
Abstract
The human uptake transporter NaCT is important for human brain development, brain function and energy metabolism and mediates the uptake of citrate and other intermediates of the tricarboxylic acid cycle from blood into neurons and hepatocytes. Mutations in the SLC13A5 gene encoding NaCT are associated with epileptic encephalopathy. To gain more insights into the transport mechanisms we analyzed the functional consequences of mutations in the SLC13A5 gene on NaCT-mediated transport function. Using HEK293 cells expressing wild-type and eight mutated NaCT proteins, we investigated the mRNA and protein amount as well as the protein localization of all NaCT variants. Furthermore, the impact on NaCT-mediated citrate uptake was measured. In addition, a structural model of the transport pore was generated to rationalize the consequences of the mutations on a structural basis. We demonstrated that all proteins were synthesized with an identical molecular weight as the wild-type transporter but several mutations (NaCTp.G219R, −p.G219E, −p.T227M, −p.L420P and −p.L488P) lead to a complete loss of NaCT-mediated citrate transport. This loss of transport activity can be explained on the basis of the developed structural model. This model may help in the further elucidation of the transport mechanism of this important uptake transporter.
Similar content being viewed by others


Introduction
Transport proteins belonging to the superfamily of SLC (solute carriers) transporters are important for the uptake of endogenous substances and xenobiotics from the extracellular space into cells. Currently, the SLC superfamily consists of 65 transporter families (SLC1–SLC65) with more than 400 different transport proteins1. The SLC13 gene family of sodium-coupled di- and tricarboxylate/sulfate transporter consists of five transporters (gene symbols SLC13A1–SLC13A5, transport proteins NaSi1, NaDC2, NaDC3, NaSUT1 and NaCT, respectively) and with orthologues found in prokaryotes and eukaryotes2. A member of the SLC13 family, the human uptake transporter NaCT (sodium-coupled citrate transporter; gene symbol SLC13A5; also known as mINDY – mammalian INDY) is expressed in the brain and the basolateral membrane of hepatocytes3. NaCT is the human orthologue of the Drosophila Indy transporter4 and mediates the uptake of citrate and other intermediates of the tricarboxylic acid (TCA) cycle5 from blood into neurons and hepatocytes. In Drosophila melanogaster and Caenorhabditis elegans, reduced expression of this transporter promotes longevity due to caloric restriction6,7,8 and long-lived flies showed an increase in mitochondrial number, a lower expression of insulin-like proteins and reduced body fat stores9. In mammals, NaCT has a broad substrate spectrum and shows an inward electrogenic sodium-coupled substrate cotransport with citrate and succinate as important endogenous substrates with high plasma concentrations5. Studies with NaCT [INDY (−/−)]-knock out mice demonstrated that deletion of this transporter mimics aspects of dietary restriction and protects these mice against insulin resistance and adiposity10. Furthermore, the absence of this transporter led to defective tooth and bone development11.
Interestingly, in addition to its role in bone development and energy metabolism, NaCT seems to be important for the development and the function of the human brain because mutations in the SLC13A5 gene are associated with epileptic encephalopathy [EE12]. EE refers to a genetically and clinically heterogeneous group of severe disorders characterized by psychomotor delay, abnormal electro-encephalograms and seizures. Thevenon et al.12 screened two families and additional unrelated individuals with early-onset EE and detected three mutations (NaCTp.G219R, −p.T227M and −p.L488P) in the SLC13A5 gene possibly affecting key residues for sodium binding. Recently, these results were confirmed and it has been demonstrated that several other mutations as well as NaCTp.G219R and −p.T227M result in a loss of citrate transport13,14 and are associated with neonatal epilepsy, developmental delay and teeth hypoplasia. The mutations NaCTp.G219R and −p.T227M and four novel mutations were examined also in COS-7 cells demonstrating the same effect of NaCTp.G219R and −p.T227M on citrate transport14. Interestingly, the phenotype may vary between individuals affected by mutations in the SLC13A5 gene as shown in a report describing the NaCTp.G219R and the novel −p.Y82C mutants15. In this report, the authors described two siblings presented with developmental delay and intractable seizures. Both mutations were found in these siblings but only one individual had focal cortical dysplasia demonstrating the heterogeneity in disease expression even when the genotype is identical.
In 2012, the first structure and proposed transport mechanism of a bacterial sodium-dependent dicarboxylate transporter has been elucidated16. Crystallization of the NaCT homologue from Vibrio cholerae (vcINDY) showed that this protein functions as a dimer, which has the shape of the letter “M” when viewed from within the membrane plane with one citrate molecule and one sodium molecule bound to each of the monomers16. The vcINDY transporter shares 26–33% sequence identity with the human SLC13 family members and catalyzes the sodium-driven uptake of succinate.
To gain more insights into the functional consequences of mutations in the SLC13A5 gene encoding the human uptake transporter NaCT, we first screened several databases and identified four mutations in the SLC13A5 gene leading to amino acid substitutions in the NaCT protein (NaCTp.G219E, −p.D243N, −p.L420P and −p.L485R). In addition, we added the three mutations (NaCTp.G219R, −p.T227M and −p.L488P) associated with EE12 into this analysis. Furthermore, one mutation known to increase NaCT-mediated citrate transport4 was included as positive control (NaCTp.F500L). In total, we investigated the functional consequences of eight mutations (Table 1) in the SLC13A5 gene leading to amino acid substitutions in the NaCT transporter on protein expression, protein localization and citrate transport. Based on the known structure of the bacterial homologue, we then generated a model of the human NaCT transporter and performed a structural analysis to rationalize the observed effects on the transport of the model substrate citrate.
Results
Analysis of genetic variants in the SLC13A5 gene
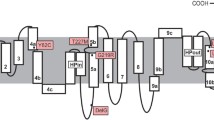
We identified several mutations in the SLC13A5 gene encoding human NaCT by database search (Table 1) and analyzed their localization in the predicted structure of the transporter (Fig. 1). The first single-nucleotide polymorphisms (SNPs) identified were rs150024888, rs142262032, rs150738356 and rs148049520, leading to the amino acid substitutions NaCTp.G219E, −p.D243N, −p.L420P and −p.L485R (Table 1), respectively. Furthermore, the mutation c.1498T > C (no rs-number) was included into this analysis as a proof of principle, because it is known that the respective amino acid exchange NaCTp.F500L results in a gain-of-function variant leading to a 2- to 3-fold stimulation of citrate uptake into NaCT-transfected cells4. Additional variants included into this study originate from the publication by Thevenon and colleagues12. They identified three mutations in the SLC13A5 gene [NaCTp.G219R (rs144332569), NaCTp.T227M (rs587777577) and NaCTp.L488P (rs587777578)] associated with autosomal epileptic encephalopathy. Because no functional characterizations of these mutations have been performed by the time of these experiments, they were included into the set of mutations analyzed in this study. A summary of all investigated mutations together with the in silico predicted functional consequences is shown in Table 1.

Transmembrane model of human NaCT with the localization of the respective mutant amino acids investigated in this study.
Analysis of transiently-transfected HEK293 cells expressing different NaCT mutant proteins
To compare the functional consequences of the investigated mutations on mRNA expression, protein amount and protein localization, quantitative RT-PCR, immunoblot analyses with total membrane fractions and plasma membrane fractions and confocal laser scanning microscopy were performed. Quantitative RT-PCR (Fig. 2A) demonstrated that SLC13A5 mRNA expression could be detected in all NaCT transfectants without significant differences, whereas SLC13A5 mRNA could not be detected in vector-transfected HEK293 cells. The immunoblot analyses (Fig. 2B,C) using the total membrane fraction (Fig. 2B) showed that NaCT protein is detectable in all total membrane fractions except in membrane fractions of vector-transfected control cells suggesting that all mutations have no effect on protein synthesis. Furthermore, all mutated NaCT proteins have the same band pattern as the wild-type NaCT protein. The protein localization analyses (Fig. 3) demonstrated that the mutations NaCTp.T227M, −p.D243N, −p.L485R and −p.F500L showed a localization directly comparable to the localization of the wild-type NaCT protein, whereas no membrane staining could be detected in control cells. Interestingly, mutations NaCTp.G219R, −p.G219E, −p.L420P and −p.L488P showed a slightly altered staining pattern with some NaCT protein retained intracellularly. This intracellular retention could be confirmed by analyzing the plasma membrane fraction (Fig. 2C). This analysis demonstrated that only in the mutants with a localization comparable to the localization of the wild-type protein (NaCTp.T227M, −p.D243N, −p.L485R and −p.F500L) NaCT protein could be detected in comparable amounts in the plasma membrane fraction whereas for all other mutants only a small amount of the protein was found in this fraction (Fig. 2C) suggesting that these mutations also affect protein localization.

Characterization of transfected HEK293 cells expressing wild-type and mutated NaCT proteins. Characterization of SLC13A5 mRNA expression (A) and of the NaCT protein amount in total membrane fractions (B) and in plasma membrane fractions (C) in transiently-transfected HEK293 cells. (A) Parental HEK293 cells were transiently-transfected with the expression plasmids encoding the wild-type NaCT transporter (wild-type) or with expression plasmids encoding variants of the NaCT protein. Parental HEK293 cells transfected with the empty expression vector served as control (control). All expression values are given in percent ± SD of the respective ß-actin mRNA expression measured under the same experimental conditions. (B) Immunoblot analysis of total membrane fractions of transiently-transfected HEK293 cells expressing different variants of the NaCT protein. Membrane isolation has been verified by staining the membrane protein pan-cadherin. Presented are the merged pictures of two immunoblots conducted under identical conditions. (C) Immunoblot analysis of plasma membrane fractions of transiently-transfected HEK293 cells expressing different variants of the NaCT protein. Membrane isolation has been verified by staining the membrane protein pan-cadherin. Presented are the merged pictures of two immunoblots conducted under identical conditions. The original immunoblots detecting NaCT in the different transient transfections are shown in the supplement (Supplement Figs 1 and 2).

Localization of wild-type and mutated NaCT proteins. Immunofluorescence analysis of NaCT protein localization in transiently-transfected HEK293 cells expressing wild-type or variant NaCT proteins. Empty vector-transfected HEK293 cells served as control.
Citrate transport by NaCT mutants
The effect of the different mutations on NaCT-mediated citrate transport was studied under established transfection and uptake conditions. Using these standardized conditions an uptake ratio of around 70-fold could be detected by comparing the uptake into wild-type transfected HEK cells with empty vector transfected HEK cells serving as control. As expected, the gain-of-function mutant NaCTp.F500L shows an increase in citrate transport of 82% (Fig. 4). A detailed kinetic analysis shown in Table 2 demonstrated that this increase in transport was due to a lower Km value resulting in a higher affinity of this mutant to citrate compared to the wild-type NaCT protein (Table 2). Furthermore, as predicted by in silico analysis (Table 1) the mutant −p.D243N has no effect on citrate transport, whereas all other mutants lead to considerably reduced or nearly abolished citrate uptake. Interestingly, the three mutants associated with autosomal epileptic encephalopathy (NaCTp.G219R, −p.T227M and −p.L488P) as well as the mutants −p.G219E and −p.L420P [with high Grantham scores17 and predicted to be probably damaging (Table 1)] showed no citrate uptake compared to the wild-type NaCT protein with uptake values comparable to the uptake values in control-transfected cells. The reduced uptake observed for mutant NaCTp.L485R was due to a higher Km value of this mutant for citrate (Table 2) compared to the Km value of the wild-type protein.

Citrate uptake mediated by wild-type and mutated NaCT proteins. Uptake of citrate (1 µM) into transiently-transfected HEK293 cells. Parental HEK293 cells were transiently-transfected and citrate uptake was measured under established experimental conditions. Cells were transfected with expression plasmids containing cDNAs encoding the wild-type NaCT protein (wild-type) or different mutant NaCT proteins. Citrate net uptake mediated by the wild-type NaCT protein was set to 100% and all net uptake values are given in relation to this uptake ± SD. ***P < 0.001
Structural modelling of wild-type and mutant NaCT proteins
In order to understand the effects of the mutations on a structural level, a three-dimensional model of the transport pore (the protein region built by the three-dimensional arrangement of the predicted transmembrane helices including residues F11-W116, R123-M159, K202-Y276, F314-G518, and K521-R549) of the NaCT protein with highlighted positions of the investigated amino acids was generated (Fig. 5). The model reveals that the residues G219, T227, L420, L485 and L488 are located within the predicted transport pore, whereas D243 is located distant from the pore and therefore, not influencing transport properties. The sidechain of F500 is oriented away from the pore and forms tight contacts with an adjacent helix. These contacts cannot be formed in the p.F500L mutant, which might lead to an overall enhanced flexibility of the NaCT structure. The residues L420 and L488 are located within helical structures and replacement by the helix breaker proline will disrupt the helical structure resulting in a reduction of transport activity as demonstrated. The effect of the p.T227M mutant is shown in detail in Fig. 5C,D. In the wild-type NaCT protein, T227 is located in immediate vicinity to the citrate molecule (Fig. 5C). In the p.T227M mutant, the sterically more extended methionine sidechain causes steric clashes with the citrate substrate (Fig. 5D) resulting in a pore too small to enable citrate transport. The effect of the G219 mutants is shown in Fig. 5E,F. The molecular modelling reveals that G219 is located at a sterically demanding position within a helix that tightly packs against the residue A113 within a second helix (Fig. 5E). Replacement by glutamate (p.G219E) or arginine [p.G219R (Fig. 5F)] leads to severe clashes with A133 of the second helix, which are predicted to disrupt the structure of the NaCT protein maybe thereby also affecting the membrane localization of the protein.

Molecular model of the human NaCT protein and localization of amino acids investigated in this study. (A) Molecular model of human NaCT indicating sites of investigated mutations. One of the subunits is shown as grey ribbon and the other subunit is shown in color gradient ranging from blue (N-terminus) to red (C-terminus). In both subunits, a citrate molecule is shown in stick presentation (atom-type coloring). The grey subunit additionally shows the position of the residues investigated by side-directed mutagenesis. These residues are shown in space-filled presentation using the following color code: G219 (yellow), T227 (orange), D243 (cyan), L420 (dark blue), L485 (red), L488 (green) and F500 (light blue). (B) Same presentation as in (A) but rotated by 90° around the horizontal axis. This corresponds to a view on the NaCT protein from the extracellular site. (C,D) Effect of the T227M mutation on the NaCT structure. In the wild-type protein, T227 (orange) is close to the substrate citrate (atom-type coloring). (D) Due to its longer sidechain M227 (orange) would form clashes with a citrate, which will no longer allow the transport of this substrate. (E,F) Effect of the G219R mutation on the NaCT structure. In the wild-type protein G219 (yellow) interacts with A133 (atom-type coloring) of an adjacent helix. (F) Due to its longer sidechain R219 (yellow) would form clashes with A133 thereby disrupting the NaCT structure.
Discussion
The aim of this study was to investigate the functional consequences of genetic variations in the SLC13A5 gene encoding the human uptake transporter NaCT (also known as mINDY) on the transport of the model substrate citrate. NaCT plays an important role in human tooth and bone development, energy metabolism and is crucial for the development and function of the human brain, because mutations in the SLC13A5 gene are associated with epileptic encephalopathy and developmental delay12,13.
Therefore, it is of special interest investigating the functional consequences of mutations and polymorphisms in the SLC13A5 gene encoding human NaCT. Genetic variations leading to amino acid substitutions were identified by database search. In addition, mutations associated with autosomal recessive encephalopathy12 were included into this analysis, three of them (NaCTp.G219R, −p.T227M and −p.L488P) analyzed by in vitro studies13,14. In total, eight mutations leading to amino acid substitutions including one known gain-of-function mutation (p.F500L) were analyzed by transient transfections and uptake studies using citrate as prototypic substrate for this uptake transporter. In addition, consequences of mutations on mRNA expression, protein amount and protein localization were investigated. Immunoblot analyses demonstrated that the molecular weights of all mutated NaCT proteins are comparable to the molecular weight of the wild-type NaCT protein (Fig. 2B) showing for all NaCT proteins the same band pattern. Interestingly, for the mutation NaCTp.G219R, this is in contrast to published data investigating this mutation13. For their analyses, Hardies and coworkers used V5-labelled NaCT proteins and an antibody directed against the V5 epitope. Whereas other mutated NaCT proteins could be detected with comparable patterns as the wild-type NaCT protein, the V5-labelled NaCTp.G219R protein was detected as a single protein band with a low molecular weight13. In our study, we used untagged NaCT proteins and analyzed total membrane fractions (Fig. 2B) of transfected HEK293 cells demonstrating that all mutated NaCT proteins showed a band pattern comparable to the band pattern of the wild-type NaCT protein, whereas in the cytosolic fraction, no NaCT protein could be detected (data not shown). Our immunoblot data were confirmed in a publication by Klotz and coworkers14 were the authors also investigated several mutations in the NaCT transporter including the NaCTp.G219R mutation. In their immunoblot analysis, all mutations showed a band pattern comparable to the wild-type NaCT protein suggesting that the altered staining pattern observed by Hardies et al. may be due to the used V5 epitope. The localization of the different NaCT proteins was studied by confocal laser scanning microscopy. This analysis demonstrated that the mutations NaCTp.T227M, −p.D243N, −p.L485R and −p.F500L showed a localization comparable to the localization of the wild-type protein, whereas the staining pattern for the mutations NaCTp.G219R, −p.G219E, −p.L420P and −p.L488P was slightly different with protein being retained intracellularly (Fig. 3). This intracellular retention was confirmed by analyzing the plasma membrane fraction (Fig. 2C) of the wild-type and of all mutant proteins. For the mutants NaCTp.G219R, −p.G219E, −p.L420P and −p.L485P only a small amount of the synthesized protein could be detected in the plasma membrane fraction suggesting that the mutations also affect membrane insertion of the mutated NaCT protein.
Standardized uptake assays using the prototypic NaCT substrate citrate demonstrated that the wild-type protein transports citrate with high uptake values (around 70-fold compared to the uptake into vector-transfected cells), whereas citrate transport mediated by the mutants −p.G219R, −p.G219E, −p.T227M, −p.L420P and −p.L488P was totally abolished (uptake values comparable to uptake values into control-transfected cells) and strongly reduced for the mutant NaCTp.L485R. On one hand, affected transport for the mutants –p.G219R, −p.G219E, −p.L420P and −p.L488P could be due to the altered localization with only a small amount of the protein being inserted into the plasma membrane. However, because wild-type NaCT has an uptake ratio for citrate of around 70-fold (comparing uptake into HEK-NaCT cells with the uptake into control-transfected HEK cells) at least a small protein amount located in the plasma membrane should be reflected in measurable uptake values. Because this is not the case, it is likely that beside the effect of the mutation on membrane localization also the transport function of the mutated NaCT protein is altered. Interestingly, for the mutation NaCTp.T227M, showing no uptake and for the mutation −p.L485R showing strongly reduced uptake, comparable amounts of protein could be detected in the plasma membrane fraction demonstrating that these mutations only affect NaCT transport function. For NaCTp.L485R this reduction was due to an altered Km value resulting in a lower affinity of this mutant to citrate compared to the affinity of the wild-type NaCT protein. As expected, transport mediated by the mutant NaCTp.F500L was stimulated in accordance to previously published data4. This increase in transport was due to a lower Km value resulting in a higher affinity of the mutated protein (Table 2) for citrate compared to the Km value of the wild-type protein. For the wild-type NaCT protein-mediated citrate transport, a Km value of 1 439 ± 270.8 µM was determined which is in the range of the previously published Km value18. Furthermore, for the mutations NaCTp.G219R and −p.T227M, these transport data are also in accordance with published data13 using also HEK293 cells and with a published analysis investigating several NaCT mutations in COS-7 cells14. Interestingly, all mutations with a large effect on NaCT-mediated transport were classified as probably damaging in the PolyPhen2 Prediction (Prediction of functional effects of human nsSNPs – http://genetics.bwh.harvad.edu/pph2) and have high Grantham scores17 (Table 1).
To rationalize the consequences of the mutations on a structural basis we established a computational model of the transport pore of the human NaCT protein and demonstrated that this model is in line with the experimentally observed transport data. Furthermore, this model is similar to the model of the SLC13 gene family member NaDC319 and highly similar [backbone RMSD (root-mean-square deviation) of 1 Å] to the NaCT model of the same working group found on their homepage (http://www.schlessingerlab.org/data/) but their model lacks the region around residue D243. Therefore, we generated a model including additional loop regions using a similar strategy. According to the dimeric oligomerization state of vcINDY, the homologous dicarboxylate transporter from Vibrio cholerae16 used as template, NaCT was also modeled as a dimer because inspection of the model suggests that dimerization is also sterically feasible for NaCT. Although a final determination of the dimerization state would require further experimental testing, none of the mutations investigated in the present study is close to the putative dimer interface and therefore the structural effects predicted for the mutations are not critically affected by the modeled oligomerization state. In the analysis of the vcINDY structure16, the authors detected one citrate molecule and one sodium molecule are bound per monomer and that conserved amino acids in the predicted transmembrane pore are important for ion binding and substrate translocation. In particular, the sodium ion binding site is highly conserved between human NaCT and vcINDY, which is also evident from a previous comparison of the human NaDC3 transporter and vcINDY19. This high conservation in the pore region between homologous residues is in line with the predicted functional consequences of the amino acid substitutions in the respective region of the NaCT transporter in this study. All amino acids investigated (except D243) are located within or near by the predicted transmembrane pore and are highly conserved among different species. A Clustal analysis demonstrated the G219, T227, D234 and L488 are found at identical positions in the alignment with NaCT proteins from human, mouse, pig, chimpanzee, macaque and zebrafish, whereas the amino acids L420 and F500 are conserved among most species with a variability only in mouse (Fig. 6A). Amino acid L485, where the mutation has a less distinct effect on citrate transport, is conserved in human, chimpanzee, macaque and pig.

Clustal analysis of NaCT proteins from different species and of members of the SLC13 family. (A) For the NaCT clustal analysis of different species the sequences NM_001143838.2 (human), HM998308.1 (chimpanzee), HM998307.1 (macaque), KF728381.1 (pig), NM_001004148.4 (mouse) and HM998309.1 (zebra fish) were used. (B) For the NaCT clustal analysis of the human SLC13 family members the sequences NM_001143838.2 (NaCT), NM_003984.3 (NaDC2), NM_022829.5 (NaDC3), NM_022444.3 (NaSI1) and NM_012450.2 (NaSUT1) were used. Clustal analysis was performed using the HUSAR program package from the German Cancer Research Center32. All investigated mutations are indicated as boxes.
L420 and L488 are located within predicted helices and replacement by helix breaking amino acids such as proline will disrupt this helical structure leading to a loss of transport activity and maybe affecting membrane insertion. Compared to the totally abolished transport of these two mutated proteins, the higher residual transport activity observed for the p.L485R mutation can most likely be attributed to the fact that the leucine sidechain is oriented away from the pore and can therefore be replaced by an arginine without causing an entire loss of structure. Amino acid residue F500 is also oriented away from the pore and the p.F500L mutation is predicted to enhance the flexibility of the NaCT structure due to poorer sidechain packing. This may be the explanation for the enhanced transport activity of this variant already published4 and shown here.
Interesting observations were made analyzing the effect of the two G219 mutants. In both mutants, NaCTp.G219R and −p.G219E, a complete loss of NaCT-mediated citrate transport activity could be observed. Molecular modelling reveals that the highly conserved glycine is located in sterically demanding position within a helix that tightly packs against A133 within a second helix (Fig. 5E,F). Due to the longer side chains of glutamate and arginine, they will form clashes with A133 thereby disrupting the protein structure, abolishing citrate transport and affecting plasma membrane insertion. The importance of this amino acid gets also obvious be analyzing both Clustal analysis (Fig. 6). G219 is not only highly conserved among NaCT proteins of different species (Fig. 6A) but is also conserved among all human SLC13 gene family members (Fig. 6B). The role of all mutations for the membrane insertion of the NaCT protein has to be analyzed in detail.
Human NaCT is mainly expressed in liver and brain20 and the consequences of NaCT dysfunction have been studied in knock-out animals. These analyses showed that the absence of NaCT in mice promotes longevity and protects from insulin resistance and adiposity10. Furthermore, NaCT deficiency led to defective tooth and bone development characterized by the absence of mature enamel, decreased bone mineral density and impaired bone formation11. In contrast to the human “knock-out” phenotype, it has not been reported that knock-out animals show evidence of epileptic activity of other neurological signs, which may be due to species differences of transporter expression or other compensatory mechanisms. Neurons are incapable of synthesizing tricarboxylic acid metabolites and are therefore dependent on the uptake of these metabolites synthesized by astrocytes21. Therefore, insufficient transport of citrate and other TCA intermediates due to mutations in NaCT may result in brain energy failure causing epileptic encephalopathy. Interestingly, the same mutations may lead to differences in disease expression. This has been demonstrated in a recent publication by Anselm and coworkers were they investigated mutations in the SLC13A5 gene identified in two siblings15. Both individuals carry the same mutations (p.G219R and p.Y82C) but only one of them had focal cortical dysplasia leading to brain surgery. These results suggest that additional factors downstream of the uptake transporter may be important for the development of this disease.
Conclusion
The human transporter NaCT, encoded by the SLC13A5 gene is important for teeth and bone development, energy metabolism, brain development and brain function by mediating the uptake of citrate and other intermediates of the tricarboxylic acid cycle from blood into cells (e.g. into hepatocytes and neurons). Mutations in the SLC13A5 gene are associated with epileptic encephalopathy and developmental delay. We analyzed eight mutations in the SLC13A5 gene leading to mutated NaCT proteins and could demonstrate that conserved amino acids within the predicted transmembrane pore are important for NaCT-mediated transport. The effect of all mutations on NaCT transport function could be explained on the structural basis of the NaCT transporter. Furthermore, some mutations may affect membrane insertion of the NaCT protein. Because HEK293 cells are not suitable in studying downstream effects of insufficient citrate uptake, additional studies using e.g. conditional knock-out mice models or in vitro studies with neural cells are required to analyze the effect of NaCT-deficiency in more detail and to explain obvious species differences.
Methods
Materials
[14C] Citrate (specific radioactivity 55 mCi/mmol) was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA). Unlabeled citrate was obtained from Fisher BioReagents, Fisher Scientific (Waltham, USA). Minimal essential medium, Dulbecco’s phosphate buffered saline, fetal bovine serum, G418-sulfate, penicillin streptomycin solution, Trypsin (0.05%)-EDTA (0.02%) solution, Lipofectamine 2000, and Optimem I were from Invitrogen/Thermo Fisher Scientific (Braunschweig, Germany). Sodium butyrate was purchased from Merck KgaA (Darmstadt, Germany). 12-Well tissue culture plates were obtained from Greiner Bio-One (Frickenhausen, Germany). Unless noted otherwise, all other chemicals were purchased from Carl Roth GmbH+ Co. KG (Karlsruhe, Germany).
Cloning and Mutagenesis of the SLC13A5 cDNA
The SLC13A5 cDNA encoding the human NaCT (mINDY) transporter was cloned by a PCR-based strategy as described18. Mutations not described before were identified by database analysis using the following databases: the NCBI-SNP database (www.ncbi.nml.nhi.gov/snp); the central mutation and SNP database (www.hgvs.org/central-mutation-snp-databases) and the genetic variation database (www.humgen.nl/snp_databases.html). Each of the selected mutations in the SLC13A5 gene was introduced separately into the expression vector pNaCT.31. Mutagenesis was performed using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, USA) according to the supplier’s instructions. Following primers were used for the mutagenesis reactions: oNaCTmutG219E: 5′-GGCCAGCATCGAGGGCACCGCCA-3′ (resulting in the plasmid pNaCT-mutG219E); oNaCTmutG219R: 5′-CGGCCAGCATCAGGGGCACCGCC-3′ (resulting in the plasmid pNaCT-mutG219R); oNaCT-mutT227M: 5′-CCACCCTGACCGGGATGGGACCCAA-3′ (resulting in the plasmid pNaCT-mutT227M); oNaCTmutD243N: 5′-CCAGATGAACGAGTTGTTTCCTCCCAGCAAGGACC-3′ (resulting in the plasmid pNaCT-mutD243N); oNaCTmutL420P: GGGCGGATTTGCTCCGGCTAAAGGATCCG-3′ (resulting in the plasmid pNaCT-mutL420P); oNaCTmutL485R: 5′-CGCTCCATCGGCCGCAATCCGCTGTAC-3′ (resulting in the plasmid pNaCT-mutL485R); oNaCTmutL488P: 5-CGGCCTCAATCCGCCGTACATCATGCTGC-3′ (resulting in the plasmid pNaCT-mutL488P); and oNaCTmutF500L: 5′-ACCCTGAGTGCCTCCCTTGCCTTCATGTTGC-3′ resulting in the plasmid pNaCT-mutF500L). Success of each mutagenesis was proofed by sequencing (LGC Genomics GmbH, Berlin, Germany).
Transient transfection
For studying the functional consequences of the mutations, HEK293 cells were transiently transfected with the respective wild-type plasmid pcNaCT.31, the empty expression vector pcDNA3.1(+) as control and the plasmids pNaCTmutG219R, pNaCTmutG219E, pNaCTmutT227M, pNaCTmutD243N, pNaCTmutL420P, pNaCTmutL485R, pNaCTmutL488P and pNaCTmutF500L encoding the different mutated NaCT protein. Transient transfection has been performed as described22.
Quantitative RT-PCR
Parental HEK293 cells were seeded at a cell density of 3 × 105 cells per well in poly-D-lysine (0.1 mg/ml) covered 12-well tissue plates. Twenty-four hours after seeding, cells were transiently transfected with the respective expression plasmid. Each transfection was performed with 1.5 µg DNA per well using Optimem I and Lipofectamine 2000 (Invitrogen/Thermo-Fisher Scientific) according to the supplier’s instructions. After a further incubation of twenty-four hours, medium containing Lipofectamine 2000 and Optimem I was changed for standard cell culture medium and cells were further incubated for additional 24 hours. Messenger RNA was isolated by using RNeasy Mini Kit (Qiagen, Düsseldorf, Germany), according to the manufacturer’s instructions. Subsequently, RNA was reverse-transcribed into sscDNA, using Transcriptor Universal cDNA Master (Roche Diagnostics, Mannheim, Germany). LightCycler-based quantitative RT-PCR (Roche Diagnostics, Mannheim, Germany) was performed with following primers: Forward primer oNaCT-RT.for (5′-CCGCCTCAATCCGCTGTACA-3′) and oNaCT-RT.rev (5′-AGTCCTGAGGAGGGTAAGGG-3′) as described23. The SLC13A5 mRNA expression in wild-type NaCT-transfected HEK293 cells, vector control-transfected HEK293 cells and in the HEK293 cells transfected with the different mutated cDNAs was calculated in relation to the expression of the housekeeping gene ß-actin and shown as relative SLC13A5 mRNA expression in percent ± SD (with n = 3 independent expression analysis followed by one-way ANOVA with Bonferroni’s multiple comparison test).
Uptake Assays
For uptake experiments, parental HEK293 cells were cultured and transiently transfected as described under quantitative RT-PCR. The uptake experiments using radiolabeled [14C]citrate were performed as described24. In brief, cells were washed with prewarmed (37 °C) uptake buffer (142 mM NaCl, 5 mM KCl, 1 mM K2HPO4, 1.2 mM MgSO4, 1.5 mM CaCl2, 5 mM glucose and 12.5 mM HEPES, pH 7.3). Subsequently, cells were incubated with 1 µM citrate in uptake buffer for 10 min at 37 °C. After incubation with the transport substrate, cells were washed three times with ice-cold uptake buffer, the cells were lysed with 0.2% SDS and the intracellular radioactivity was measured using a liquid scintillation counter (TriCarb 2800, Perkin Elmer Life Science GmbH, Rodgau-Jügesheim, Germany). Kinetic parameters (Km and Vmax values) of wild-type NaCT and of the mutants NaCT −p.L485R and p.F500L (showing altered citrate transport) were determined with the same experimental setup using 1 µM, 100 µM, 1000 µM and 500 µM citrate. All uptake experiments were performed on two separate days in triplicates each (n = 6). In each well, parental HEK293 cells were separately transfected and subsequently used for the uptake experiments.
Confocal laser scanning immunofluorescence analysis
Immunofluorescence was performed as described previously22. In brief, parental HEK293 cells were seeded at a cell density of 3.5 × 105 cells per well in poly-D-lysine (0.1 mg/ml) covered 12-well tissue plates. Each well contained an autoclaved cover slip for higher adherence of the cells. After 24 hours, cells were transiently transfected with 1.5 µg DNA per well of the respective expression plasmids using Lipofectamine 2000 and Optimem I, according to the manufacturer’s instructions. Forty-eight hours after seeding, medium was changed and 24 hours later, cells were washed with Tris-buffered saline (TBS). After fixation in ice-cold 70% methanol, the cells were incubated with TBS/Triton (0.4%) for 10 min. Subsequently, the cells were incubated with 2% BSA/H2O for 45 min and then 90 min with a dilution (1:350 in 2% BSA/H2O) of monoclonal mouse anti-human-SLC13A5 antibody (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) at room temperature. Afterwards, the cells were washed with TBS-Triton (0.1%) and TBS – Tween (0.05%), followed by incubation in a dilution (1:1 000 in 2% BSA/H2O) of the secondary antibody Cy2-coupled goat anti-mouse IgG (Dianova, Hamburg, Germany,) for 30 min at 37 °C. Subsequently, the cells were washed with TBS – Tween (0.05%), TBS Triton (0.1%), TBS and H2O. Then, the cover slips were fixated on microscope slides with mounting medium (Thermo Scientific, Dreieich, Germany). The immunofluorescence was detected with confocal laser scanning microscopy (Axiovert 100 M, Carl Zeiss GmbH, Jena, Germany). The images were sighted with the Zeiss LSM Image Browser version 4.2.0.121.
Plasma membrane protein expression
The influence of the respective mutant on protein expression and localization was investigated using an immunoblot analysis after isolation of the total membrane protein fraction and of the plasma membrane fraction. For isolation of the total membrane fraction, 5 × 106 parental HEK293 cells were seeded in 75 cm2 cell culture flasks for 24 hours under normal culture conditions. After these 24 hours, cells were transiently transfected with the respective wild-type or mutant expression plasmid or with the empty expression vector (32 µg plasmid/flask) as described above, grown for additional 24 hours before the HEK293 cell medium was changed. After additional 24 hours, cells were harvested and the plasma membrane fraction was prepared using the ProteoExtract Transmembrane Protein extraction Kit (Novagene, Merck-Company, Darmstadt, Germany) according to the manufacturer’s instructions. In brief, cells were washed two times with PBS at 4 °C and detached using a cell scraper. Then cells were transferred into a 15 mL falcon tube and centrifuged for 5 min at 1 000 g and 4 °C. The cell pellet was resuspended in 1 mL extraction buffer 1 supplemented with 5 µL protease inhibitor cocktail (supplied with the kit) and incubated for 10 min at 4 °C with gentle shaking. After centrifugation (5 min at 1000 g and 4 °C) the pellet (containing the membrane fraction) was resuspended in 200 µL extraction buffer 2B, supplied with 5 µL protease cocktail and incubated for 45 min at room temperature. After centrifugation (15 min at 16 000 g and 4 °C) the supernatant, containing the total membrane protein fraction was transferred to a fresh tube and the protein amount was calculated using a bicinchoninic acid assay (BCA Protein Assay Kit, Thermo Fisher Scientific, Rockford, Illinois, USA). For the isolation of the plasma membrane fraction the minute plasma membrane protein isolation kit (MoBi Tec, Goettingen, Germany) was used according to the manufacturer’s instructions. In brief, transiently-transfected cells were detached using a cell scraper, pelleted and resuspended in 500 µL buffer A. After incubation on ice, cellsuspension was vortexed and applied onto the filter unit. After centrifugation (30 s with 14 000 rpm) the pellet was vortexed again, centrifugalized, the supernatant was transferred into a fresh tube and centrifugalized again for 30 min with 16 000 rpm obtaining the total membrane fraction. This fraction was resuspended in 200 µL buffer B and intracellular vesicles were separated by centrifugation (5 min with 10 000 rpm). The supernatant was transferred into a fresh tube and the plasma membrane fraction was obtained by adding 1.6 mL ice-cold PBS and centrifuged again for 30 min with 16 000 rpm. The pellet containing the plasma membrane fraction was resuspended in PBS (with 0.5% Triton-100) and used for the immunoblot analyses.
Laemmli buffer (5×) and 20 µg of the respective protein (total membrane fraction or plasma membrane fraction) were mixed and denaturated at 95 °C for 5 min and then separated on 10% SDS-polyacrylamid gel. Subsequently, the proteins were transferred to Protran Nitrocellulose Membrane (Schleicher & Schuell, BioScience GmbH, Whatman Group, Dassel, Germany) and incubated with the primary monoclonal mouse anti-human-SLC13A5 antibody (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) in a dilution of 1:500 for one hour at room temperature. After washing with PBS-Tween, the secondary goat-anti mouse antibody was incubated in a dilution of 1:2 000 for 30 min at room temperature. Imaging was performed with the Chemidoc XRS imaging system (Biorad, Munich, Germany) using ECL Western Blotting Detection Reagents (GE Healthcare). Membrane protein isolation was verified by stripping the membrane and restaining the immunoblot with an antibody directed against the membrane marker protein pan-cadherin (Sigma-Aldrich, Taufkirchen, Germany) under the same experimental conditions with a 1:250 dilution.
Molecular Modelling
The structure of the human NaCT transporter was modeled using the structure of a homologous dimeric dicarboxylate transporter from Vibrio cholerae as a template [PDB code: 4F3516]. Both proteins exhibit a sequence similarity of 58%, which allowed to generate a NaCT model by homology modelling techniques. We noted that there already exists a model of NaCT (deposited on http://www.schlessingerlab.org/data/). However, this model lacks the region around residue D243 and therefore, we decided to generate a model including additional loop regions using a strategy similar to the one described25. Modelling was performed using Modeller9.1026 based on the INDY-NaCT sequence alignment published in16. The resulting model comprised residues F11-W116, R123-M159, K202-Y276, F314-G518, and K521-R549 of NaCT. In addition to the peptide chain itself, a sodium ion and a citrate molecule present in each subunit of the template crystal structure were also modeled in NaCT at the respective structure positions. The resulting model was verified using PROCHECK27 and WHATCHECK28 and revealed a good stereochemistry and no steric clashes. A comparison with the model by Schlessinger revealed a backbone RMSD (Root-mean-square-deviation) of 1.0 Å indicating that both models are highly similar. Mutations were modeled with SWISS-Model29 by selecting the lowest-energy sidechain rotamer for each mutated residue followed by 100 steps of energy minimization. Structure analysis and visualization was performed with visual molecular dynamics [VMD30] or RasMol (www.rasmol.org).
Data analysis
The topology of the human NaCT transporter was predicted using the program TopPred1.1031. Net uptake values of citrate uptake were calculated by subtracting the uptake into vector-transfected cells from the uptake into NaCT-transfected (wild-type or mutated NaCT protein) cells. The citrate uptake was normalized to the protein concentrations of the cell lysate. Finally, all net uptake values of the mutant NaCT proteins are given in percent of citrate uptake into wild-type NaCT-transfected cells. Changes in transport rates were analyzed using one-way ANOVA with Bonferroni´s multiple comparison test. Affinity constants (Km values) and maximal transport velocity (Vmax values) were calculated using Michaelis-Menten kinetics. A p-value of ≤0.05 was required for statistical significance. All calculations were performed using GraphPad Prim 5.01.The Clustal analyses were performed using the HUSAR program package32 from the German Cancer Research Center, Heidelberg.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Hediger, M. A. et al. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Arch 447, 465–468 (2004).
Pajor, A. M. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch 466, 119–130, https://doi.org/10.1007/s00424-013-1369-y (2014).
Inoue, K., Zhuang, L., Maddox, D. M., Smith, S. B. & Ganapathy, V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J Biol Chem 277, 39469–39476, https://doi.org/10.1074/jbc.M207072200 (2002).
Inoue, K., Zhuang, L., Maddox, D. M., Smith, S. B. & Ganapathy, V. Human sodium-coupled citrate transporter, the orthologue of Drosophila Indy, as a novel target for lithium action. Biochem J 374, 21–26, https://doi.org/10.1042/BJ20030827 (2003).
Zwart, R., Peeva, P. M., Rong, J. X. & Sher, E. Electrophysiological characterization of human and mouse sodium-dependent citrate transporters (NaCT/SLC13A5) reveal species differences with respect to substrate sensitivity and cation dependence. J Pharmacol Exp Ther 355, 247–254, https://doi.org/10.1124/jpet.115.226902 (2015).
Neretti, N. et al. Long-lived Indy induces reduced mitochondrial reactive oxygen species production and oxidative damage. Proc Natl Acad Sci USA 106, 2277–2282, https://doi.org/10.1073/pnas.0812484106 (2009).
Schwarz, F. et al. Knockdown of Indy/CeNac2 extends Caenorhabditis elegans life span by inducing AMPK/aak-2. Aging (Albany NY) 7, 553–567 (2015).
Wang, P. Y. et al. Long-lived Indy and calorie restriction interact to extend life span. Proc Natl Acad Sci USA 106, 9262–9267, https://doi.org/10.1073/pnas.0904115106 (2009).
Rogers, R. P. & Rogina, B. Increased mitochondrial biogenesis preserves intestinal stem cell homeostasis and contributes to longevity in Indy mutant flies. Aging (Albany NY) 6, 335–350 (2014).
Birkenfeld, A. L. et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab 14, 567–567, https://doi.org/10.1016/j.cmet.2011.09.005 (2011).
Irizarry, A. R. et al. Defective enamel and bone development in sodium-dependent citrate transporter (NaCT) Slc13a5 deficient mice. PloS One 12, e0175465, https://doi.org/10.1371/journal.pone.0175465 (2017).
Thevenon, J. et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet 95, 113–120, https://doi.org/10.1016/j.ajhg.2014.06.006 (2014).
Hardies, K. et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 138, 3238–3250, https://doi.org/10.1093/brain/awv263 (2015).
Klotz, J., Porter, B. E., Colas, C., Schlessinger, A. & Pajor, A. M. Mutations in the Na+/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol Med 22, https://doi.org/10.2119/molmed.2016.00077 (2016).
Anselm, I., MacCuaig, M., Prabhu, S. B. & Berry, G. T. Disease heterogeneity in Na+/Citrate cotransporter deficiency. JIMD Rep. 31, 107–111, https://doi.org/10.1007/8904_2016_546 (2016).
Mancusso, R., Gregorio, G. G., Liu, Q. & Wang, D. N. Structure and mechanism of a bacterial sodiumdependent dicarboxylate transporter. Nature 491, 622–626, https://doi.org/10.1038/nature11542 (2012).
Tavtigian, S. V., Greenblatt, M. S., Lesueur, F. & Byrnes, G. B. IARC Unclassified Genetic Variants Working Group. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 29, 1327–1336, https://doi.org/10.1002/humu.20892 (2008).
von Loeffelholz, C. et al. The human longevity gene homolog INDY and Interleukin-6 interact in hepatic lipid metabolism. Hepatology 66, 616–630, https://doi.org/10.1002/hep.29089 (2017).
Schlessinger, A., Sun, N. N., Colas, C. & Pajor, A. M. Determinants of substrate and cation transport in the human Na+/dicarboxylate cotransporter NaDC3. J Biol Chem 289, 16998–17008, https://doi.org/10.1074/jbc.M114.554790 (2014).
Bergeron, M. J., Clemencon, B., Hediger, M. A. & Markovich, D. SLC13 family of Na+-coupled di- and tricarboxylate/ sulfate transporters. Mol Aspects Med 34, 299–312, https://doi.org/10.1016/j.mam.2012.12.001 (2013).
Rodrigues, T. B., Valette, J. & Bouzier-Sore, A. K. 13C NMR spectroscopy applications to brain energy metabolism. Frontiers in neuroenergetics 5, 9, https://doi.org/10.3389/fnene.2013.00009 (2013).
Glaeser, H., Mandery, K., Sticht, H., Fromm, M. F. & König, J. Relevance of conserved lysine and arginine residues in transmembrane helices for the transport activity of organic anion transporting polypeptide 1B3. Br J Pharmacol 159, 698–708, https://doi.org/10.1111/j.1476-5381.2009.00568.x (2010).
Huber, S. et al. Esters of Bendamustine are by far more potent cytotoxic agents than the parent compound against human sarcoma and carcinoma cells. PloS One 10, e0133743, https://doi.org/10.1371/journal.pone.0133743 (2015).
Seithel, A. et al. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab Dispos 35, 779–786, https://doi.org/10.1124/dmd.106.014407 (2007).
Colas, C., Pajor, A. M. & Schlessinger, A. Structure-Based identification of inhibitors for the SLC13 Family of Na+/Dicarboxylate cotransporters. Biochemistry 54, 4900–4908, https://doi.org/10.1021/acs.biochem.5b00388 (2015).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 47(5), 1–32, https://doi.org/10.1002/0471250953.bi0506s47 (2014).
Laskowski, R. A., Macarthur, M. W., Moss, D. S. & Thornton, J. M. Procheck - a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26, 283–291, https://doi.org/10.1107/S0021889892009944 (1993).
Hooft, R. W. W., Vriend, G., Sander, C. & Abola, E. E. Errors in protein structures. Nature 381, 272–272, https://doi.org/10.1038/381272a0 (1996).
Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18, 2714–2723, https://doi.org/10.1002/elps.1150181505 (1997).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J Mol Graph Model 14, 33–38, https://doi.org/10.1016/0263-7855(96)00018-5 (1996).
Claros, M. G. & von Heijne, G. TopPred II: an improved software for membrane protein structure predictions. Comput Appl Biosci 10, 685–686 (1994).
Senger, M., Glatting, K. H., Ritter, O. & Suhai, S. X-HUSAR, an X-based graphical interface for the analysis of genomic sequences. Comput Methods Programs Biomed 46, 131–141 (1995).
Acknowledgements
We thank Katrin Singer and Claudia Hoffman for excellent technical assistance and Eileen Socher for preparing the structure presentation of the NaCT transporter. The present work was performed in fulfillment of the requirements for obtaining the degree “Dr. med.” at the Friedrich-Alexander-Universität Erlangen-Nürnberg for Stefan Selch. ALB was supported by a grant from the DFG (BI1292/4-2, IRTG2251).
Author information
Authors and Affiliations
Contributions
S.S. contributed to the research design, data analysis and drafting this article, A.C. contributed to research design and revised the article, H.S. contributed to research design, modelling and data analysis, A.L.B. contributed to research design and drafting of the article, M.F.F. contributed to research design and manuscript writing and J.K. contributed to research design, data analysis and manuscript writing. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Selch, S., Chafai, A., Sticht, H. et al. Analysis of naturally occurring mutations in the human uptake transporter NaCT important for bone and brain development and energy metabolism. Sci Rep 8, 11330 (2018). https://doi.org/10.1038/s41598-018-29547-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29547-8
- Springer Nature Limited
This article is cited by
-
The citrate transporters SLC13A5 and SLC25A1 elicit different metabolic responses and phenotypes in the mouse
Communications Biology (2023)
-
Role of sodium dependent SLC13 transporter inhibitors in various metabolic disorders
Molecular and Cellular Biochemistry (2023)