
Abstract
Solanum trilobatum L. is an important medicinal plant in traditional Indian system of medicine belonging to Solanaceae family. However, non-availability of genomic resources hinders its research at the molecular level. We have analyzed the S. trilobatum leaf transcriptome using high throughput RNA sequencing. The de novo assembly of 136,220,612 reads produced 128,934 non-redundant unigenes with N50 value of 1347 bp. Annotation of unigenes was performed against databases such as NCBI nr database, Gene Ontology, KEGG, Uniprot, Pfam, and plnTFDB. A total of 60,097 unigenes were annotated including 48 Transcription Factor families and 14,490 unigenes were assigned to 138 pathways using KEGG database. The pathway analysis revealed the transcripts involved in the biosynthesis of important secondary metabolites contributing for its medicinal value such as Flavonoids. Further, the transcripts were quantified using RSEM to identify the highly regulated genes for secondary metabolism. Reverse-Transcription PCR was performed to validate the de novo assembled unigenes. The expression profile of selected unigenes from flavonoid biosynthesis pathway was analyzed using qRT-PCR. We have also identified 13,262 Simple Sequence Repeats, which could help in molecular breeding. This is the first report of comprehensive transcriptome analysis in S. trilobatum and this will be an invaluable resource to understand the molecular basis related to the medicinal attributes of S. trilobatum in further studies.
Similar content being viewed by others


Introduction
Solanum trilobatum L. is one of the important medicinal plants belonging to family Solanaceae, more commonly available in Southern part of India. S. trilobatum is a prickly diffuse, bright green perennial herb, 2–3 m in height, mostly found in dry places, and grows like a weed along roadsides and wastelands1. In traditional Indian system of medicine, it is broadly used to treat many respiratory disease conditions. Its extract is used to treat conditions like chronic bronchitis and tuberculosis2,3. A pilot study, on clinical efficacy of S. trilobatum and Solanum xanthocarpum, confirmed the usefulness of these herbs in bronchial asthma. The reports confirmed its response to be equivalent to deriphylline but less than that of salbutamol, which is the preferred choice of drugs for the treatment of bronchial asthma4,5. S. trilobatum is also reported to have shown different activities like anti-oxidative activity, hepatoprotective activity6,7,8, anti-inflammatory activity9, anti-microbial activity10 and anti-tumoral activity11. The ethanol extract of S. trilobatum have shown a hypoglycaemic effect in alloxan-induced diabetic rats through antioxidant defense mechanism12. The majority of secondary metabolites, attributed to medicinal properties are mainly present in its leaves, including alkaloids such as soladunalinidine and tomatidine. S. trilobatum is reported to contain various chemical compounds as sobatum, β-solamarine, solasodine, solaine, and diosogenin13,14. Moreover, sobatum, purified from S. trilobatum, is found to be effective in suppressing drug-induced toxicity in rats15. Despite the well-established role of S. trilobatum in traditional Indian medicine, the genetics and genomics of this medicinal plant are least explored. As for the genomic resources, only 56 nucleotide sequences are available at National Center for Biotechnology Information (NCBI) database for S. trilobatum (as accessed on February 1, 2018). An in-depth study of S. trilobatum transcriptome is needed for analysis and characterization of its functional genes. This would help to achieve large-scale production of drugs via molecular breeding, transgenic technology, and metabolic engineering. RNA Sequencing-based de novo assembly is a well-developed approach to understanding transcriptomes of non-model plants with limited genomic information. Moreover, RNA-Seq is a cost-effective tool, offers much data with better coverage and sufficient sequence depth for de novo assembly of transcriptomes. In the past few years, there has been a significant increase in utilizing RNA-Seq for discovery and identification of functional genes involved in the biosynthesis of active compounds in plants16. In this study, we report the transcriptome from the leaf of Solanum trilobatum using high throughput next-generation sequencing for the first time. The high-quality reads were de novo assembled into unique transcripts, which were then extensively evaluated and annotated to identify the putative pathways and genes responsible for its medicinal properties.
Materials and Methods
Plant material and RNA isolation
Solanum trilobatum L. collected from Guduvanchery, Kancheepuram district, Tamil Nadu was taxonomically identified by the Centre for Floristic Research, Madras Christian College, Tambaram, Chennai, with field no. 523. The mature leaves from top and middle parts of the healthy plant during its flowering stage were collected and used for total RNA extraction immediately after collection using modified CTAB method17. The extracted total RNA was treated with DNase A and purified using RNeasy MinElute clean up kit (Qiagen Inc., GmbH, Germany). The quality was assessed using NanodropLite spectrophotometer (Thermo Scientific, Wilmington, Delaware, USA) and Qubit 2.0 (Invitrogen, Carlsbad, California, USA). The RNA integrity value was measured using Bioanalyzer 2100 (Agilent Technologies, Santa Clara, California, USA). The purified total RNA was used for sequencing library preparation.
Library preparation and illumina sequencing
The total RNA was made ribosomal RNA free using Ribo-Zero rRNA removal kit (Illumina Inc., Singapore) and the remaining fraction was purified and eluted. The purified RNA was disrupted into short fragments using fragmentation buffer; these short fragments are used as a template for first strand cDNA synthesis using superscript II reverse transcriptase (Invitrogen, Carlsbad, California, USA) followed by second strand synthesis and purification. The purified double-stranded cDNA was polyadenylated, and adapter-ligated for paired-end library preparation. The adaptor primers were used for amplification of the library for the enrichment of the cDNA fragments. Caliper LabChip GX using HT DNA High Sensitivity Assay Kit (Caliper Life Sciences Inc., USA) was used for library quality assessment. The library was hybridized on a flowcell, and clonal clusters were generated on cBOT using TruSeq PE Cluster Kit v3-cBot-HS (Illumina Inc., USA). Sequencing was carried out on Illumina Hiseq. 2500 using TruSeq v3-HS kit to generate 100 bp paired-end reads (Illumina Inc., USA).
De novo assembly and clustering
The raw paired-end reads were quality assessed by FastQC v0.11.218. Pre-processing of raw reads was performed with Cutadapt v1.7.119 and Sickle v1.33 tools20 for adapter trimming and quality filtering respectively. Reads with Phred score >=30 were retained. The filtered reads were further used for transcriptome assembly using Trinity21. Trinity, a de novo assembler consists of Inchworm, Chrysalis, and Butterfly modules, which are applied sequentially to process RNA-seq raw reads into full-length transcripts. The process begins with Inchworm which generates full-length transcripts from raw reads based on default k-mer values. The Chrysalis clusters the contigs generated by Inchworm and prepares de bruijn graph for each cluster. Finally, butterfly processes individual graphs reporting full-length transcripts for alternatively spliced isoforms. The redundancy of the assembled contigs was removed using CD-HIT v4.5.422.
Assessment of gene completeness
The gene completeness analysis was performed by using the TRAPID tool (http://bioinformatics.psb.ugent.be/webtools/trapid). The analysis was carried out by comparing the unigene transcripts against PLAZA 2.5 green plants clade database23 with an E-value of <1E-5 for significant similarity search and annotation of unigenes. The completeness of unigenes was assessed by considering one or more hits in TRAPID database24 for “full length”, “quasi full length” or “partial” based on length of the open reading frame (ORF).
Functional annotation and classification
The de novo assembled sequences of S. trilobatum were compared against plant non-redundant (nr) protein database at National Centre for Biotechnology Information (NCBI) using BLASTX tool from stand alone BLAST+ package with an E-value parameter not greater than 1E-05 for identification of best significant match. The BLASTX results were further imported to Blast2GO suite25 for retrieving Gene Ontology (GO) terms of assembled unigenes and for mapping, the annotation was further continued with unique enzyme codes (EC) and Kyoto Encyclopedia of Genes and Genomes (KEGG) maps. Moreover KEGG Automated Annotation Server (KAAS) was also used for pathway mapping in addition to Blast2GO. GO terms are precisely defined as controlled vocabulary which can be used to describe functions of genes or gene products. The assembled transcripts based on the retrieved GO terms were classified into three categories viz. Biological process, Molecular function, and Cellular component. The pathway maps were determined from KEGG database with an E-value of 1E-05.
Prediction of transcription factor families
The prediction of transcription factor (TF) families, in S. trilobatum transcriptome, was done using Plant Transcription Factor Database26 (plnTFDB v4.0; http://planttfdb.cbi.pku.edu.cn/prediction.php).
Identification of simple sequence repeats (SSRs)
The MicroSAtellite Identification tool (MISA) was used for identification of SSRs from assembled unigenes of S. trilobatum27. The parameters were set to identify perfect di-, tri-, tetra-, penta- and hexa nucleotide motifs with a minimum thresholds of 6, 5, 5, 5 and 5 repeats respectively.
Transcript quantification
The estimation of unigene abundance was determined using RNA-Seq by Expectation-Maximization (RSEM) tool, which quantifies transcript level abundance from RNA Seq data. RSEM first generates and preprocesses a set of reference transcript sequences and then aligns reads to reference transcripts followed by estimation of transcript abundances. RSEM calculates fragments per kilo base per million (FPKM) and transcripts per million (TPM) values for the assembled individual unigenes from S. trilobatum28. FPKM and TPM values are calculated to understand the expression levels of unigenes involved in biosynthetic pathways of secondary metabolites.
Validation by reverse transcription PCR
In order, to experimentally validate de novo transcriptome assembly, some of the assembled unigenes of S. trilobatum which share sequence similarity to various secondary metabolite biosynthetic pathway genes were selected for reverse transcription-PCR. All the primers were designed from final assembled sequences, and actin (house keeping gene) was used a positive control. The primer sequences are given in Supplementary Table S1. The reverse transcription-PCR products were electrophoresed on 1% agarose gel.
Gene expression analysis by qRT-PCR
The quantitative gene expression analysis was performed using QuantStudio 5 Real-Time PCR System (Thermo Scientific, Wilmington, Delaware, USA) and QuantiNova SYBR Green PCR Kit (Qiagen Inc., GmbH, Germany). For each primer pair, a control reaction without a template was also included. Elongation factor 1-alpha (ef1a) from Solanum trilobatum was used as an internal reference gene for normalization and estimation of gene expression. The data from qRT-PCR data was analysed using comparative Ct (2−ΔΔCt) method. Fold change in gene expression was calculated as 2−ΔΔCt using ΔCt values29. All the experiments were repeated using three technical and two biological replicates. Gene specific oligonucleotides used for qRT-PCR analysis are provided in Supplementary Table S2.
Results and Discussion
Illumina paired-end sequencing and De novo assembly
A total of 136,220,612 raw reads were generated from Solanum trilobatum leaf transcriptome that accounts for about 35.4 GB of paired-end sequencing data. The raw data were deposited at National Centre for Biotechnology Information (NCBI) Short Read Archive (SRA) database under the accession number SRP132765. The pre-processing of raw reads was done for removal of adaptor sequences and low-quality reads (Phred score <30) and a total of 124,413,306 clean reads (Phred score >=30) with GC content 39.21% were retained. Trinity assembler was employed for de novo assembly of short read sequences. A total of 144,580 assembled transcripts were generated with a mean size of 823 bp. The assembled transcripts were further clustered, using CD-HIT, into 128,934 unigenes with a mean length of 745 bp and N50 contig length of 1347 bp. The transcriptome assembly details are given in Table 1. Among unigenes, the minimum length was 201 bp and maximum length 19956 bp. The length distribution of the unigenes is given in Fig. 1.

Overview of unigene length distribution from Solanum trilobatum leaf transcriptome.
Assessment of gene completeness
Gene completeness analysis using TRAPID tool revealed the presence of 17521 (13.6%) full-length unigenes, 9517 (7.4%) quasi full length and 12433 (9.6%) partial coding unigenes. Moreover, there were 89463 (69.4%) unigenes that did not match to any protein in PLAZA green plants clade database.
Functional annotation of unigenes
The assembled unigenes of S. trilobatum were annotated for sequence similarity search and comparison using BLASTX against plant non-redundant protein database at NCBI with an E-value cut off 1E-5. The BLASTX results showed 60,097 annotated unigenes, and 68,837 remained non-annotated unigenes out of 128,934 assembled transcripts. Among annotated unigenes, 1.5% were uninformative hits (e.g., Predicted: Uncharacterized or hypothetical proteins) due to inadequate S. trilobatum genome information in public databases. BLASTX results were imported to BLAST2GO suite for further annotation. The unigenes showed top-hit species similarity with Solanum tuberosum (38.08%), Solanum pennellii (14.69%), Capsicum annuum (11.22%), Solanum lycopersicum (9.61%), Nicotiana tabacum (7.36%) and others (Fig. 2). The results indicate that S. trilobatum is more closely related to S. tuberosum. The annotations against proteins in Pfam database showed 35,141 significant hits. Moreover, 61986, 30427, and 14490 unigenes were annotated against the Uniprot, GO and KEGG databases respectively (Table 2). BLAST2GO is a preferable tool for large-scale functional annotation and data mining for sequencing data of non-model species. BLASTX results were imported to BLAST2GO tool for mapping to retrieve GO terms and then to retrieve EC numbers. The KEGG pathway annotation for assembled unigenes of S. trilobatum was performed using BLAST2GO and KAAS against KEGG database to unravel molecular interaction networks and metabolic pathways. (Supplementary Table S3).

BLASTX top hit species distribution of Solanum trilobatum unigenes against plant nr database.
Functional classification of unigenes
Gene Ontology (GO) classification was used to classify assembled unigenes based on annotations. Using BLAST2GO based on GO annotation, 30427 unigenes were assigned to one or more GO terms, which were allocated to three major categories and 97 subcategories. In biological function, category 38,714 unigenes were assigned to 44 classes. This category includes proteins highly involved in the biosynthetic process (4671 unigenes), nucleobase-containing compound metabolic process (3808 unigenes), cellular protein modification process (3704 unigenes) and cellular process (3201 unigenes). In molecular function, category 34,917 unigenes were assigned to 26 classes and includes proteins highly encoded in nucleotide binding (5826 unigenes), hydrolase activity (4093 unigenes), binding (3847 unigenes) and catalytic activity (3698 unigenes). Cellular components were grouped into 27 classes with most assignments to a membrane (7773 unigenes), nucleus (3242 unigenes), plastid (2519 unigenes) and cytoplasm (2114 unigenes) (Fig. 3) (Supplementary Table S4).

GO classification of Solanum trilobatum unigenes. The Histogram shows the results of unigene classification under three major categories of GO terms.
Biological pathway analysis
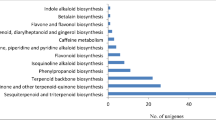
The active biochemical pathways in S. trilobatum were identified by mapping the unigenes against KEGG pathway database using KAAS and BLAST2GO. In total, 14,490 (11.23%) unigenes were annotated using KEGG database and assigned to 138 pathway maps. There were 14,212 unigenes assigned to metabolic pathway category including nucleotide metabolism, carbohydrate metabolism and biosynthesis of secondary metabolites with 2849 (20%), 2277 (16%) and 2050 (14%) unigene respectively (Fig. 4a). The unigenes representing secondary metabolism were further divided into 17 sub-categories, where in “flavonoid biosynthesis” pathway was found to be encoded by highest number of unigenes (1348 unigenes) followed by “caffeine metabolism” (260 unigenes) “tropane, piperidine and pyridine alkaloid” biosynthesis (123 unigenes), “phenylpropanoid biosynthesis”(72 unigenes), “isoquinoline alkaloid” biosynthesis (48 unigenes) (Fig. 4b).

KEGG pathway unigene assignments. (a) Based on metabolic pathway. (b) Based on secondary metabolite biosynthesis.
Secondary metabolite biosynthesis
Phenylpropanoid biosynthesis genes
Phenylpropanoids comprise a large group of plant-based natural compounds derived from phenylalanine30. Phenylpropanoids are responsible for plant responses to biotic and abiotic stimuli31. Phenylpropanoid biosynthesis starts with the formation of Cinnamic acid from Phenylalanine, which gets converted into Cinnamoyl-CoA, p-Coumaryl-CoA, p-Coumarylquinic acid, Caffeoyl quinic acid, Caffeoyl-CoA, Feruloyl-CoA, and Sinapoyl-CoA. Caffeoyl quinic acid (Chlorgenic acid) is a well established soluble phenylpropanoid in Solanaceae and also plays a role as defense compound or as a potential antioxidant32. In this study, KEGG analysis revealed the presence of 11 genes which are involved in the biosynthesis of various compounds of this pathway. The major genes identified were Phenylalanine ammonia lyase (EC: 4.3.1.24, EC: 4.3.1.25), Trans cinnamate-4-monooxygenase (EC: 1.14.13.11), Cinnamoyl-CoA CoA reductase (EC: 1.2.1.44) and 4-Coumarate CoA ligase (EC: 6.2.1.12). The presence of all these enzymes compliments the phytotherapeutic properties of S. trilobatum (Fig. 5).

Phenylpropanoid biosynthesis pathway from Solanum trilobatum by KEGG analysis showing the different identified enzymes (one color for each Enzyme Code or EC). KEGG pathway map 00940 is adapted here from http://www.kegg.jp/kegg/kegg1.html. The KEGG database has been described previously60,61,62.
Analysis of flavonoid biosynthesis genes
The coloring pigment of most flowers, fruits, and seeds are flavonoids. They are widely distributed in plants and classified into 6 major subgroups: chalcones, flavones, flavonols, flavandiols, anthocyanins and proanthocyandins, one more subgroup is found in some species, the aurones. Flavonoids are synthesized through Phenylpropanoid pathway, by transformation of phenylalanine to p-Coumaryl-CoA, which actually enters into the flavonoid biosynthesis pathway. The first enzyme specific to this pathway is Chalcone Synthase, which produces chalcone scaffolds used for the formation of other flavonoids33. In the present study, Chalcone synthase (EC: 2.3.1.74) and Chalcone isomerase (EC: 5.5.1.6) enzymes were identified and are responsible for the formation of Naringenin from p-Coumaroyl-CoA. The enzymes required for conversion of Naringenin to produce apiforol by flavonone-4-reductase (EC: 1.1.1.234), eriodictyol by flavonoid-3′5′-hydroxylase (EC: 1.14.1388) and dihydrotricetin by flavonoid 3′-monooxygenase (EC: 1.14.13.88) were also identified. Moreover, the enzyme 6′-deoxychalcone synthase (EC: 2.3.1.170) responsible for converting p-Coumaroyl CoA to Isoliquiritigenin for production of butein is also identified in our dataset where Chalcone isomerase further converts butein to butin. The flavonone 4- reductase also acts on eriodictyol to produce a bioactive compound luteoforol. The enzyme Naringenin-3-dioxygenase (EC: 1.14.11.9), found in our data set, acts on pinocembrin to produce pinobanksin which is further acted by Flavanol synthase (EC: 1.14.11.23) to convert it into galingin, Pinocembrin also gets converted to Pinostrobin. Naringenin-3-dioxygenase acts on liquiritigenin to produce garbanzol and also acts on eriodictoyl to produce dihydroquercetin or taxifolin which is further converted to quercetin by Flavonol synthase enzyme were identified in S. trilobatum leaf transcriptome dataset. Moreover, Naringenin 3-dioxygenase acts on dihydrotricetin to convert it into dihydromyricetin which is finally converted to myrecitin by Flavonol synthase. A similar set of flavonoid pathway genes has been already reported in leaf transcriptomes of endangered medicinal plant Chlorophytum borivilianum34 and also in the leaf tissue of medicinal plant Phyllanthus amarus35. These reports support our findings in the present dataset and also suggest how flavonoid pathway is complimenting for medicinal properties of S. trilobatum. The current study identified the genes in the biosynthesis of various flavonoids like butein, butin, pinostrobin, naringenin, galingin, garbanzol, dihydrofisetin (futin), eriodictyol, homoeriodictyol, kaempferol, apiforol, luteoforol, myricetin, dihydroquercetin (taxifolin) and cyanidin.
One of the study reported that butin has aromatase inhibitory action whereas butein shows its effect against breast and lung cancer36,37,38. Pinostrobin has been reported to have antiviral property39 and it also possesses antioxidant and chemoprotective activity40. Naringenin, a flavonone flavonoid, has been shown to possess marked antioxidant and anti-inflammatory properties41. Naringenin has been reported to have a protective effect on carbon tetracholride induced acute nephrotoxicity in mouse models42. Naringenin is reported to be an effective chemotherapeutic agent for prostate cancer43. It has also been reported that S. trilobatum possesses neuroprotective role against pathology of Parkinson’s disease44. Galangin is considered to be the potential candidate for new drugs against Alzheimer’s disease45. It also possesses hepatoprotective and anticancer properties46,47. Quercetin is one of the important flavonoids reported in our dataset, it possess antioxidant, also shows anti-carcinogenic and hepatoprotective properties48. Luteoforol, quercetin, and myricetin are reported to have anti microbial properties49,50. The flavonol dihydrofisetin (fustin) is reported to show protective role in neuronal cell death51. The Kaempferol also identified in our dataset is a well-known phytoestrogen reported to induce osteoblastic differentiation52. Kaempferol is also reported to possess hepatoprotective, antioxidant and anticancer effects53,54,55. Dihydroquercetin (Taxifolin) reported in our data acts as a potential chemopreventive agent56. Cyanidin is also reported as potent inhibitor of EGFR, shutting off downstream signalling cascades57 (Fig. 6). A summary of major genes involved in phenylpropanoid and flavonoid pathways has been presented in Table 3.

Flavonoid biosynthesis pathway from Solanum trilobatum by KEGG analysis showing the different identified enzymes (one color for each Enzyme Code or EC). KEGG pathway map 00941 is adapted here from http://www.kegg.jp/kegg/kegg1.html. The KEGG database has been described previously60,61,62.
Discovery of simple sequence repeats (SSRs)
Microsatellites or SSRs are tandem repeats of DNA sequences abundantly present throughout the genome58 and are widely useful for molecular- assisted selection (MAS) in plant breeding and improvement programs. We analyzed 128,934 assembled transcripts from S. trilobatum using MISA tool and a total of 13,262 SSRs were identified including 561 SSRs in compound formation. Among the analyzed sequences 11,727 contained SSRs and 1,303 sequences showed the presence of more than one SSR. The statistical distribution of SSRs is given in Table 4. The most abundant repeat motif was found to be tri-nucleotide repeats with 2661(54.65%) followed by di-nucleotide repeats 2045 (42%), tetra-nucleotide 88 (1.81%), penta-nucleotide 26 (0.53%) and hexa-nucleotide 49 (1.01%). SSRs with five tandem repeats (1,840) were most common in S. trilobatum and are followed by six repeats (1,376) and seven repeats (665). Among di-nucleotide repeats, AG/CT (44.94%) followed by AT/AT (28.75%), AC/GT (25.72%), CG/CG (0.59%). In case of tri-nucleotide repeats highest frequency occurred in AAC/GTT (31.94%), followed by AAG/CTT (29.20%), ATC/ATG (9.66%), AGC/CTG (8.45%) and other motifs were almost uniformly distributed (Fig. 7).

Summary of SSR types identified in Solanum trilobatum leaf transcriptome.
Transcription factors from S. trilobatum
Transcription factors (TFs) play a crucial role in regulation of secondary metabolites by regulating the expression of related genes of biosynthetic pathways. The identification of such TFs will benefit us in understanding gene regulatory networks. TFs known to regulate plant secondary metabolism include R2R3-MYB, bHLH proteins like CrMYC2, AP2/ERF family proteins, WRKY, NAC, DOF, bZIP, HD-ZIP and TFIIIA zinc finger TFs59. We identified a total of 654 unitranscripts representing 48 TF families. Among the represented families, AP2 family TFs (47 unitranscripts) were the most abundant followed by ARF (43), ARR-B (41), B3 (39), BBR-BPC (36), BES1 (32), bHLH (28), bZIP (26) and C2H2 (24). Among the annotated TF unigenes, TFs related to Secondary metabolism were AP2, bHLH, bZIP, Dof, MYB, MYB related, WRKY and ZF-HD (Fig. 8) (See Supplementary Table S5).

Transcription factors identified from Solanum trilobatum leaf transcriptome.
Transcript quantification
The estimation of transcript abundance or expression levels of de novo assembled unigenes from S. trilobatum leaf transcriptome was calculated based on FPKM and TPM values using RSEM. The FPKM and TPM values for each unigene and isoforms of our data set are given in Table S6 (See Supplementary Information). The flavonoid biosynthesis pathway is highly represented as per our data analysis and also includes phenylpropanoid biosynthesis pathway as its backbone. So, understanding the transcript abundance of unigenes involved in phenylpropanoid and flavonoid biosynthesis pathways would be of great importance. In phenylpropanoid pathway, the TPM values of key enzymes involved are 56.01, 26.25 and 7 for Phenylalanine Ammonia Lyase (PAL), Cinnamate 4-hydroxylase/trans-cinnamate 4-monooxygenase and Cinnamoyl-CoA reductase respectively. In case of flavonoid biosynthesis pathway, the key genes with highest TPM values are Chalcone isomerase, Flavonol synthase and Coumaroylquinate 3′-monooxygenasewith 11.02, 10.85, 10.62 respectively and the gene encoding for flavonoid 3′, 5′-hydroxylaseobtained lowest TPM value of 1.58 (Table 3). The list of top most abundant unigenes for de novo assembled S. trilobatum transcriptome is given in Table 5. The list includes genes mostly from chloroplast, and our data set is a leaf transcriptome so it’s expected to have these genes abundantly reflected in our transcriptome data analysis.
Validation by reverse transcription PCR
The secondary metabolite genes of S. trilobatum were selected for performing Reverse Transcription-PCR to validate the assembled unigenes from leaf transcriptome. We selected a total of seven genes involved in secondary metabolite biosynthesis such as Phenylalanine ammonia lyase (EC: 4.3.1.24) from Phenylpropanoid biosynthesis pathway; Chalcone Isomerase (EC: 5.5.1.6), Flavonol synthase (EC: 1.14.11.23), Naringenin-3-dioxygenase (EC: 1.14.11.9) and Coumaroylquinate 3′-monooxygenase (EC: 1.14.13.36) from flavonoid biosynthesis pathway; Squalene monooxygenase (EC: 1.14.14.17) from Steroid biosynthesis pathway. We performed Reverse transcription PCR for above mentioned unigenes with Actin as positive control (Fig. 9). Experimental confirmation of the gene expression data authenticates the functionally annotated transcriptome assembly.

Reverse Transcription PCR analysis of selected unigenes of Solanum trilobatum leaf transcriptome. M is GeneRuler DNA ladder mix (Thermo Fisher Scientific, Waltham, MA), Lane 1–7 are amplicons of Phenylalanine ammonia lyase (2396 bp), Chalcone isomerase (998 bp), Flavonol synthase (1227 bp), Naringenin 3-dioxygenase (1215 bp), Coumaroylquinate 3′-monooxygenase (1673 bp), Squalene monooxygenase (1785 bp) and Actin (480 bp) respectively.
Gene expression analysis of Solanum trilobatum leaf tissue
The qRT-PCR was introduced to analyze the expression pattern of selected flavonoid biosynthesis genes and to validate the transcriptome assembly in S. trilobatum leaf. The Selected transcripts included Chalcone Isomerase (EC: 5.5.1.6), Flavonol Synthase (EC: 1.14.11.23), Naringenin 3-dioxygenase (EC: 1.14.11.9) and Flavonone 4-reductase (EC: 1.1.1.234). Based on our qRT-PCR results, the selected genes displayed different expression profiles, where Chalcone isomerase, Flavonol synthase and Naringenin 3-dioxygenase showed significant up-regulation in leaf tissue when compared to stem as control, with Chalcone isomerase showing maximum up-regulation and also Flavonone-4 reductase showing significant down regulation in the leaf tissue (See Supplementary Table S7). The qRT-PCR expression profiles of selected genes showed significant agreement with our transcriptomic data hence validating the de novo assembly of Solanum trilobatum leaf tissue (Fig. 10).

Validation of gene expression by qRT-PCR. CHA is Chalcone Isomerase, FLS (Flavonol Synthase), NAR (Naringenin 3-dioxygenase) and FLR is Flavonone 4- reductase.
Conclusion
The main aim of our study was to analyze the transcriptome of Solanum trilobatum using Illumina high throughput sequencing platform. In order to facilitate molecular research in Solanum trilobatum, we characterized the leaf transcriptome for identification of unitranscripts involved in biosynthesis of secondary metabolites, since several medicinal attributes are affiliated to leaf tissue of this plant. Our results suggested that Flavonoid biosynthesis pathway is highly represented in the leaf transcriptome and is possibly contributing for the medicinal attributes of this plant. The predicted biosynthetic pathway is going to serve as lead for identification and isolation of medicinally important phytocompounds. We have also quantified the expression levels of transcripts for various important metabolic pathways. We have validated the de novo assembly by amplifying randomly selected unigenes by Reverse Transcription PCR method and also performed gene expression analysis of selected key genes from flavonoid biosynthesis pathway by qRT-PCR. This is the first report on leaf transcriptome assembly and analysis of Solanum trilobatum and it will serve as an important resource for studying molecular mechanisms involved in biosynthesis of its medicinal compounds.
References
Sahu, J., Rathi, B., Koul, S. & Khosa, R. L. Solanum trilobatum (Solanaceae) – An Overview. Journal of Natural Remedies. 13(2), 76–80 (2013).
Kanchana, A. & Balakrishnan, M. Anti-Cancer effect of saponins isolated from Solanum trilobatum leaf extract and induction of apoptosis in human layrnx cancer cell lines. Int. J. Pharm Sci. 3(4), 356–364 (2011).
Ram, J. & Baghel, M. S. Clinical efficacy of Vyaghriharitaki Avaleha in the management of chronic bronchitis. Ayu. 36, 50–5 (2015).
Govindan, S., Viswanathan, S., Vijayasekaran, V. & Alagappan, R. A pilot study on the clinical efficacy of Solanum xanthocarpum and Solanum trilobatum in bronchial asthma. Journal of Ethnopharmacology. 66, 205–210 (1999).
Govindan, S., Viswanathan, S., Vijayasekaran, V. & Alagappan, R. Further Studies on the Clinical Efficacy of Solanum xanthocarpum and Solanum trilobatum in Bronchial Asthma. Phytother. Res. 18, 805–809 (2004).
Sini, H. & Devi, K. S. Antioxidant activities of the choloroform extract of Solanum trilobatum. Pharm. Biol. 42, 462–466 (2004).
Moula, S. J., Ganapathy, V. & Chennam, S. S. Effect of Solanum trilobatum on hepatic drug metabolizing enzymes during diethylnitrosamine-induced hepatocarcinogenesis promoted by Phenobarbital in rat. Hepatology Research. 37, 35–49 (2007).
Kumar, G., Sukalingam, K. & Xu, B. Solanum trilobatum L. Ameliorate Thioacetamide-Induced Oxidative Stress and Hepatic Damage in Albino Rats. Antioxidants. 6(3), 68 (2017).
Emmanuel, S., Ignacimuthu, S., Perumalsamy, R. & Amalraj, T. Anti-inflammatory activity of Solanum trilobatum. Fitoterapia. 77, 611–612 (2006).
Prasad, S. D., Sugnanam, M. K. & Rajanala, V. Screening of anti-bacterial activity of Solanum trilobatum L. Seed extract against dental pathogens. Asian Journal of Plant Science and Research. 5(2), 34–37 (2015).
Mohananan, P. V. & Devi, K. S. Cytotoxic potential of the preparations from Solanum trilobatum and the effect of sobatum on tumour reduction in mice. Cancer Letters. 110, 71–76 (1996).
Doss, A. & Anand, S. P. Free Radical Scavenging Activity of Solanum trilobatum Linn. On Alloxan - Induced Diabetic Rats. Biochem Anal Biochem. 1, 115 (2012).
Chinthana, P. & Ananthi, T. J. Protective Effect of Solanum nigrum and Solanum trilobatum aqueous leaf extract on lead induced neurotoxicity in albino mice. Chem. Pharma. Res. 4(1), 72–74 (2012).
Balakrishnan, P., Ansari, T., Gani, M., Subrahmanyam, S. & Shanmugam, K. Aperspective on bioactive compounds from Solanum trilobatum. J. Chem. Pharm. Res. 7(8), 507–512 (2015).
Vijaimohan, K., Mallika, J. & Shyamala, D. C. S. Chemoprotective Effect of Sobatum against Lithium-Induced Oxidative Damage in Rats. J Young Pharm. 2(1), 68–73 (2010).
Abdelrahman, M. et al. RNA-sequencing-based transcriptome and biochemical analyses of steroidal saponin pathway in a complete set of Allium fistulosum—A. cepa monosomic addition lines. PLOS ONE. 12 ( 8 ) (2017)
Chang, S., Puryear, J. & Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Rep. 11, 113–116 (1993).
Andrews, S. FastQC: a quality control tool for high throughput sequence data (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1), 10–12 (2011).
Joshi, N. A. & Fass, J. N. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (2011).
Grabherr, M. G. et al. Full length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 22, 1658–9 (2006).
Proost, S. et al. PLAZA: a comparative genomics resource to study gene and genome evolution in plants. The Plant Cell Online. 21(12), 3718–3731 (2009).
Van Bel, M. et al. TRAPID: an efficient online tool for the functional and comparative analysis of de novo RNA-Seq transcriptomes. Genome Biology. 14(12), 134 (2013).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research. 36, 3420–3435 (2008).
Pérez-Rodríguez, P. et al. PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, 822–827 (2009).
Thiel, T., Michalek, W., Varshney, R. K. & Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 106(3), 411–22 (2003).
Bo, L. & Colin, N. D. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 12, 323 (2011).
Rao, X., Huang, X., Zhou, Z. & Lin, X. An improvement of the 2(−delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 3, 71–85 (2013).
Michal, G. Biochemical Pathways: An Atlas of Biochemistry and Molecular Biology. Heidelberg: Spektrum Akademischer (1999).
Thomas, V. Phenylpropanoid biosynthesis. Molecular Plant. 3(1), 2–20 (2010).
Petersen, M. et al. Evolution of rosmarinic acid biosynthesis. Phytochemistry. 70, 1663–1679 (2009).
Ferreyra, M. L. F., Rius, S. P. & Paula, C. Flavonoids: biosynthesis, biological functions, and biotechnological applications. Frontiers in plant sciences. 3 ( 222) (2012).
Kalra, S. et al. De Novo Transcriptome Sequencing Reveals Important Molecular Networks and Metabolic Pathways of the Plant, Chlorophytum borivilianum. PLoS ONE (2013).
Bose, M. A. & Chattopadhyay, S. Sequencing, De novo Assembly, Functional Annotation and Analysis of Phyllanthus amarus Leaf Transcriptome Using the Illumina Platform. Front. Plant Sci. 6, 1199 (2016).
Park, M. H. et al. Inhibitory effect of Rhusverniciflua Stokes extract on human aromatase activity; butin is its major bioactive component. Bioorg. Med. Chem. Lett. 24, 1730–1733 (2014).
Cho, S. G., Woo, S. M. & Ko, S. G. Butein suppresses breast cancer growth by reducing a production of intracellular reactive oxygen species. J. Exp. Clin. Cancer Res (2014).
Seo, Y. H. & Jeong, J. H. Synthesis of butein analogues and their anti-proliferative activity against gefitinib-resistant non-small cell lung cancer (NSCLC) through Hsp90 inhibition. Bull. Korean Chem. Soc. 35, 1294–1298 (2014).
Wu, N. et al. Activity investigation of pinostrobin towards herpes simplex virus-1 as determined by atomic force microscopy. Phytomedicine. 18, 110–118 (2011).
Fahey, J. W. & Stephenson, K. K. Pinostrobin from honey and Thai ginger (Boesenbergia pandurata): a potent flavonoid inducer of mammalian phase 2 chemoprotective and antioxidant enzymes. J. Agric. Food Chem. 50, 7472–7476 (2002).
Amr, A. F., Waleed, H. A., Ahmed, Z. & Gomaa, W. Protective effect of naringenin against gentamicin-induced nephrotoxicity in rats. Environmental Toxicology and Pharmacology. 38(2), 420–429 (2014).
Hermenean, A., Ardelean, A., Stan, M. & Dinischiotu, A. Protective Effects of Naringenin on Carbon Tetrachloride - Induced Acute Nephrotoxicity in Mouse Kidney. Chemico-Biological Interactions. 205, 138–147 (2013).
Lin, E., Zhang, X., Wang, D., Hong, S. & Li, L. Naringenin modulates the metastasis of human prostate cancer cells by down regulating the matrix metalloproteinases −2/−9 via ROS/ERK1/2 pathways. Bangladesh J Pharmacol. 9, 419–27 (2014).
Lou, H. et al. Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology. 79, 380–388 (2014).
Guo, A. J. et al. Galangin, a flavonol derived from Rhizoma Alpiniae Officinarum, inhibits acetylcholinesterase activity in vitro. Chem. Biol. Interact. 187, 246–248 (2010).
Wang, X. et al. Antifibrotic activity of galangin, a novel function evaluated in animal liver fibrosis model. Environ. Toxicol. Pharmacol. 36, 288–295 (2013).
Zhang, W., Tang, B., Huang, Q. & Hua, Z. Galangin inhibits tumor growth and metastasis of B16F10 melanoma. J. Cell. Biochem. 114, 152–161 (2013).
Ji, L. et al. Quercetin prevents pyrrolizidine alkaloid clivorine-induced liver injury in mice by elevating body defense capacity. PLoS ONE (2014).
Ramesh, N. et al. Antibacterial activity of luteoforol from Brideliacrenulata. Fitoterapia. 72(4), 409–411(2001).
Rashed, K., Ciric, A., Glamoˇclija, J. & Sokovi´c, M. Antibacterial and antifungal activities of methanol extract and phenolic compounds from Diospyros virginiana L. Ind. Crop. Prod. 59, 210–215 (2014).
Park, B. C. et al. Protective effects of fustin, a flavonoid from Rhusverniciflua Stokes, on 6-hydroxydopamine-induced neuronal cell death. Exp. Mol. Med. 39, 316–326 (2007).
Guo, A. J. et al. Kaempferol as a flavonoid induces osteoblastic differentiation via estrogen receptor signaling. Chinese Medicine. 7 ( 10 ) (2012).
Shakya, G., Manjini, S., Hoda, M. & Rajagopalan, R. Hepatoprotective role of kaempferol during alcohol- and 1PUFA-induced oxidative stress. J. Basic Clin. Physiol. Pharmacol. 25, 73–79 (2014).
Huang, Y. B. et al. Anti-oxidant activity and attenuation of bladder hyperactivity by the flavonoid compound kaempferol. Int. J. Urol (2014).
Dang, Q. et al. Kaempferol suppresses bladder cancer tumor growth by inhibiting cell proliferation and inducing apoptosis. Mol. Carcinog. 54, 831–840 (2015).
Lee, S. B., Cha, K. H., Selenge, D., Solongo, A. & Nho, C. W. The Chemopreventive Effect of Taxifolin Is Exerted through ARE-Dependent Gene Regulation. Biol Pharm Bull. 30(6), 1074–9 (2007).
Meiers, S. et al. The Anthocyanidins Cyanidin and Delphinidin Are Potent Inhibitors of the Epidermal Growth-Factor Receptor. J. Agric. Food Chem. 49(2), 958–962 (2001).
Liu, S.-R., Li, W.-Y., Long, D., Hu, C.-G. & Zhang, J.-Z. Development and Characterization of Genomic and Expressed SSRs in Citrus by Genome-Wide Analysis. PLoS ONE. 8 ( 10 ) (2013).
Bhattacharyya, D., Sinha, R., Hazra, S., Datta, R. & Chattopadhyay, S. De novo transcriptome analysis using 454 pyrosequencing of the Himalayan Mayapple, Podophyllum hexandrum. BMC Genomics. 14, 748 (2013).
Kanehisa Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Acknowledgements
This study was financially supported by SRM-DBT Partnership Platform for Contemporary Research Services and Skill Development in Advanced Life Sciences Technologies (Order No. BT/PR12987/INF/22/205/2015). The High-Performance Computing Cluster Facility (Genome Server) at SRM Institute of Science and Technology (SRMIST) was used for the transcriptome assembly and analysis. We also acknowledge Indian Council of Medical Research (ICMR), New Delhi for Junior Research Fellowship (Ref. No. 3/1/3/JRF-2015/HRD-LS/76/93193/116).
Author information
Authors and Affiliations
Contributions
P.N. conceived the study, designed the experiments, guided the data analysis and reviewed the manuscript; A.L. analysed the data, prepared figures and designed the manuscript; S.K.P. and A.L. collected the samples and performed experiments. All authors contributed the manuscript at various stages.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lateef, A., Prabhudas, S.K. & Natarajan, P. RNA sequencing and de novo assembly of Solanum trilobatum leaf transcriptome to identify putative transcripts for major metabolic pathways. Sci Rep 8, 15375 (2018). https://doi.org/10.1038/s41598-018-33693-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33693-4
- Springer Nature Limited
Keywords
This article is cited by
-
Transcriptome sequencing of medical herb Salvia Rosmarinus (Rosemary) revealed the phenylpropanoid biosynthesis pathway genes and their phylogenetic relationships
Molecular Biology Reports (2024)
-
Transcriptome Analysis of Different Sections of Rhizome in Polygonatum sibiricum Red. and Mining Putative Genes Participate in Polysaccharide Biosynthesis
Biochemical Genetics (2022)
-
De novo transcriptome analysis of Justicia adhatoda reveals candidate genes involved in major biosynthetic pathway
Molecular Biology Reports (2022)
-
Comparative evaluation of full-length isoform quantification from RNA-Seq
BMC Bioinformatics (2021)
-
Transcriptome and de novo analysis of Rosa xanthina f. spontanea in response to cold stress
BMC Plant Biology (2021)