized neurons without an axon (0, red), polarized neurons with a single axon (1, blue) and neurons with multiple axons (>1, green) is shown (Student’s t-test and two-way ANOVA; n = 3, independent experiments with >150 neurons per experiment for; values are means ± s.e.m, **p < 0.01; ***p < 0.001 compared to control and as indicated.
The expression of constitutively active Plexin-A1Δsema has an effect that is similar to that of a Rap1B knockdown and leads to the loss of axons. We tested if this phenotype can be rescued by the expression of active Rap1BV12. As reported before11, expression of Rap1BV12 increased the number of hippocampal neurons with multiple axons (53 ± 1%) compared to controls (17 ± 1%) while expression of Plexin-A1Δsema increased the number of unpolarized neurons (51 ± 2%, control: 11 ± 1%; Fig. 6a,b). The suppression of axon formation by PlexinA1Δsema could be rescued by co-expression of Rap1BV12, which increased the percentage of polarized neurons with a single axon from 43 ± 1% (only PlexinA1Δsema) to 63 ± 4% (PlexinA1Δsema and Rap1BV12, Fig. 6b). These results indicate that Plexin-A1 acts upstream of Rap1 GTPases and restricts their activity.

Active Rap1B recues the suppression of axon formation by constitutively active Plexin-A1. (a) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green) and Plexin-A1Δsema (PlxnA1-CA), Rap1BV12 or a combination of both and analyzed at 3 d.i.v. by staining with an anti-MAP2 (red, dendrites) and the Tau-1 antibody (blue, axon). pBK-CMV and pSHAG-1 were transfected as control. Representative images of transfected neurons are shown. The scale bar is 25 μm. (b) The percentage of unpolarized neurons without an axon (0, red), polarized neurons with a single axon (1, blue) and neurons with multiple axons (>1, green) is shown (Student’s t-test and two-way ANOVA; n = 3, independent experiments with >150 neurons per experiment; values are means ± s.e.m, **p < 0.01; ***p < 0.001 compared to control and as indicated).
To confirm that Rap1 GTPases act downstream of Plexin-A1 we tested if the formation of supernumerary axons caused by the knockdown of Plexin-A1 can be prevented by the knockdown of Rap1B. After transfection of hippocampal neurons with the shRNA vector directed against Plexin-A1, 48 ± 1% of the transfected neurons extended multiple axons (Fig. 7a,b). This number was reduced to 14 ± 1% when the shRNA against Rap1B was cotransfected with the Plexin-A1 shRNA and the majority of the neurons (70 ± 3%) extended a single Tau-1 positive axon. These results show that Rap1B is required for the induction of supernumerary axons by the knockdown of Plexin-A1. Taken together, our results show that Plexin-A1 acts upstream of Rap1B and is required to restrict its activity in hippocampal neurons.

Rap1B is required for the induction of supernumerary axons by the knockdown of Plexin-A1 in hippocampal neurons. (a) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green) and Plexin-A1Δsema (PlxnA1-CA), Rap1BV12, pSHAG-1 (control) or a combination of both and analyzed at 3 d.i.v. by staining with an anti-MAP2 (red, dendrites) and the Tau-1 antibody (blue, axon). Representative images of transfected neurons are shown. The scale bar is 25 μm. (b) The percentage of unpolarized neurons without an axon (0, red), polarized neurons with a single axon (1, blue) and neurons with multiple axons (>1, green) is shown (Student’s t-test and two-way ANOVA; n = 3, independent experiments with >150 neurons per experiment for; values are means ± s.e.m, **p < 0.01; ***p < 0.001 compared to control and as indicated).
Rap1B acts downstream of Sema3A in hippocampal neurons
Sema3A-deficient neurons extend multiple axons similar to the phenotype of neurons transfected with a vector for constitutively active Rap1BV12. To investigate if Rap1B acts downstream of Sema3A we tested if the knockdown of Rap1B prevents the formation of supernumerary axons after knockdown of Sema3A. 32 ± 5% of the transfected neurons extended multiple axons (Fig. 8) that were also positive for Rap1 after the knockdown of Sema3A (Suppl. Fig. S4). This number was reduced to 18 ± 2% with the majority of the neurons (67 ± 6%) extending a single Tau-1 positive axon when Rap1B was knocked down together with Sema3A. These results show that the formation of supernumerary axons in Sema3A-deficient neurons depends on Rap1B.

Rap1B acts downstream of Sema3A in hippocampal neurons. (a) Hippocampal neurons from E18 rat embryos were transfected with vectors for GFP (green), an shRNA against Sema3A (S3A RNAi) or pcDNA6.2-GW/EmGFP-miR (control) and an shRNA against Rap1B (R1B RNAi) or pSHAG-1 (control) as indicated. Neurons were analyzed at 3 d.i.v. by staining with an anti-MAP2 (red, dendrites) and the Tau-1 antibody (blue, axon). Representative images of transfected neurons are shown. The scale bar is 25 μm. (b) The percentage of unpolarized neurons without an axon (0, red), polarized neurons with a single axon (1, blue) and neurons with multiple axons (>1, green) is shown (Student’s t-test and two-way ANOVA; n = 3, independent experiments with >150 neurons per experiment for; values are means ± s.e.m, ***p < 0.001 compared to control as indicated).
Discussion
Our results suggest that Sema3A acts as an autocrine signal through Plexin-A1, which restricts the activity of Rap1 and thereby prevents the formation of supernumerary axons. Hippocampal neurons from Sema3a−/− knockout mice form multiple axons in culture. Suppressing Plexin-A1 function by a dominant-negative construct or a knockdown had a similar effect while expression of constitutively active Plexin-A1 blocked axon formation. The defects in neuronal polarity were rescued by the knockdown of Rap1B and the expression of active Rap1BV12, respectively, which is consistent with the activity of Plexin-A1 as a GAP for Rap120,21. These results indicate that Rap1 GTPases act downstream of Plexin-A1 and are required for its function to suppress the formation of supernumerary axons.
Our results are consistent with previous reports that Sema3A suppresses axon formation48,49. Hippocampal neurons express Sema3A44,55 that remains tightly bound to the surface of producing cells after secretion by binding to proteoglycans51. A knockdown of Sema3A induces the formation of supernumerary axons only in transfected neurons while non-transfected neurons in the same culture are not affected. The co-expression of an RNAi-resistant Sema3A rescues the Sema3A knockdown and prevents the formation of supernumerary axons. These results suggest that Sema3A acts primarily in a cell-autonomous manner as an autocrine factor as described for other cell types56,57,58,59,60. Sema3a knockout neurons extend multiple long neurites that are initially positive for both axonal and dendritic markers at 3 d.i.v. before they become axons by 4 d.i.v. while most neurons extend multiple axons already at 3 d.i.v. after expression of dominant-negative Plexin-A1 and knockdown of Plexin-A1 or Sema3A. The major reason for this difference in the time course probably is the time point at which Sema3A/Plexin-A1 signaling is inhibited. Sema3A acts like a global inhibitory signal that suppresses axon formation48,61. It is absent from the beginning in knockout neurons while it is transiently produced after a knockdown, which may explain why knockout neurons initially extend neurites with both dendritic and axonal properties before these become axons.
In contrast to Plexin-A1, we did not observe defects in neuronal polarity after expression of dominant-negative Plexin-B1. The analysis of Plexin-B1 suggested that it acts as a GAP for R-Ras and M-Ras27,28,29,30. Different Ras GTPases have been implicated in axon formation29,62,63. The activation of Plexin-B1 by Sema4D down-regulates the activity of R- and M-Ras in the growth cones of axons and dendrites of cultured hippocampal neurons, respectively27,30,31,32. However, neither single nor double knockouts of Ras GTPases show defects in brain development64, indicating that they are not required for neuronal polarity in vivo or that their loss is compensated by other Ras GTPases. In vivo studies indicate that B-type plexins perform different functions during neuronal development22,65,66,67,68,69. Knockout of Plxnb1 and Plxnb2 results in a reduced proliferation of neural progenitors and cortical thinning, which could indicate a function in regulating the orientation of the mitotic spindle as shown for epithelial cells22,67,69. Further studies are required to elucidate which GTPases are regulated by the different plexins in vivo and what determines their specificity. It also remains to be investigated whether other GAPs are involved in mediating the function of Sema3A or the regulation of Rap1 GTPases during neuronal polarization.
Unlike in hippocampal neurons, axon formation was not affected in cortical neurons. The differential effects of the Sema3a knockout may result from differences in the expression of semaphorin receptors and other semaphorins like Sema3C may act in the developing neocortex. Sema3C is expressed in migrating neurons in the cortex and its overexpression interferes with their polarization and radial migration43. Sema3A could be required specifically in hippocampal neurons to suppress axon formation. A difference between hippocampus and cortex was observed also for the Rap1a;Rap1b knockout1. The conditional knockout of Rap1 GTPases at different time points showed that an inactivation late during neuronal polarization interferes with axon formation in the hippocampus but not in the cortex. The hippocampus-specific functions of Sema3A and Rap1 GTPases could be linked to the extended time that hippocampal neurons remain in the multipolar phase of migration, which may require additional mechanisms to delay axon formation70.
Materials and Methods
Sema3a knockout mice
Sema3a+/− mice71 were maintained in a C57Bl/6 background. Genotyping was performed using the primers 5′-ATGGTTCTGA TAGGTGAGGC ATGG-3′, 5′-GTTCTGCTCC CGGCTCTAAA TCTC-3′ and 5′-AGGCAAACTA TGCAAACGG AAAG-3′72. Mice were housed at four to five per cage with a 12 h light/dark cycle (lights on from 07:00 to 19:00 h) at constant temperature (23 °C) with ad libitum access to food and water. All animal protocols were carried out in accordance with the relevant guidelines and regulations and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen.
Cell culture and transfection
Cultures of hippocampal and cortical neurons were prepared from E17 Sema3a mouse embryos as described before1. Hippocampal neurons from E18 rat embryos were prepared and transfected by calcium phosphate co-precipitation as described previously1. Dissociated neurons were plated at a density as 65,000 cells per well of a 24-well plate containing cover slips coated with poly-L-ornithine (15 μg/ml, SigmaAldrich). Neurons were cultured at 37 °C and 5% CO2 for 3–6 days in Neurobasal medium (Invitrogen) with supplements. An excess of the expression and knockdown vectors were combined with a small amount of pEGFP-N3 (Clontech) to label transfected neurons.
Immunofluorescence staining
Neurons were fixed with 4% paraformaldehyde/15% sucrose in phosphate buffered saline (PBS) for 15 min at RT and permeabilized with 0.1% Triton X-100/0.1% Na-Citrate/PBS for 10 min on ice. Cells were incubated in 10% normal goat serum (PAN Biotech) in PBS for 1 h at RT, stained with primary antibody overnight at 4 °C and secondary antibody for 90 min, and mounted using Mowiol (SigmaAldrich). Neuronal morphology and axon formation were analyzed as described before using a Zeiss Axiophot microscope equipped with a Visitron CCD camera and the SPOT Advanced Imaging software or a Zeiss LSM 700 using the ZEN (black edition) software1. Image analysis was done using ImageJ 1.45 s (NIH), ZEN (black edition) and Adobe Photoshop CS5. All statistical data are means ± s.e.m. from at least three times independent experiments. Statistical significance was determined using the Student’s t-test.
Antibodies
The following antibodies were used: mouse Tau-1 (Chemicon, MAB3420, 1:200), mouse anti-MAP2 (SigmaAldrich, M4403, 1:1500), rabbit anti-MAP2 (Abcam, ab32454, 1:1000), mouse anti-Ankyrin-G (Antibodies Inc., 75–146, 1:100), rabbit anti-Plexin-A1 (Abcam, ab23391, 1:1000), mouse anti-GFP (Covance, MMS-118P, 1:1000), mouse anti-FLAG M2 (SigmaAldrich, F3165, 1:1000), anti-Rap1 (Upstate, #07–916, 1:200), anti-Sema3A (Abcam, ab23393, 1:200), SMI-312 (BioLegend, 837904, 1:200) and goat secondary antibodies labeled with Alexa-350 (Molecular Probes, 1:200), −488 (1:800) or −594 (1:800). Nuclei were stained with Hoechst 33342 (Molecular Probes, 1:6000).
Plasmids
Published shRNA vectors were used for a knockdown of different A-type plexins (target sequences: Plexin-A1: 5′-CCGTATTTAC AAGCTGTCG-3′; Plexin-A2: 5′-GCGCAAGTCT AGGGAAAAT-3′; Plexin-A3: 5′-GTGCGGGTTC GGCCTAATA-3′; Plexin-A4: 5′-AGATGCTGCT TATAGAC TA-3′)41. The Plexin-A1 shRNA is specific for the rat sequence41. For rescue experiments, an expression vector for murine Plexin-A119 that contains mismatches in the shRNA target site compared to the rat sequence was used. The shRNA vector targeting Rap1B and the expression vectors for Rap1BV12 and Flag-Sema3A have been described before11,73. A vector for RNAi-resistant Rap1B (Rap1B-res) was constructed by site-directed mutagenesis of the shRNA target site11 to introduce mismatches using the QuikChange Site-Directed Mutagenesis kit (Stratagene) with the oligonucleotides 5′-GTTGTAGGAA AAGAACAGGG TCAAAACCTA GCAAGACAG-3′ and 5′-CTGTCT TGCT AGGTTTTGAC CCTGTTCTTT TCCTACAAC-3′. The miRNA vector targeting Sema3A with the target sequence 5′-TTCCGGGAAC CAACAACTATT-3′ was generated using the BLOCK-iT Pol II miRNA Expression Vector Kit (Invitrogen), inserted into pcDNA6.2-GW/EmGFP-miR and confirmed by sequencing. A vector for RNAi-resistant FLAG-Sema3A (Sema3A-res) was constructed by site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis kit (Stratagene) with the oligonucleotides 5′-GAAATGACCG TCTTCCGTGA ACCGACAACC ATTTCAGCAATG-3′ and 5′-CATTGCTGAA ATGGTTGTCG GTTCACGGAA GACGGTCATT TC-3′. pEGFP-N3 (Clontech) was used as transfection control. The dominant-negative Plexin-A1 construct pBK-CMV-VSV-Plexin-A1Δcyt with a deletion of the intracellular domain and the constitutively active pBK-CMV-VSV-Plexin-A1Δsema with a deletion of the semaphorin domain were constructed as described before19,39,52,54.
Transfection of HEK 293T cells and Western blot
HEK 293 T cells were transfected using the calcium phosphate co-precipitation method as described previously74. Transfected HEK 293 T cells were lysed in Tris/HCl 50 mM, pH 7,4, NaCl 150 mM, DTT 1 mM, MgCl2 1,5 mM, EDTA 4 mM, glycerol 10% (v/v), Triton X-100 1% (v/v), cOmplete protease inhibitor (Sigma-Aldrich) and expression of Sema3A analyzed by Western blot using horseradish peroxidase conjugated secondary antibodies (Dianova, 1:3000). Peroxidase activity was visualized by the enhanced chemiluminescence detection system (Uptima, Interchim UP99619A) using the ChemiDocTM MP imaging system (Bio-Rad).
Quantification
The establishment of neuronal polarity was quantified by counting the number of transfected (GFP-positive) neurons that did not extend an axon (unpolarized neuron), formed a single axon positive for Tau-1 (polarized neuron), multiple axons positive for Tau-1 (multiple axons) or multiple axons positive for Tau-1 and MAP2 (indeterminate phenotype). Statistical analyses were done using the GraphPad Prism 6.0 software. Statistical significance was calculated for at least three independent experiments using two-way ANOVA with Tukey’s multiple comparison test and Student’s t-Test for parametric data sets. Significance was defined as: p > 0.05, n.s.; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
References
Shah, B. et al. Rap1 GTPases Are Master Regulators of Neural Cell Polarity in the Developing Neocortex. Cereb Cortex 27, 1253–1269 (2017).
Franco, S. J., Martinez-Garay, I., Gil-Sanz, C., Harkins-Perry, S. R. & Muller, U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 69, 482–497 (2011).
Jossin, Y. & Cooper, J. A. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat Neurosci 14, 697–703 (2011).
Ye, T., Ip, J. P., Fu, A. K. & Ip, N. Y. Cdk5-mediated phosphorylation of RapGEF2 controls neuronal migration in the developing cerebral cortex. Nat Commun 5, 4826 (2014).
Shah, B. & Püschel, A. W. In vivo functions of small GTPases in neocortical development. Biol Chem 395, 465–476 (2014).
Funahashi, Y., Namba, T., Nakamuta, S. & Kaibuchi, K. Neuronal polarization in vivo: Growing in a complex environment. Curr Opin Neurobiol 27, 215–223 (2014).
Namba, T. et al. Extracellular and Intracellular Signaling for Neuronal Polarity. Physiol Rev 95, 995–1024 (2015).
Namba, T. et al. Pioneering axons regulate neuronal polarization in the developing cerebral cortex. Neuron 81, 814–829 (2014).
Sakakibara, A. & Hatanaka, Y. Neuronal polarization in the developing cerebral cortex. Front Neurosci 9, 116 (2015).
Schelski, M. & Bradke, F., Neuronal polarization: From spatiotemporal signaling to cytoskeletal dynamics. Mol Cell Neurosci (2017).
Schwamborn, J. C. & Püschel, A. W. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci 7, 923–929 (2004).
Bilasy, S. E. et al. Dorsal telencephalon-specific RA-GEF-1 knockout mice develop heterotopic cortical mass and commissural fiber defect. Eur J Neurosci 29, 1994–2008 (2009).
Maeta, K. et al. Crucial Role of Rapgef2 and Rapgef6, a Family of Guanine Nucleotide Exchange Factors for Rap1 Small GTPase, in Formation of Apical Surface Adherens Junctions and Neural Progenitor Development in the Mouse Cerebral Cortex. eNeuro 3, ENEURO.0142-16.2016 (2016).
Shah, B. & Püschel, A. W. Regulation of Rap GTPases in mammalian neurons. Biol Chem 397, 1055–1069 (2016).
Voss, A. K., Krebs, D. L. & Thomas, T. C3G regulates the size of the cerebral cortex neural precursor population. EMBO J 25, 3652–3663 (2006).
Voss, A. K. et al. C3G regulates cortical neuron migration, preplate splitting and radial glial cell attachment. Development 135, 2139–2149 (2008).
Hota, P. K. & Buck, M. Plexin structures are coming: opportunities for multilevel investigations of semaphorin guidance receptors, their cell signaling mechanisms, and functions. Cell Mol Life Sci 69, 3765–3805 (2012).
Pascoe, H. G., Wang, Y. & Zhang, X. Structural mechanisms of plexin signaling. Prog Biophys Mol Biol 118, 161–168 (2015).
Rohm, B., Rahim, B., Kleiber, B., Hovatta, I. & Püschel, A. W. The semaphorin 3A receptor may directly regulate the activity of small GTPases. FEBS Lett 486, 68–72 (2000).
Wang, Y., Pascoe, H. G., Brautigam, C. A., He, H. & Zhang, X. Structural basis for activation and non-canonical catalysis of the Rap GTPase activating protein domain of plexin. Elife 2, e01279 (2013).
Wang, Y. et al. Plexins are GTPase-activating proteins for Rap and are activated by induced dimerization. Sci Signal 5, ra6 (2012).
Worzfeld, T. et al. Genetic dissection of plexin signaling in vivo. Proc Natl Acad Sci USA 111, 2194–2199 (2014).
Yang, T. & Terman, J. R. 14-3-3epsilon couples protein kinase A to semaphorin signaling and silences plexin RasGAP-mediated axonal repulsion. Neuron 74, 108–121 (2012).
Jongbloets, B. C. & Pasterkamp, R. J. Semaphorin signalling during development. Development 141, 3292–3297 (2014).
Koropouli, E. & Kolodkin, A. L. Semaphorins and the dynamic regulation of synapse assembly, refinement, and function. Curr Opin Neurobiol 27, 1–7 (2014).
Worzfeld, T. & Offermanns, S. Semaphorins and plexins as therapeutic targets. Nat Rev Drug Discov 13, 603–621 (2014).
Oinuma, I., Ito, Y., Katoh, H. & Negishi, M. Semaphorin 4D/Plexin-B1 stimulates PTEN activity through R-Ras GTPase-activating protein activity, inducing growth cone collapse in hippocampal neurons. J Biol Chem 285, 28200–28209 (2010).
Oinuma, I., Katoh, H. & Negishi, M. Molecular dissection of the semaphorin 4D receptor plexin-B1-stimulated R-Ras GTPase-activating protein activity and neurite remodeling in hippocampal neurons. J Neurosci 24, 11473–11480 (2004).
Oinuma, I., Katoh, H. & Negishi, M. R-Ras controls axon specification upstream of glycogen synthase kinase-3beta through integrin-linked kinase. J Biol Chem 282, 303–318 (2007).
Saito, Y., Oinuma, I., Fujimoto, S. & Negishi, M. Plexin-B1 is a GTPase activating protein for M-Ras, remodelling dendrite morphology. EMBO Rep 10, 614–621 (2009).
Tasaka, G., Negishi, M. & Oinuma, I. Semaphorin 4D/Plexin-B1-mediated M-Ras GAP activity regulates actin-based dendrite remodeling through Lamellipodin. J Neurosci 32, 8293–8305 (2012).
Ito, Y., Oinuma, I., Katoh, H., Kaibuchi, K. & Negishi, M. Sema4D/plexin-B1 activates GSK-3beta through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep 7, 704–709 (2006).
Yukawa, K. et al. Sema4A induces cell morphological changes through B-type plexin-mediated signaling. Int J Mol Med 25, 225–230 (2010).
Yoo, S. K. et al. Plexins function in epithelial repair in both Drosophila and zebrafish. Nat Commun 7, 12282 (2016).
Ueda, Y. et al. Sema3e/Plexin D1 Modulates Immunological Synapse and Migration of Thymocytes by Rap1 Inhibition. J Immunol 196, 3019–3031 (2016).
Goshima, Y., Yamashita, N., Nakamura, F. & Sasaki, Y. Regulation of dendritic development by semaphorin 3A through novel intracellular remote signaling. Cell Adh Migr 10, 627–640 (2016).
He, Z. & Tessier-Lavigne, M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 90, 739–751 (1997).
Kolodkin, A. L. et al. Neuropilin is a semaphorin III receptor. Cell 90, 753–762 (1997).
Tamagnone, L. et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99, 71–80 (1999).
Winberg, M. L. et al. Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell 95, 903–916 (1998).
Chen, G. et al. Semaphorin-3A guides radial migration of cortical neurons during development. Nat Neurosci 11, 36–44 (2008).
Sasaki, Y. et al. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 35, 907–920 (2002).
Wiegreffe, C. et al. Bcl11a (Ctip1) Controls Migration of Cortical Projection Neurons through Regulation of Sema3c. Neuron 87, 311–325 (2015).
Chedotal, A. et al. Semaphorins III and IV repel hippocampal axons via two distinct receptors. Development 125, 4313–4323 (1998).
Polleux, F., Giger, R. J., Ginty, D. D., Kolodkin, A. L. & Ghosh, A. Patterning of cortical efferent projections by semaphorin-neuropilin interactions. Science 282, 1904–1906 (1998).
Polleux, F., Morrow, T. & Ghosh, A. Semaphorin 3A is a chemoattractant for cortical apical dendrites. Nature 404, 567–573 (2000).
Ruediger, T. et al. Integration of opposing semaphorin guidance cues in cortical axons. Cereb Cortex 23, 604–614 (2013).
Shelly, M. et al. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron 71, 433–446 (2011).
Nishiyama, M. et al. Semaphorin 3A induces CaV2.3 channel-dependent conversion of axons to dendrites. Nat Cell Biol 13, 676–685 (2011).
Lerman, O., Ben-Zvi, A., Yagil, Z. & Behar, O. Semaphorin3A accelerates neuronal polarity in vitro and in its absence the orientation of DRG neuronal polarity in vivo is distorted. Mol Cell Neurosci 36, 222–234 (2007).
De Wit, J., De Winter, F., Klooster, J. & Verhaagen, J. Semaphorin 3A displays a punctate distribution on the surface of neuronal cells and interacts with proteoglycans in the extracellular matrix. Mol Cell Neurosci 29, 40–55 (2005).
Takahashi, T. et al. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell 99, 59–69 (1999).
Wu, K. Y. et al. Semaphorin 3A activates the guanosine triphosphatase Rab5 to promote growth cone collapse and organize callosal axon projections. Sci Signal 7, ra81 (2014).
Takahashi, T. & Strittmatter, S. M. PlexinA1 autoinhibition by the plexin sema domain. Neuron 29, 429–439 (2001).
Nakamura, F. et al. Increased proximal bifurcation of CA1 pyramidal apical dendrites in sema3A mutant mice. J Comp Neurol 516, 360–375 (2009).
Foley, K. et al. Semaphorin 3D autocrine signaling mediates the metastatic role of annexin A2 in pancreatic cancer. Sci Signal 8, ra77 (2015).
Bagci, T., Wu, J. K., Pfannl, R., Ilag, L. L. & Jay, D. G. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene 28, 3537–3550 (2009).
Fukuda, T. et al. Sema3A regulates bone-mass accrual through sensory innervations. Nature 497, 490–493 (2013).
Hamm, M. J., Kirchmaier, B. C. & Herzog, W. Sema3d controls collective endothelial cell migration by distinct mechanisms via Nrp1 and PlxnD1. J Cell Biol 215, 415–430 (2016).
Man, J. et al. Sema3C promotes the survival and tumorigenicity of glioma stem cells through Rac1 activation. Cell Rep 9, 1812–1826 (2014).
Arimura, N. & Kaibuchi, K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci 8, 194–205 (2007).
Yoshimura, T. et al. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem Biophys Res Commun 340, 62–68 (2006).
Fivaz, M., Bandara, S., Inoue, T. & Meyer, T. Robust neuronal symmetry breaking by Ras-triggered local positive feedback. Curr Biol 18, 44–50 (2008).
Esteban, L. M. et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol 21, 1444–1452 (2001).
Azzarelli, R. et al. An antagonistic interaction between PlexinB2 and Rnd3 controls RhoA activity and cortical neuron migration. Nat Commun 5, 3405 (2014).
Deng, S. et al. Plexin-B2, but not Plexin-B1, critically modulates neuronal migration and patterning of the developing nervous system in vivo. J Neurosci 27, 6333–6347 (2007).
Daviaud, N., Chen, K., Huang, Y., Friedel, R. H. & Zou, H. Impaired cortical neurogenesis in plexin-B1 and -B2 double deletion mutant. Dev Neurobiol 76, 882–899 (2016).
Hirschberg, A. et al. Gene deletion mutants reveal a role for semaphorin receptors of the plexin-B family in mechanisms underlying corticogenesis. Mol Cell Biol 30, 764–780 (2010).
Xia, J. et al. Semaphorin-Plexin Signaling Controls Mitotic Spindle Orientation during Epithelial Morphogenesis and Repair. Dev Cell 33, 299–313 (2015).
Kitazawa, A. et al. Hippocampal pyramidal neurons switch from a multipolar migration mode to a novel “climbing” migration mode during development. J Neurosci 34, 1115–1126 (2014).
Taniguchi, M. et al. Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron 19, 519–530 (1997).
Schlomann, U., Schwamborn, J. C., Muller, M., Fassler, R. & Puschel, A. W. The stimulation of dendrite growth by Sema3A requires integrin engagement and focal adhesion kinase. J Cell Sci 122, 2034–2042 (2009).
Adams, R. H., Lohrum, M., Klostermann, A., Betz, H. & Püschel, A. W. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J 16, 6077–6086 (1997).
Yang, R., Kong, E., Jin, J., Hergovich, A. & Puschel, A. W. Rassf5 and Ndr kinases regulate neuronal polarity through Par3 phosphorylation in a novel pathway. J Cell Sci 127, 3463–3476 (2014).
Acknowledgements
We thank Maria Wenning and Ina Kowsky for technical assistance and Drs. T. Yagi, M. Taniguchi, T. Holtmaat and J. Verhaagen for providing us with the Sema3a+/− mice. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through the Cells-in-Motion Cluster of Excellence (EXC 1003 - CiM) and with a fellowship to N.W. by the China Scholarship Council.
Author information
Authors and Affiliations
Contributions
A.W.P. conceived the study, N.W., P.D. and J.C. performed the experiments, N.W., P.D., J.C. and A.W.P. contributed to the experimental design and interpretation of the results, and N.W. and A.W.P. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, N., Dhumale, P., Chiang, J. et al. The Sema3A receptor Plexin-A1 suppresses supernumerary axons through Rap1 GTPases. Sci Rep 8, 15647 (2018). https://doi.org/10.1038/s41598-018-34092-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34092-5
- Springer Nature Limited
Keywords
This article is cited by
-
Metabotropic signaling within somatostatin interneurons controls transient thalamocortical inputs during development
Nature Communications (2024)
-
Biallelic and monoallelic variants in PLXNA1 are implicated in a novel neurodevelopmental disorder with variable cerebral and eye anomalies
Genetics in Medicine (2021)
-
Semaphorin 3A controls enteric neuron connectivity and is inversely associated with synapsin 1 expression in Hirschsprung disease
Scientific Reports (2020)
-
Expression of a novel brain specific isoform of C3G is regulated during development
Scientific Reports (2020)