
Abstract
Dysfunction in dopamine (DA) systems is a prominent feature in schizophrenia patients and may result from the abnormal development of mesencephalic (mes)DA systems. Maternal immune activation (MIA) and developmental vitamin D (DVD)-deficiency both induce schizophrenia-relevant dopaminergic abnormalities in adult offspring. In this study, we investigated whether maternal administration of the vitamin D hormone (1,25OHD, VITD) could prevent MIA-induced abnormalities in DA-related behaviors and mesDA development. We administrated the viral mimetic polyriboinosinic-polyribocytidylic (poly (I:C)) simultaneously with 1,25OHD and/or their vehicles, to pregnant mouse dams at gestational day 9. Maternal treatment with VITD prevented MIA-induced hypersensitivity to acute DA stimulation induced by amphetamine, whereas it failed to block prepulse inhibition deficiency in MIA-exposed offspring. MIA and VITD both reduced fetal mesDA progenitor (Lmx1a + Sox2+) cells, while VITD treatment increased the number of mature (Nurr1 + TH+) mesDA neurons. Single-cell quantification of protein expression showed that VITD treatment increased the expression of Lmx1a, Nurr1 and TH in individual mesDA cells and restored normal mesDA positioning. Our data demonstrate that VITD prevents abnormal dopaminergic phenotypes in MIA offspring possibly via its early neuroprotective actions on fetal mesDA neurons. Maternal supplementation with the dietary form of vitamin D, cholecalciferol may become a valuable strategy for the prevention of MIA-induced neurodevelopmental abnormalities.
Similar content being viewed by others


Introduction
Today schizophrenia is conceptualized as a polygenetic disorder possibly triggered by diverse environmental factors. Epidemiological studies implicate a growing number of pre- and perinatal environmental risk factors, including fetal hypoxia, obstetric complications, maternal infection and/or immune activation (MIA), and certain nutritional deficiencies such as developmental vitamin D (DVD)-deficiency1,2,3,4. These factors combine to affect the normal trajectory of brain development in ways that remain opaque.
Dopamine dysregulation has long been proposed as an important pathophysiological feature of schizophrenia. Molecular imaging studies suggest that neurochemical abnormalities of presynaptic dopamine (DA) uptake and/or release may be core to the psychotic component in most schizophrenic patients5. We have proposed that early alterations in the trajectory of developing DA neurons may represent a first step in the journey towards the dopaminergic changes observed in schizophrenia6.
Animal models of MIA and DVD-deficiency provide support for this hypothesis7,8,9,10. Not only do these models capture DA-related behavioral and cognitive deficits relevant to schizophrenia7,8,9,10,11,12,13,14, but also they suggest that dopaminergic alterations originate in early fetal development14,15,16. DA neurons are born very early around 5 weeks post conception (1st trimester) in the human embryonic mesencephalon17, which is roughly equivalent to gestational day (GD) 9.5 in mice18,19. MIA on GD 9 affects the genesis of mesencephalic dopamine (mesDA) neurons and affects the expression of genes crucial for their establishment, including sonic hedgehog (SHH) and fibroblast growth factor 8 (FGF8)14. MIA also changes the fetal expression of nuclear receptor related 1 protein (Nurr1), which is essential for mesDA differentiation and maintenance11,14. In DVD-deficient rat fetuses, the ratio of DA neurons between the two major mesDA neuronal subgroups (the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA)), are altered16. DVD-deficiency in rats also reduces the expression of Nurr1 and tyrosine hydroxylase (TH), the rate-limiting enzyme in DA synthesis in mesDAs16,20,21,22. MIA and DVD-deficiency in rodents also lead to altered DA metabolisms in neonatal brains13,23,24,25.
MIA is also a risk factor for other psychiatric conditions most noticeably autism11,26. Using a MIA model that is based on maternal treatment with the viral mimetic polyriboinosinic-polyribocytidylic (poly (I:C)), we have recently shown that when the active vitamin D hormone (1,25OHD) (here on referred to as VITD) is administered to MIA-exposed dams simultaneously with poly(I:C), it prevents certain MIA-induced behavioral deficits in associative learning, stereotyped digging and social interaction in juvenile offspring27. Such behavioural disruptions are consistent but not exclusive to autism-related phenotypes. In the present study, we investigated whether administration of VITD to MIA-exposed dams also prevents DA-related behavioral alterations in MIA offspring. We further explored the effects of VITD and MIA on the expression of factors involved in early mesDA neuron maturation. To this aim, we established an automated image analysis method for the identification and measurement of individual mesDA cells. This method allowed us to assess the acute effects of MIA and VITD treatments on mesDA neurogenesis and maturation at the single-cell level. GD11 was chosen to examine post-MIA effects at this time point, which allowed us to assess both active mesDA neurogenesis and the early stages of mesDA neuron maturation. We show that although MIA and VITD both reduced mesDA progenitor numbers, VITD treatment increased mature mesDA neuron number and increased the expression of key mesDA differentiation factors. Therefore, one plausible neuroprotective action of VITD may be its pro-differentiating role in mesDA neurogenesis in the MIA-exposed fetuses.
Materials and Methods
Animals
Female and male breeder C57BL6/N mice were obtained from Charles River Laboratories (Sulzfeld, Germany) at the age of 12–14 weeks. They were kept in regular open cage systems (Macrolon Type-III, Turnhout, Belgium) and had had a libitum access to water and standard rodent chow (Kliba 3430, Kaiseraugst, Switzerland) that contained 1000IU cholecalciferol/kg. Breeding began after 2 weeks of acclimatization to the animal holding rooms. Animals were housed and tested under a reversed light–dark cycle (lights on from 19h00 to 07h00). All procedures described in the present study had been approved by the Cantonal Veterinarian’s Office of Zurich, and all efforts were made to minimize the number of animals used and their suffering.
Experimental groups
The active hormonal form of VITD (1α, 25-dihydroxy vitamin D3 = 1,25OHD; solid powder, Calbiochem, EMD Millipore, Cat No 679101-50UG, Switzerland) was prepared as described previously27. Sterile corn oil (Sigma–Aldrich, Switzerland) was used as vehicle (VEH). Poly(I:C) (potassium salt; Sigma–Aldrich, Switzerland) was prepared using sterile, pyrogen-free 0.9% NaCl (saline), and injected as described previously28. Saline (CON) was used as the vehicle for poly (I:C).
C57BL6/N female mice were subjected to a timed-mating procedure as described previously28. Pregnant dams on GD 9 were first injected subcutaneously with VITD (400 ng/kg/2 ml) or vehicle (VEH), and then immediately injected intravenously with either poly(I:C) (POL, 5 mg/kg; calculated based on the pure form) or saline solution (CON) at a volume of 5 ml/kg as described previously27,28. Thus, the experiment consisted of 4 groups: CON-VEH, CON-VITD, POL-VEH, POL-VITD. VITD dose was chosen based on pilot studies that showed no effects on AMPH-mediated locomotion or PPI (Fig. S1).
Behavioral Studies
Weaned offspring were housed in same-sex groups of 3–4 animals per cage28. Behavioral testing was conducted at 10 weeks of age. To minimize possible confounds arising from litter effects29, only 1 male offspring per litter was randomly selected. Hence, the number of offspring in each experimental group was equal to the number of treated dams. The group sizes were as follows: N(CON-VEH) = 11, N(CON-VITD) = 8, N(POL-VEH) = 11, and N(POL-VITD) = 8. All offspring were first subjected to prepulse inhibition (PPI), followed by amphetamine (AMPH) locomotion two to three days later. All behavioral tests were performed in the dark phase of the light-dark cycle.
Prepulse Inhibition of the Acoustic Startle Reflex
PPI was conducted using startle chambers (San Diego Instruments, California, USA) as described previously (for details see suppl methods)27,28. PPI was indexed by percentage inhibition of the startle response obtained in pulse-alone trials by the following expression: 100% × (1 − [mean reactivity on prepulse-plus- pulse trials/mean reactivity on pulse-alone trials]). In addition, the animals’ reactivity to pulse-alone trials (i.e., startle reactivity) and prepulse-alone trials (i.e., prepulse-induced reactivity) were also measured and analyzed.
Spontaneous and Amphetamine-induced Locomotor Activity
Testing conditions, lighting and amphetamine dose were all as previously described11,28. Briefly, the animals were first acclimatized to the open field for 20 min and were then injected with sterile 0.9% saline and immediately returned to the same arenas for another 20 min. Subsequently, the animals were injected intraperitoneally (i.p.) with D-amphetamine sulfate (=AMPH; Sigma-Aldrich, Switzerland) at dose of 2.5 mg/kg (5 ml/kg) and monitored for a period of 60 min.
Fetal Tissue Collection and Immunohistochemistry
A second cohort of pregnant dams were prepared and assigned to poly(I:C), VITD, or appropriate vehicle on GD 9 as described above. The number of dams assigned to each treatment group was N(CON-VEH) = 5, N(CON-VITD) = 4, N(POL-VEH) = 5, and N(POL-VITD) = 7. Two fetuses per dam were randomly collected (see below), so that the number of fetus in each group was n(CON-VEH) = 10, n(CON-VITD) = 8, N(POL-VEH) = 10, and N(POL-VITD) = 14.
Fetal mesDA development was assessed at GD11 for the following reasons. First, at this age, the four major mesDA subpopulations we wished to assess (see below) are all present30. Second, by GD 11 the elevation in maternal inflammatory cytokines and chemokines induced by MIA has returned to baseline levels31,32,33, thus removing any active cytokine-mediated cellular effects. Third, the half-life of 1,25OHD is 4–6 hours34. Therefore, it is likely the active agent would have been eliminated after 48 hrs.
To collect the fetuses, dams were decapitated without prior anesthesia. The uterus was exposed and collected fetuses were immersion-fixed in 4% paraformaldehyde overnight. Fixed brains were soaked in 30% sucrose solution prior to embedding (Tissue-Tek, Emgrid, Australia). The mesencephalon cryosections were obtained in a one in three series. Sections were immunohistochemically processed simultaneously to eliminate variability. One series was processed for Sox2/Lmx1a, and a second series for Nurr1/TH. Primary antibodies were: rabbit anti-Sox2 (1:200, Millipore Chemicon, Australia), goat anti-Lmx1a (1:100, Santa Cruz Biotechnology, USA), sheep anti-TH (1:100, Novus Biologicals, USA), and rabbit anti-Nurr1 (1:500, Santa Cruz Biotechnology, USA). Fluorophore-conjugated secondary antibodies were diluted 1:1000 (Thermo Fisher Scientific, Australia). Nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI, 1:1000, Sigma-Aldrich, Australia).
Image acquisition and quantitative immunochemistry
Immunostaining was assessed using a confocal microscope (Tie, Nikon Inc.) equipped with a Spectral Applied Research’s Diskovery spinning disk module with an inverted spinning disk. Images were captured and stitched using NIS software (Nikon Inc.). Images were acquired using 60x oil N.A. 1.4 objective (CFI Apo Lamda/W.D. 0.14 mm), providing a pixel size 0.092 × 0.092 µm2. Four mesDA subpopulations were assessed: mesDA progenitors (Lmx1a+Sox2+), post-mitotic (Lmx1a+Sox2−) mesDA neurons, immature (Nurr1+TH−) mesDA neurons, and mature (Nurr1+TH+) mesDA neurons. Briefly, images were background-corrected using ImageJ software and were then analyzed using CellProfiler software (Broad Institute, Massachusetts, USA). This allowed us to quantify cell number, mediolateral/dorsoventral positions, individual protein expression (mean gray value per cell), and nuclear morphological parameters (for image processing and quantitative immunochemistry details see Fig. S2).
Statistical Analysis
All data were analyzed using parametric analysis of variance (ANOVA). Percent PPI was analyzed using a 2 × 2 × 3 × 3 (POL treatment × VITD treatment × pulse level × prepulse level) ANOVA. In the AMPH sensitivity test, the total distance moved was expressed as a function of 5-min bins and was analyzed by 2 × 2 × 4 (POL treatment × VITD treatment × 5-min bins) repeated-measure ANOVAs for the initial habituation and saline-treatment phases, and by 2 × 2 × 12 (POL treatment × VITD treatment × 5-min bins) repeated-measure ANOVA for the subsequent AMPH treatment phase. The quantitative immunochemistry data were analyzed using a 2 × 2 (POL treatment × VITD treatment) ANOVA using a repeated-measure design (repeated for section intervals). The distribution data were analyzed using a 2 × 2 (POL treatment × VITD treatment) ANOVA using a repeated-measure design (repeated for bins). Following these initial ANOVAs, restricted ANOVAs or Dunnett’s Multiple Comparison Tests were conducted whenever appropriate. Statistical significance was set at p < 0.05.
Results
Effect of maternal VITD treatment on MIA-induced behaviors
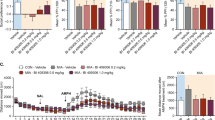
Consistent with previous studies14,28,32, MIA impaired PPI in adult offspring (main overall effect of MIA: F(1,34) = 4.63, p < 0.05; Fig. 1A). The MIA-induced effect on PPI was driven primarily by a marked reduction the 110 dBA pulse condition (main effect of MIA treatment at the 110 dBA condition: F(1,34) = 13.19, p < 0.001), but not in the 100 or 120 dBA conditions (100 dBA: F(1,34) = 0.67, p = 0.42; 120 dBA: F(1,34) = 3.04, p = 0.09). Maternal VITD treatment did not prevent MIA-induced PPI impairments (Fig. 1A).

The effects of single and combined MIA and VITD on prepulse inhibition and amphetamine sensitivity in adult offspring. (A) Prepulse inhibition (PPI) of the acoustic startle reflex was used to investigate sensorimotor gating in adult offspring. %PPI is shown for three startling amplitudes (100, 110 and 120 dBA, which are noted as P-100, P-110 and P-120, respectively) and prepulse intensities (71, 77 and 83 dBA, corresponding to +6, +12 and +18 dBA above background white noise). Poly(I:C) exposure impaired P-110dBA PPI. The bar plots represent the mean %PPI for all prepulse and pulse levels. All values were medians ±SEM. *p < 0.05, reflecting the significant main effect of MIA on mean % PPI; ***p < 0.001, reflecting the significant main effect of MIA in the P-110 condition. (B) The line plot shows total distance travelled in an open field per bin (=5 min) during initial habituation and subsequent saline administration periods, followed by the amphetamine (AMPH; 2.5 mg/kg, i.p.) administration phase. *p < 0.05, reflecting the significant difference between POL-VEH and CON-VEH offspring (AMPH bins 1–9); +p < 0.05, reflecting the significant difference between POL-VEH and POL-VitD offspring (AMPH bins 11 and 12). All values were means ± SEM.
In contrast, maternal VITD treatment prevented the emergence of AMPH hypersensitivity in offspring exposed to MIA. In line with previous findings14,23,28,32, AMPH-induced locomotor activity was increased in MIA offspring (Fig. 1B). This effect of MIA was blocked when pregnant dams were co-treated with VITD, leading to a significant three-way interaction between POL treatment, VITD treatment, and bins (F(11,34) = 1.83, p < 0.05). Subsequent post-hoc group comparisons at each individual 5-min bin confirmed a significant increase in POL-VEH mediated locomotion relative to CON-VEH offspring (all p < 0.05; Fig. 1B), and between POL-VEH and POL-VitD offspring at bin 11 and 12 (p’s < 0.05; Fig. 1B). Neither MIA nor maternal VITD treatment affected locomotor activity in the initial habituation and saline treatment phases of the test (Fig. 1B).
MIA and VITD treatments both reduce mesDA progenitor number
At GD11, mesDA progenitors are still proliferating18,19. Here we examined mesDA progenitors co-expressing the mesDA specification factor Lmx1a and the neural stem cell transcription factor Sox2 (Fig. 2A–D and A’–D’). Sox2 is absent in post-mitotic Lmx1a + mesDAs (Fig. 2C–D and C’–D’). There was a significant interaction between MIA and maternal VITD treatment on the number of mesDA progenitors (Lmx1a+Sox2 +) (F(1,36) = 5.19, p < 0.05; Fig. 2E). Post hoc comparisons revealed that mesDA progenitors were significantly reduced in MIA-treated fetuses (POL-VEH) (F(1,36) = 6.68, p < 0.05), VITD-treated fetuses (CON-VITD) (F(1,36) = 8.12, p < 0.05) and MIA-VITD co-treated fetuses (POL-VITD) (F(1,36) = 4.51, p < 0.05) relative to controls (CON-VEH). Hence, MIA and VITD both reduced mesDA progenitor numbers in GD11 fetuses.

The effects of single and combined MIA and VITD on early mesencephalic dopamine (mesDA) cell number. (A–D) Fate mapping of developing mesDA cells in a medial coronal mesencephalic (MES) sections at gestational day (GD) 11. Markers are the nuclear dye DAPI (blue, A), mesDA specification factor Lmx1a (green, B), neuronal progenitor maker Sox2 (red, C), and a channel-merged image (D). (A’–D’) Higher magnification images of the dashed outlined boxes representing the floor plate (FP) from (A–D) accordingly. Lmx1a specifically marked mesDA cells in the FP dorsoventrally from ventricular zone (VZ), intermediate zone (IZ) to mantle zone (MZ). MesDA progenitors (Lmx1a+Sox2+) were primarily located in the proliferative VZ, in contrast, post-mitotic (Lmx1a+Sox2−) mesDAs were mostly located in IZ and MZ. (E) The cell number of these two mesDA subpopulations were counted using CellProfiler software at 60 µm intervals along the anterior-posterior (A–P) axis. The numbers of mesDA progenitors (Lmx1a+Sox2+) were reduced by all treatments relative to the control (CON-VEH) (p’s < 0.05). (F) Post-mitotic (Lmx1a+Sox2−) mesDA number was not altered by any treatment. (G–J) Fate mapping of post-mitotic mesDA subgroups that are characterized by Nurr1 and TH in medial coronal MES sections at GD11. Markers are the nuclear dye DAPI (blue, G), Nurr1 (magenta, H), TH (green, I), and a channel-merged image (J). (G’–J’) Higher magnification images of the dashed outlined boxes representing FP from (G–J) accordingly. Immature Nurr1+ mesDAs were primarily located in the IZ and MZ. Mature (Nurr1+ TH+) mesDAs were primarily located in MZ. (K) There were no significant differences of immature mesDA number among treatment groups (p’s > 0.05). (L) VITD treatment itself increased mature (Nurr1+ TH+) mesDA number compared to its vehicle (VEH) (p < 0.05). VITD treatment itself (CON-VITD) particularly increased mature mesDA number in the posterior MES compared to control (CON-VEH) (p’s < 0.05). All values were means ± SEM. *P < 0.05. n.s., represents not statistically significant. Scale bars: 100 µm (A–D and G–J); 50 µm (A’–D’ and G’–J’).
Maternal VITD treatment increases the number of mature mesDAs
Consistent with previous studies18, immature (Nurr1+TH−) cells were found in the IZ, whereas mature (Nurr1+TH+) cells were largely restricted to the MZ (Fig. 2G–J and G’-J’). MIA and VITD had no effect on the number of immature mesDAs (p’s > 0.05; Fig. 2K). However, there was a significant main effect of maternal VITD treatment on mature mesDAs (F(1,40) = 5.574, p < 0.05; Fig. 2L). Significant interactions of MIA x VITD treatments with section position was also found along the anterior-posterior (A-P) axis of the MES (F(6,240) = 2.941, p < 0.05). Post hoc comparison revealed that VITD treatment itself (CON-VITD) increased mature mesDA number relative to control (CON-VEH), particularly in the posterior MES (p’s < 0.05).
MIA and VITD treatments affect the position of mesDA progenitors and mature mesDAs
CellProfiler allowed us to position individual mesDAs in the floor plate (FP). We assessed the average mediolateral (ML) position, (x distance from the midline of the coronal MES section, x0) and average dorsoventral (DV) position (y distance from the most ventral point of the ventricle, y0) (Fig. 3A,B).

The effects of single and combined MIA and VITD treatments on the coronal positioning of mature mesencephalic dopamine neurons (mesDAs). (A) Schematic image of registered floor plate (FP) in a medial coronal mesencephalic (MES) section with mediolateral (ML, x) and dorsoventral (DV, y) positions of cells. The coordinates (x0, y0) were chosen as the most ventral point of ventricle (V) along the midline (*). The black dot represents the mean center positioning (x n , y n ) of a representative mesDA nucleus (ellipse). The ML positioning (x) of mesDA cells was measured bilaterally as the absolute distance from the center of mesDA nucleus (x n ) to the coordinate (x0) = ABS (x n −x0). The DV positioning (y) of mesDA cell was calculated as the distance from the center of mesDA nucleus (y n ) to the coordinate (y0) = (y0−y n ). (B) An example of mesDA positioning in the FP. Yellow dots represent mesDA progenitors (Lmx1a+Sox2+), and green dots represent post-mitotic (Lmx1a+Sox2−) mesDAs. (C) A representative FP section showing cells that were triple-labeled by DAPI (blue), Lmx1a (green) and Sox2 (red). No significant effects of MIA or VITD treatment were noticeable for the mean ML (D) or DV (E) positioning of mesDA progenitors (p’s > 0.05). (F) There were no significant effects of MIA or VITD treatments on the mean ML positioning of post-mitotic (Lmx1a+Sox2−) mesDAs (p’s > 0.05). (G) There were significant interactions between MIA or VITD treatments on the average DV positioning of post-mitotic (Lmx1a+Sox2−) (p < 0.05). MIA treatment (POL-VEH) reduced the DV positioning of post-mitotic mesDAs compared to its control (CON-VEH) (p < 0.05). (H) A representative FP of a coronal MES section showing cells that were triple-labeled by DAPI (blue), Nurr1 (magenta) and TH (green). There were no significant effects of MIA or VITD treatment for the mean ML (I) or DV (J) positioning of immature (Nurr1+TH−) mesDAs in the FP (p’s > 0.05). (K) MIA treatment (POL) reduced the mean ML positioning of mesDAs relative to CON (p < 0.05). (L) There were significant interactions between MIA x VITD treatments in the average DV positioning of mature (Nurr1+TH+) mesDAs (p < 0.05). Post hoc comparison revealed MIA treatment (POL-VEH) decreased the mean DV positioning relative to the control (CON-VEH) (p < 0.05). Additionally, the co-treatment of VITD restored the mean DV positioning of mature mesDAs in POL-VITD group compared to POL-VEH group (p < 0.05). All values were means ± SEM. *p < 0.05. n.s. represents not statistically significant. Scale bars: 50 µm.
MIA or VITD had no effect on the ML or DV positioning of mesDA progenitors (Lmx1a + Sox2+) in the FP (p’s > 0.05; Fig. 3C–E). There were also no significant effects of MIA or VITD treatments on the average ML positioning of post-mitotic (Lmx1a + Sox2−) mesDAs (p’s > 0.05; Fig. 3F). There was significant interactions between MIA and VITD treatments on the average DV positioning of post-mitotic (Lmx1a + Sox2−) (F(1,36) = 4.616, p < 0.05; Fig. 3G). Subsequent post hoc comparison revealed that MIA treatment (POL-VEH) reduced the DV positioning of post-mitotic mesDAs compared to its control (CON-VEH) (F(1,36) = 5.997, p < 0.05).
There were no significant main effects or interactions of MIA or VITD treatments on the average ML or DV positioning of immature (Nurr1 + TH−) mesDAs in the FP (p’s > 0.05; Fig. 3H–J). In contrast, there was a significant main effect of MIA treatment on the mean ML positioning of mature (Nurr1+TH+) mesDAs (F(1,40) = 5.962, p < 0.05; Fig. 3K). In addition, there were significant interactions between MIA x VITD treatments in the average DV positioning of mature (Nurr1+TH+) mesDAs (F(1,40) = 5.128, p < 0.05; Fig. 3L). Post hoc comparisons showed that MIA treatment (POL-VEH) reduced the DV positioning of mature mesDAs compared to its control (CON-VEH) (F(1,40) = 5.871, p < 0.05). Most intriguingly, abnormal DV positioning of mature mesDAs in MIA fetuses was restored by the co-administration of VITD (POL-VITD) (F(1,40) = 6.125, p < 0.05).
VITD treatment increases the expression of Lmx1a, Nurr1 and TH proteins in mesDAs
mesDA proliferation, specification and differentiation are tightly regulated by the transcription factors Sox2, Lmx1a and Nurr118,19. CellProfiler analysis allowed us to quantify the protein expression of these factors in individual mesDA cells by measuring the mean fluorescence intensity of these factors.
Sox2 expression
MIA and VITD had no effect on the mean intensity of Sox2 fluorescence in individual mesDA progenitors (p’s > 0.05; Fig. 4B). Distribution analysis also confirmed Sox2 staining was constant across all treatments (p’s > 0.05; Fig. 4C).
Lmx1a expression
There was a significant main effect of VITD treatment on the mean intensity of Lmx1a expression (F(1,36) = 5.891, p < 0.05), and significant interactions between MIA x VITD treatments (F(1,36) = 7.993, p < 0.05; Fig. 4E). Post hoc comparisons revealed that Lmx1a expression was significantly higher in individual cells of CON- VITD fetuses compared to CON-VEH (p < 0.01) and the other two treatment groups (p’s < 0.05). Supporting this, the distribution analysis of Lmx 1a+ demonstrated that more CON-VITD mesDA cells expressed higher levels of Lmx1a compared to other treatment groups (p’s < 0.05; Fig. 4F).

The effects of single and combined MIA and VITD treatment on the expression of factors important in the development of mesDA neurons. (A) A representative floor plate (FP) of coronal MES section showing mesDA cells expressing Sox2 (red). (B) Neither MIA or VITD treatment altered the mean intensity of Sox2 in mesDA progenitors (Lmx1a+Sox2+) (p’s > 0.05). (C) Bar plots represent the average fluorescence of Sox2 in mesDA progenitors among treatment groups. Distribution analysis representing a nonlinear regression to predict the Gaussian distribution of mesDA progenitor number relative to the intensity of Sox2 expression. (D) A representative FP of coronal MES section showing post-mitotic mesDA cells expressing Lmx1a (green). (E) VITD treatment significantly increased the mean intensity of Lmx1a in mesDAs compared with other treatment groups (p’s < 0.05). (F) Bar plots represent the average expression of Lmx1a among treatment groups. Distribution analysis showed VITD treatment itself increased the number of cells expressing higher levels of Lmx1a compared to other treatment groups (p’s < 0.05). (G) A representative FP of coronal MES section showing post-mitotic mesDA cells expressing Nurr1 (magenta). (H) VITD treatment significantly increased the expression of Nurr1 in post-mitotic (Nurr1+) mesDAs (p < 0.05). (I) Bar plots represent the average fluorescence of Nurr1 among treatment groups. Distribution analysis showed VITD treatment increased the number of cells expressing higher levels of Nurr1 compared to VEH (p < 0.05). (J) A representative FP of coronal MES section showing mature mesDAs expressing TH (green). (K) VITD treatment increased the average fluorescence of TH in mature (Nurr1+TH+) mesDAs. (L) Bar plots represent the average TH mean intensity in mature mesDAs among treatment groups. The distribution analysis verified that VITD treatment increased the number of mature (Nurr1+TH+) mesDAs that expressed higher levels of TH compared to VEH (p < 0.01). All values were means ± SEM. *p < 0.05, **p < 0.01. Scale bars: 50 µm.
Nurr1 expression
There was a significant main effect of VITD treatment on Nurr1 expression in individual post-mitotic mesDAs (F(1,40) = 14.451, p < 0.01; Fig. 4H). Supporting this, the distribution analysis revealed significant main effects of VITD treatment where more mesDAs had greater Nurr1 expression (F(19, 684) = 6.243, p < 0.01; Fig. 4I).
TH Expression
There was a significant main effect of VITD treatment on TH expression in individual mature (Nurr1+TH+) mesDAs (F(1,40) = 12.817, p < 0.01; Fig. 4K). Supporting this, the distribution analysis revealed significant main effects of VITD treatment where more mature mesDAs had greater TH expression (F(19,684) = 5.301, p < 0.01; Fig. 4L).
Discussion
VITD abolishes MIA-induced sensitivity to AMPH
Rodent models of MIA and DVD-deficiency both exhibit early molecular and behavioural abnormalities consistent with altered DA signaling6,7,10,35, suggesting a common pathogenic pathway6. In support of this hypothesis, the present study shows that co-administration of VITD during gestation prevents the MIA-induced potentiation of AMPH sensitivity in adult offspring. It thus appears that the process leading to AMPH hypersensitivity in MIA offspring is initiated during early fetal development, such that interventions targeting the initiation process (e.g., through co-administration of VITD during gestation) are capable of preventing the subsequent establishment of this phenotype.
The failure of VITD to block MIA-induced impairments in PPI may seem counterintuitive at first, given that dopaminergic mechanisms have also been implicated in this association11,36. More specifically, previous studies found that acute and chronic administration of DA receptor antagonists in adulthood and adolescence, respectively, mitigated PPI deficits in adult MIA offspring11,36. While these findings clearly point to an involvement of the DA system in the adolescent and/or adult expression of PPI, they are not informative with regards to the underlying developmental processes occurring prior to adolescence. Based on the present data, we propose that MIA-induced alterations in PPI and AMPH sensitivity have a differential ontogeny. This hypothesis is also consistent with our previous findings showing that (i) whilst DVD-deficiency induces locomotor sensitivity to AMPH13, it does not impair PPI12, and (ii) MIA-induced PPI impairments are not critically dependent on the mesDA differentiation and maintenance factor, Nurr137, the latter of which we find to be deregulated in the fetal brain after DVD-deficiency or MIA.
The acute elevation of inflammatory cytokines is proposed as one of the primary pathways leading to neuropsychiatric phenotypes in MIA offspring35,38. Given the well-known anti-inflammatory actions of vitamin D, this could well represent its neuroprotective mechanism39. However, we have previously reported that VITD co-treatment in this model failed to affect poly(I:C)-induced elevations in IL-6, IL-1ß and TNF alpha, in either maternal serum or fetal brain27. Although there may be other inflammatory factors/processes involved40, our current findings suggest an alternative neuroprotective mechanism. Based on our novel data presented here, we believe that maternal VITD may exert its beneficial effects through actions on the developing DA system.
Both MIA and VITD decrease mesDA progenitor numbers, but only VITD increases mature mesDA number
All maternal treatments reduced mesDA progenitor (Lmx1a+Sox2+) numbers. There are likely to be distinct mechanisms for these effects, and consequences thereof as well. Whilst the MIA-induced reduction in mesDA progenitor cells was accompanied by the subsequent emergence of behavioral deficits, maternal VITD was without consequences on offspring behavior per se. Mechanistically, the effects of MIA on mesDA progenitor numbers likely involve the inhibition of neural progenitor formation via signaling at toll-like 3 receptors (TLR3), which are expressed early in the embryonic brain41. The pro-inflammatory cytokines induced by MIA can inhibit neural proliferation as well42,43,44.
Contrary to MIA, maternal VITD may reduce mesDA progenitor numbers by means of shifting the progenitor cells to a more differentiated phenotype. Indeed, the pro-differentiation properties of VITD in neurons and in the brain are well described. In vitro VITD treatment differentiates neural stem cells into neurons45; SH-SY5Y cells into DA neurons21,22 and inhibits neural progenitor formation46. In contrast, the absence of vitamin D during gestation leads to effects that are diametrically opposite to those reported here, namely an increase in mitosis across multiple brain regions47,48 and enhanced neural progenitor formation46.
Importantly, our study further shows that VITD and MIA have divergent effects on mature (Nurr1+TH+) mesDA neuron number. Although not significant, MIA clearly appeared to reduce (Nurr1 + TH+) mesDA cell number. In contrast, VITD increased (Nurr1+TH+) mesDA number, particularly for the more posterior mesDAs (Fig. 2L). A VITD-mediated restoration of TH cell number therefore represents one plausible mechanism for the normalization of DA-mediated behaviours in MIA exposed offspring.
VITD restores MIA-induced alterations in the distribution of mesDAs
For post-mitotic neurons, we show that MIA alters the mean DV positioning of post-mitotic (Lmx1a+Sox2−) mesDAs and the ML positioning of mature (Nurr1+TH+) mesDAs. VITD co-treatment did not rescue these deficits. However when examining the DV positioning of mature mesDAs, we show a profound MIA-induced reduction in DV positioning, which was fully prevented by VITD. Importantly, VITD by itself had no effect on ML or DV positioning of any mesDA neuron type. Therefore, the correction of early mis-positioning of mesDA neurons by VITD could represent another plausible mechanism for the normalization of DA-mediated behaviours in MIA-exposed offspring.
VITD regulates protein expression of key mesDA factors
VITD treatment increased the expression of all mesDA-related proteins examined. In contrast, MIA by itself had no significant effects on the expression of these proteins.
Here, we show that VITD treatment upregulates the expression of Lmx1a in developing mesDAs. Lmx1a is a convergent upstream molecule from SHH-Wnt pathways that specify mesDA lineage49,50,51. SHH and Wnt signaling are affected in MIA and DVD-deficiency models respectively14,21.
Nurr1 a direct upstream target of Lmx1a promotes mesDA differentiation49,50,52. MIA and DVD-deficiency reduce the mRNA expression of Nurr1 in the fetal brain14,15. This is consistent with present cell counting data, which indicates a recognizable (but not significant) reduction in Nurr1+ cell number in MIA embryos. At an individual cell level, however, the remaining cells have normal Nurr1 protein expression. Therefore, the likely explanation for previous findings showing reduced Nurr1 mRNA expression in MIA fetuses is a reduction in cell number14. Importantly, genetic deficiency of Nurr1 has previously been shown to induce AMPH hypersensitivity in a similar way to MIA53. Therefore the promotion of Nurr1 expression by VITD may represent a third mechanism for the prevention of AMPH hypersensitivity in MIA exposed offspring.
Lmx1a and Nurr1 regulate the expression of TH54,55. We have shown VITD elevates TH mRNA and protein in neuroblastomas in culture21,22. Again consistent with these findings the absence of VITD in DVD-deficiency leads to the downregulation of TH in fetal rodent brains16,20. Here, we verified that VITD increases the expression of TH in vivo.
In summary VITD increases the expression of the mesDA transcription factors (Lmx1a and Nurr1) and the rate-limiting enzyme in DA synthesis (TH) in developing mesDA cells. The expression of these three factors in control mesDAs clearly increases with differentiation state (Fig S4). Therefore VITD acts to differentiate the remaining mesDAs in MIA-exposed embryos. This represents a highly plausible mechanism for the prevention of DA-related behavioural deficits induced by MIA. Given that VITD treatment itself has no effect on the behaviours examined here and those examined previously27, we conclude that the accelerated maturation effects of VITD treatment alone on mesDAs is benign.
Limitations
Although our findings provide more data for the translational relevance of introducing dietary intervention during early pregnancy for the prevention of developmental psychiatric diseases, the direct supplementation of the hormonal form of vitamin D, 1,25OHD, to pregnant women would adversely affect calcium in the developing embryo and therefore is not clinically feasible. Therefore preclinical replication studies are now warranted using the safe to use dietary form of vitamin D, cholecalciferol. In this study, we have only investigated one developmental time point and only male foetuses. In future studies, it will be obviously important to examine the entire ontogeny of mesDA systems in both sexes to understand long-term outcomes.
Conclusion
This study represents a small but growing number of preclinical studies showing that the trajectory of adverse MIA-induced outcomes for adult brain function can be diminished or averted by dietary interventions. Two such studies have employed dietary interventions in MIA-exposed juvenile offspring with either a ketogenic diet or glucoraphanin to reverse certain MIA-induced behavioural phenotypes of relevance to psychiatry56,57. Of closer relevance to our study Weiser and colleagues have supplemented MIA dams with docosahexaenoic acid and prevented the onset of autistic-like phenotypes58. We recently reported that maternal treatment with VITD blocked the emergence of MIA-induced deficits in cognitive and social behavioural phenotypes of relevance to autism in offspring27. Using a mouse model of MIA, the present study is the first to reveal preventive effects of maternal VITD treatment on AMPH hypersensitivity, a phenotype with relevance to the positive symptoms of schizophrenia59,60,61.
We further identify three plausible pathways via which VITD may act to restore or support normal DA development in the event of MIA. First, MIA retarded the positioning of mesDAs particularly in the DV dimension, whereas VITD treatment restored this at least for TH+ mesDA neurons. Second, MIA appeared to reduce TH+ cell number, while at the same time, VITD increased TH+ mesDA cells. Third, VITD increased the expression of proteins consistent with a more differentiated mesDA neuron. These three processes likely reflect an accelerated differentiation of DA neurons induced by VITD, which in turn may have protected against the emergence of AMPH hypersensitivity in adult MIA offspring. By contrast, MIA-induced PPI deficiency does not seem to critically involve the same developmental pathways.
This data adds to a steadily increasing literature indicating developing DA systems may be particularly vulnerable to adverse environmental factors such as MIA and DVD-deficiency and represent a plausible convergent early mechanistic pathway to psychiatric disorders such as schizophrenia.
References
Di Forti, M., Lappin, J. M. & Murray, R. M. Risk factors for schizophrenia - All roads lead to dopamine. European Neuropsychopharmacology 17, 101–107, https://doi.org/10.1016/j.euroneuro.2007.02.005 (2007).
van Os, J., Kenis, G. & Rutten, B. P. F. The environment and schizophrenia. Nature 468, 203–212, https://doi.org/10.1038/nature09563 (2010).
Brown, A. S. The environment and susceptibility to schizophrenia. Progress in Neurobiology 93, 23–58, https://doi.org/10.1016/j.pneurobio.2010.09.003 (2011).
Owen, M. J., Sawa, A. & Mortensen, P. B. Schizophrenia. The Lancet 388, 86–97, https://doi.org/10.1016/S0140-6736(15)01121-6 (2016).
Weinstein, J. J. et al. Pathway-Specific Dopamine Abnormalities in Schizophrenia. Biological Psychiatry 81, 31–42, https://doi.org/10.1016/j.biopsych.2016.03.2104 (2017).
Eyles, D., Feldon, J. & Meyer, U. Schizophrenia: do all roads lead to dopamine or is this where they start? Evidence from two epidemiologically informed developmental rodent models. Translational Psychiatry 2, e81, https://doi.org/10.1038/tp.2012.6 (2012).
Meyer, U. Prenatal Poly(I:C) exposure and other developmental immune activation models in rodent systems. Biological Psychiatry 75, 307–315, https://doi.org/10.1016/j.biopsych.2013.07.011 (2014).
Eyles, D. & Brown, J. Mackay-Sim, a., McGrath, J. & Feron, F. Vitamin d3 and brain development. Neuroscience 118, 641–653, https://doi.org/10.1016/S0306-4522(03)00040-X (2003).
Eyles, D. W., Burne, T. H. J. & McGrath, J. J. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Frontiers in neuroendocrinology 34, 47–64, https://doi.org/10.1016/j.yfrne.2012.07.001 (2013).
Schoenrock, S. A. & Tarantino, L. M. Developmental vitamin D deficiency and schizophrenia: the role of animal models. Genes, brain, and behavior 15, 45–61, https://doi.org/10.1111/gbb.12271 (2016).
Vuillermot, S., Weber, L., Feldon, J. & Meyer, U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience 30, 1270–1287, https://doi.org/10.1523/JNEUROSCI.5408-09.2010 (2010).
Kesby, J. P., Burne, T. H. J., McGrath, J. J. & Eyles, D. W. Developmental vitamin D deficiency alters MK 801-induced hyperlocomotion in the adult rat: An animal model of schizophrenia. Biological psychiatry 60, 591–596, https://doi.org/10.1016/j.biopsych.2006.02.033 (2006).
Kesby, J. P. et al. Developmental vitamin D deficiency alters dopamine-mediated behaviors and dopamine transporter function in adult female rats. Psychopharmacology 208, 159–168, https://doi.org/10.1007/s00213-009-1717-y (2010).
Meyer, U., Engler, A., Weber, L., Schedlowski, M. & Feldon, J. Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience 154, 701–709, https://doi.org/10.1016/j.neuroscience.2008.04.031 (2008).
Cui, X., Pelekanos, M., Burne, T. H. J., McGrath, J. J. & Eyles, D. W. Maternal vitamin D deficiency alters the expression of genes involved in dopamine specification in the developing rat mesencephalon. Neuroscience letters 486, 220–223, https://doi.org/10.1016/j.neulet.2010.09.057 (2010).
Luan, W. et al. Developmental Vitamin D (DVD) Deficiency Reduces Nurr1 and TH Expression in Post-mitotic Dopamine Neurons in Rat Mesencephalon. Molecular Neurobiology, https://doi.org/10.1007/s12035-017-0497-3 (2017).
Nelander, J., Hebsgaard, J. B. & Parmar, M. Organization of the human embryonic ventral mesencephalon. Gene expression patterns: GEP 9, 555–561, https://doi.org/10.1016/j.gep.2009.10.002 (2009).
Blaess, S. & Ang, S.-L. Genetic control of midbrain dopaminergic neuron development. Wiley interdisciplinary reviews. Developmental biology 4, 113–134, https://doi.org/10.1002/wdev.169 (2015).
Arenas, E., Denham, M. & Villaescusa, J. C. How to make a midbrain dopaminergic neuron. Development (Cambridge, England) 142, 1918–1936, https://doi.org/10.1242/dev.097394 (2015).
Hawes, J. E. et al. Maternal vitamin D deficiency alters fetal brain development in the BALB/c mouse. Behavioural Brain Research 286, 192–200, https://doi.org/10.1016/j.bbr.2015.03.008 (2015).
Cui, X., Pertile, R., Liu, P. & Eyles, D. W. Vitamin D regulates tyrosine hydroxylase expression: N-cadherin a possible mediator. Neuroscience 304, 90–100, https://doi.org/10.1016/j.neuroscience.2015.07.048 (2015).
Pertile, R. A. N., Cui, X. & Eyles, D. W. Vitamin D signaling and the differentiation of developing dopamine systems. Neuroscience 333, 193–203, https://doi.org/10.1016/j.neuroscience.2016.07.020 (2016).
Zuckerman, L. & Weiner, I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. Journal of Psychiatric Research 39, 311–323, https://doi.org/10.1016/j.jpsychires.2004.08.008 (2005).
Romero, E., Guaza, C., Castellano, B. & Borrell, J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Molecular Psychiatry 15, 372–383, https://doi.org/10.1038/mp.2008.44 (2010).
Kesby, J. P. et al. Developmental vitamin D deficiency alters multiple neurotransmitter systems in the neonatal rat brain. International Journal of Developmental Neuroscience 62, 1–7, https://doi.org/10.1016/j.ijdevneu.2017.07.002 (2017).
Vuillermot, S. et al. Prenatal Immune Activation Interacts with Genetic Nurr1 Deficiency in the Development of Attentional Impairments. 32, 436–451, https://doi.org/10.1523/JNEUROSCI.4831-11.2012 (2012).
Vuillermot, S., Luan, W., Meyer, U. & Eyles, D. Vitamin D treatment during pregnancy prevents autism-related phenotypes in a mouse model of maternal immune activation. Molecular Autism 8, 9, https://doi.org/10.1186/s13229-017-0125-0 (2017).
Meyer, U., Feldon, J., Schedlowski, M. & Yee, B. K. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neuroscience and biobehavioral reviews 29, 913–947, https://doi.org/10.1016/j.neubiorev.2004.10.012 (2005).
Spencer, S. J. & Meyer, U. Perinatal programming by inflammation. Brain, Behavior, and Immunity 63, 1–7, https://doi.org/10.1016/j.bbi.2017.02.007 (2017).
Bodea, G. O. et al. Reelin and CXCL12 regulate distinct migratory behaviors during the development of the dopaminergic system. Development (Cambridge, England) 141, 661–673, https://doi.org/10.1242/dev.099937 (2014).
Brown, A. J., Ritter, C. S., Holliday, L. S. & Knutson, J. C. & Strugnell, S. a. Tissue distribution and activity studies of 1,24-dihydroxyvitamin D2, a metabolite of vitamin D2 with low calcemic activity in vivo. Biochemical pharmacology 68, 1289–1296, https://doi.org/10.1016/j.bcp.2004.06.015 (2004).
Meyer, U. et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. The Journal of neuroscience: the official journal of the Society for Neuroscience 26, 4752–4762, https://doi.org/10.1523/JNEUROSCI.0099-06.2006 (2006).
Arrode-Brusés, G. & Brusés, J. L. Maternal immune activation by poly I:C induces expression of cytokines IL-1β and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. Journal of neuroinflammation 9, 83, https://doi.org/10.1186/1742-2094-9-83 (2012).
Knutson, J. C., Levun, L. W., Valliere, R., Bishop, C. W. & International, B. C. Pharmacokinetics and Systemic Effect on Calcium Homeostasis of la,24-Dihydroxyvitamin D2, in Rats. 53, 829–837 (1997).
Knuesel, I. et al. Maternal immune activation and abnormal brain development across CNS disorders. Nature Reviews Neurology 10, 643–660, https://doi.org/10.1038/nrneurol.2014.187 (2014).
Meyer, U., Spoerri, E., Yee, B. K., Schwarz, M. J. & Feldon, J. Evaluating early preventive antipsychotic and antidepressant drug treatment in an infection-based neurodevelopmental mouse model of schizophrenia. Schizophrenia Bulletin 36, 607–623, https://doi.org/10.1093/schbul/sbn131 (2010).
Vuillermot, S., Feldon, J. & Meyer, U. Nurr1 is not essential for the development of prepulse inhibition deficits induced by prenatal immune activation. Brain, Behavior, and Immunity 25, 1316–1321, https://doi.org/10.1016/j.bbi.2011.06.012 (2011).
Estes, M. L. & McAllister, A. K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 353, 772–777, https://doi.org/10.1126/science.aag3194 (2016).
Hewison, M. An update on vitamin D and human immunity. Clinical Endocrinology 76, 315–325, https://doi.org/10.1111/j.1365-2265.2011.04261.x (2012).
Choi, G. B. et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939, https://doi.org/10.1017/CBO9781107415324.004 (2016).
Lathia, J. D. et al. Toll-Like Receptor 3 Is a Negative Regulator of Embryonic Neural Progenitor Cell Proliferation. The Journal of Neuroscience 28, 13978–13984, https://doi.org/10.1523/JNEUROSCI.2140-08.2008 (2008).
Wang, X. et al. Interleukin-1 β mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. 36, 343-354, https://doi.org/10.1016/j.mcn.2007.07.005 (2007).
Utani, A. Interleukin-6 and Neural Stem Cells: More Than Gliogenesis. Seikagaku 82, 327–331, https://doi.org/10.1091/mbc.E08 (2010).
Ernardino, L. I. B., Gasse, F. A. A., Ilva, B. R. S., Erreira, R. A. F. & Rade, S. O. G. Tumor Necrosis Factor-a Modulates Survival, Proliferation, and Neuronal Differentiation in Neonatal Subventricular Zone Cell Cultures. 2361–2371, https://doi.org/10.1634/stemcells.2007-0914 (2008).
Shirazi, H. A., Rasouli, J., Ciric, B., Rostami, A. & Zhang, X. 1,25-dihydroxyvitamin D3 enhances neural stem cell proliferation and oligodendrocyte differentiation. 98, 240–245, https://doi.org/10.1016/j.yexmp.2015.02.004.1 (2016).
Cui, X., McGrath, J. J., Burne, T. H. J., Mackay-Sim, A. & Eyles, D. W. Maternal vitamin D depletion alters neurogenesis in the developing rat brain. International journal of developmental neuroscience: the official journal of the International Society for Developmental Neuroscience 25, 227–232, https://doi.org/10.1016/j.ijdevneu.2007.03.006 (2007).
McGrath, J., Eyles, D., Mowry, B., Yolken, R. & Buka, S. Low maternal vitamin D as a risk factor for schizophrenia: a pilot study using banked sera. Schizophrenia Research 63, 73–78, https://doi.org/10.1016/S0920-9964(02)00435-8 (2003).
Ko, P., Burkert, R., McGrath, J. & Eyles, D. Maternal vitamin D3 deprivation and the regulation of apoptosis and cell cycle during rat brain development. Brain research. Developmental brain research 153, 61–68, https://doi.org/10.1016/j.devbrainres.2004.07.013 (2004).
Chung, S. et al. Wnt1-lmx1a forms a novel autoregulatory loop and controls midbrain dopaminergic differentiation synergistically with the SHH-FoxA2 pathway. Cell stem cell 5, 646–658, https://doi.org/10.1016/j.stem.2009.09.015 (2009).
Hoekstra, E. J. et al. Lmx1a Encodes a Rostral Set of Mesodiencephalic Dopaminergic Neurons Marked by the Wnt/B-Catenin Signaling Activator R-spondin 2. PLoS ONE 8, 1–12, https://doi.org/10.1371/journal.pone.0074049 (2013).
Nakatani, T., Kumai, M., Mizuhara, E., Minaki, Y. & Ono, Y. Lmx1a and Lmx1b cooperate with Foxa2 to coordinate the specification of dopaminergic neurons and control of floor plate cell differentiation in the developing mesencephalon. Developmental biology 339, 101–113, https://doi.org/10.1016/j.ydbio.2009.12.017 (2010).
Saucedo-Cardenas, O. et al. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proceedings of the National Academy of Sciences of the United States of America 95, 4013–4018 (1998).
Rojas, P., Joodmardi, E., Hong, Y., Perlmann, T. & Ogren, S. O. Adult mice with reduced Nurr1 expression: an animal model for schizophrenia. Molecular psychiatry 12, 756–766, https://doi.org/10.1038/sj.mp.4001993 (2007).
Jacobs, F. M. J. et al. Retinoic acid-dependent and -independent gene-regulatory pathways of Pitx3 in meso-diencephalic dopaminergic neurons. Development (Cambridge, England) 138, 5213–5222, https://doi.org/10.1242/dev.071704 (2011).
Veenvliet, J. V. & Smidt, M. P. Molecular mechanisms of dopaminergic subset specification: fundamental aspects and clinical perspectives. Cellular and molecular life sciences: CMLS 71, 4703–4727, https://doi.org/10.1007/s00018-014-1681-5 (2014).
Matsuura, A. et al. Dietary glucoraphanin prevents the onset of psychosis in the adult offspring after maternal immune activation. Scientific reports 8, 2158, https://doi.org/10.1038/s41598-018-20538-3 (2018).
Ruskin, D. N., Murphy, M. I., Slade, S. L. & Masino, S. A. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PloS one 12, e0171643, https://doi.org/10.1371/journal.pone.0171643 (2017).
Weiser, M. J. et al. Dietary docosahexaenoic acid alleviates autistic-like behaviors resulting from maternal immune activation in mice. Prostaglandins, leukotrienes, and essential fatty acids 106, 27–37, https://doi.org/10.1016/j.plefa.2015.10.005 (2016).
Laruelle, M. & Abi-Dargham, a. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. Journal of psychopharmacology (Oxford, England) 13, 358–371, https://doi.org/10.1177/026988119901300405 (1999).
Laruelle, M. The role of endogenous sensitization in the pathophysiology of schizophrenia: Implications from recent brain imaging studies. Brain Research Reviews 31, 371–384, https://doi.org/10.1016/S0165-0173(99)00054-5 (2000).
Abi-Dargham, A., van de Giessen, E., Slifstein, M., Kegeles, L. S. & Laruelle, M. Baseline and Amphetamine-Stimulated Dopamine Activity Are Related in Drug-Naïve Schizophrenic Subjects. Biological Psychiatry 65, 1091–1093, https://doi.org/10.1016/j.biopsych.2008.12.007 (2009).
Acknowledgements
Funding bodies. This work was supported by a National Health and Medical research council grant (APP 1057883) to DWE and UM; additional financial support was received from the Swiss National Science Foundation (grant number 310030_169544) awarded to UM. We acknowledge the support of the Histology and Microscopy Facility, and the Animal Facility of Queensland Brain institute of the University of Queensland.
Author information
Authors and Affiliations
Contributions
W.L., S.V., U.M. and D.W.E. designed research; W.L., L.A.H. and S.V. performed research and analyzed the data; W.L., L.A.H., U.M. and D.W.E. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luan, W., Hammond, L.A., Vuillermot, S. et al. Maternal Vitamin D Prevents Abnormal Dopaminergic Development and Function in a Mouse Model of Prenatal Immune Activation. Sci Rep 8, 9741 (2018). https://doi.org/10.1038/s41598-018-28090-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-28090-w
- Springer Nature Limited
This article is cited by
-
Vitamin D3 improves iminodipropionitrile-induced tic-like behavior in rats through regulation of GDNF/c-Ret signaling activity
European Child & Adolescent Psychiatry (2024)
-
Early activation of cellular stress and death pathways caused by cytoplasmic TDP-43 in the rNLS8 mouse model of ALS and FTD
Molecular Psychiatry (2023)
-
Interaction of the pre- and postnatal environment in the maternal immune activation model
Discover Mental Health (2023)
-
Prenatal hypoxia alters the early ontogeny of dopamine neurons
Translational Psychiatry (2022)
-
How do established developmental risk-factors for schizophrenia change the way the brain develops?
Translational Psychiatry (2021)