
Abstract
Cytochrome c oxidase catalyzes reduction of O2 to H2O at a catalytic site that is composed of a copper ion and heme group. The reaction is linked to translocation of four protons across the membrane for each O2 reduced to water. The free energy associated with electron transfer to the catalytic site is unequal for the four electron-transfer events. Most notably, the free energy associated with reduction of the catalytic site in the oxidized cytochrome c oxidase (state O) is not sufficient for proton pumping across the energized membrane. Yet, this electron transfer is mechanistically linked to proton pumping. To resolve this apparent discrepancy, a high-energy oxidized state (denoted O H ) was postulated and suggested to be populated only during catalytic turnover. The difference between states O and O H was suggested to be manifested in an elevated midpoint potential of CuB in the latter. This proposal predicts that one-electron reduction of cytochrome c oxidase after its oxidation would yield re-reduction of essentially only CuB. Here, we investigated this process and found ~5% and ~6% reduction of heme a3 and CuB, respectively, i.e. the apparent redox potentials for heme a3 and CuB are lower than that of heme a.
Similar content being viewed by others

Introduction
The electrochemical proton gradient across the inner mitochondrial or bacterial cytoplasmic membrane in aerobic organisms is maintained by a series of membrane-bound redox-driven proton pumps. The final electron acceptor, O2, binds to cytochrome c oxidase (CytcO) at a bimetallic heme a3-CuB catalytic site where it is reduced by four electrons to water (Fig. 1a). During turnover of CytcO, electrons are transferred one-by-one from a water-soluble or membrane-associated cyt. c consecutively to the primary electron acceptor, CuA, the secondary acceptor heme a and the catalytic site. The CytcO links this electron transfer to proton pumping from the more negative (N) to the more positive (P) side of the membrane with an average stoichiometry of one pumped proton per electron transferred to the catalytic site. Thus, the overall reaction is:
where the subscripts P and N refer to the two sides of the membrane. The free energy for reduction of O2 by cyt. c is ~2 eV, which is used to move 8 charges across the membrane; 4 protons that are pumped across the membrane and, 4 electrons and 4 protons that are transferred from opposite sides of the membrane resulting in a charge separation (summarized in1).

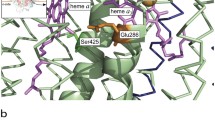
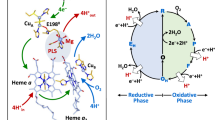
Experimental system and the studied reaction. (a) CytcO accommodates four redox-active centers: CuA, heme a and the catalytic site composed of heme a3 and CuB, that upon reduction each bind one electron. The cyt. c-CytcO electrostatic complex accommodates in total five electrons in the reduced state. Two proton-uptake pathways, called D and K are indicated in the figure, but not discussed in the text. (b) Turnover of CytcO. One electron at a time is donated from cyt. c to CytcO. The initial state is the “relaxed” oxidized state. Consecutive addition of two electrons yields the two-electron reduced catalytic site which binds O2 to form the peroxy state PM. Reduction of PM yields F, which is then reduced to form the “activated” oxidized state, OH. This state is then reduced by cyt. c. (c) Reaction studied in this work. The oxidized cyt. c-CytcO complex is reduced by 5 electrons. Upon initiation of the reaction (with a laser flash, not shown), O2 binds to the catalytic site and an electron is transferred from heme a to the catalytic site to form the peroxy state, PR. This step is followed in time by formation of the ferryl state, F, which occurs over the same time scale as electron equilibration among CuA/cyt. c and heme a. Electron transfer from this equilibrium to the catalytic site leads to formation of state OH. Over a longer time scale of ~0.1 s an electron equilibrates between the CytcO molecules via the bound cyt. c to eventually yield oxidized CytcO. Note that proton-transfer reactions are not indicated in the Figure.
A number of distinct intermediate states have been identified and characterized during CytcO turnover (Fig. 1b). Consecutive transfer of two electrons to the oxidized CytcO (denoted O) yields formation of the one- (denoted E) and two- (denoted R) electron reduced catalytic site. When heme a3 and CuB are reduced, O2 binds to the heme iron after which the O-O bond is broken to form a ferryl state called “peroxy” for historical reasons (denoted P M ). Transfer of an additional electron and a proton to P M , results in formation of the ferryl state, F. Further transfer of an electron and a proton to F yields the oxidized CytcO (O H , see below). The overall process has been reviewed in the past1,2,3,4,5,6,7,8,9,10,11,12,13,14 and it is summarized in Fig. 1b.
Early studies indicated that the free energy from O2 reduction is not released evenly in the four electron-transfer reactions, but the majority of this free energy was found to be liberated in the P M → F and F → O reaction steps15. Furthermore, elegant studies performed by Wikström suggested that proton pumping is coupled only to these two steps16, which stimulated Wikström and colleagues to propose a detailed molecular model based on these findings17. According to this model, two protons would be pumped in each of the reactions P M → F and F → O, while no proton pumping would be observed upon reduction of the CytcO, i.e. O → E → P M . Later, a novel kinetic approach was used to obtain data18 that resulted in re-evaluation of the model to yield a scenario where one proton is pumped in each of the four reaction steps that involve electron transfer to the catalytic site19.
A key aspect of the new model was postulation of a “high-energy” metastable oxidized form of the CytcO that would presumably differ in structure from the oxidized “as isolated” (O) state. The state is denoted O H and it is assumed to be formed as a product of the F → O H reaction (below we denote by O H the oxidized state that is formed after re-oxidation of the reduced CytcO independently of the properties of this state)18. According to this scenario part of the free energy released in P M → F and F → O H would be conserved in the O H state to be used during subsequent reduction of the CytcO, i.e. the reaction sequence O H → E → P M would be more exergonic than O → E → P M . The question arises: what is the difference between states O and O H ?
Postulation of state O H also found support in an experimental observation: when starting from the fully oxidized CytcO (O) the free energy associated with electron transfer from cyt. c to heme a3-CuB, before O2 binds, is too small to drive proton translocation (see Kaila et al.1). Consequently, one possible difference between the “as isolated” state O and state O H could be that in the latter the midpoint potentials of heme a3 and CuB are higher than those in state O19,20. Indeed, data from experimental21 and theoretical22 studies suggested that the midpoint potential of CuB is significantly elevated in state O H . Furthermore, Blomberg and Siegbahn found that the inherent midpoint potential of CuB is high during catalytic turnover, and that in the resting oxidized state a slow protonation process may cause a significant decrease of the proton-coupled reduction potential23. As discussed in the paper by Belevich et al.21, there are conflicting experimental data regarding possible differences in the CuB midpoint potentials between states O and O H . For example, Jancura et al.24 did not observe any spectral or kinetic differences between the O H and O forms of CytcO. Brand et al.25 prepared CytcO in the O and O H states, respectively, and then added one electron to each of the samples. They found that the extent of electron transfer from heme a to the catalytic site was relatively small and the same in both states, which indicated that the midpoint potential of CuB was not raised in state O H .
In the present study, we investigated the reaction of the reduced Rhodobacter sphaeroides CytcO with O2 in the absence and presence, respectively, of cyt. c at low ionic strength. In the latter case a cyt. c-CytcO electrostatic complex is formed (see Fig. 1a,c). This arrangement contrasts the earlier studies in that it resembles the in vivo situation where electrons are continuously fed into CytcO from the donor cyt. c. The cyt. c-CytcO complex accommodates five electrons. Upon binding of O2 four electrons are transferred to form H2O yielding the oxidized CytcO, presumably in state O H . Then the “fifth electron” can equilibrate among the five sites cyt. c, CuA, heme a, heme a3 and CuB26,27,28. We reasoned that if in state O H the CuB midpoint potential would be elevated, this fifth electron would be transferred to CuB. The data show that this was not the case and in ~90% of CytcOs the fifth electron was distributed among sites cyt. c, CuA and heme a with the largest fraction at heme a.
Results and Discussion
Spectral analysis
To determine whether or not binding of cyt. c to CytcO affects the spectral properties of the hemes, we recorded the reduced - oxidized difference spectra of each of the isolated proteins (cyt. c and CytcO) as well as of a mixture of the two at low ionic strength (i.e. the cyt. c-CytcO complex). As seen in Fig. 2, the sum of the cyt. c and CytcO difference spectra was essentially equal to that of the mixture, except for a small increase in the 550 nm absorbance peak upon forming the cyt. c-CytcO complex.

Absorbance difference spectra. A comparison of reduced minus oxidized difference spectra of the CytcO-cyt. c complex (black) and the sum of the reduced minus oxidized difference spectra of CytcO and cyt. c (red), respectively. Spectra of the oxidized states were recorded first and then the atmosphere in the cuvette was replaced for N2 after which the samples were reduced with ascorbate (2 mM) and hexaammineruthenium(II) chloride (1 μM). Reduction of the samples was followed in time by recording spectra over ~2 hours until no further changes were observed. The data to the right of the axis break have been multiplied by a factor of five. The concentrations of CytcO and cyt. c were ~3 μM and ~5 μM, respectively, in 50 mM HEPES (pH 7.5), 0.05% DDM and 100 µM EDTA.
Reaction of CytcO with O2
The reduced CytcO carries four electron equivalents. Thus, upon reaction with O2 the CytcO becomes fully oxidized while O2 is reduced to H2O. The anaerobic samples were mixed with an O2-saturated solution in a stopped-flow apparatus. After ~200 ms the CO ligand was dissociated by means of a short laser flash, which initiates the reaction of the reduced CytcO with O2. In the absence of cyt. c (black traces in Fig. 3) the reaction exhibits a number of distinct kinetic steps, while populating states in which O2 is progressively reduced and protonated to eventually form H2O. The details of the reaction sequence have been described in the past1,4,5,29,30 and it is outlined in Fig. 1c.

Kinetics of absorbance changes upon reaction with O2. The four-electron reduced CytcO (black traces) or the five-electron-reduced cyt. c-CytcO complex (red traces) was mixed with an O2-saturated buffer solution. About 200 ms after mixing with the O2-containing buffer, at time = 0, the CO-ligand was removed by a laser flash. The four-electron complex becomes oxidized over a time scale of 8 ms (left-hand side boxes). At 445 nm (a) the main contribution is from redox changes at hemes a and a3. At 605 nm (b) the main contribution is from redox changes at heme a (80%) and the remaining fraction originates from changes at heme a3. At 830 nm (c) the main contribution is from CuA where an increase in absorbance is associated with oxidation. At 550 nm (d) the main contribution is from redox changes at cyt. c. At 445 nm and 605 nm, the rapid change in absorbance at t = 0 is associated with CO dissociation. It is followed in time by a decrease in absorbance associated with binding of O2 (τ ≅ 10 μs), formation of the P R state (τ ≅ 40 μs) and oxidation of the CytcO (τ ≅ 1.5 ms). The P R → F reaction is not seen at these wavelengths. At 830 nm two components are seen with time constants of 200 µs (P R → F) and 1.5 ms (F → O). Oxidation of cyt. c occurs over the same time scale. In the presence of cyt. c absorbance changes attributed to the CytcO (panels a–c) were smaller because the redox sites were re-reduced by cyt. c during the course of the reaction. Over a time scale of ~500 ms (right-hand side boxes) all redox sites become oxidized. The small increase in absorbance over this time scale in the absence of cyt. c is due to small fractional re-reduction of CytcO by ascorbate. Experimental conditions after mixing: 1.1 μM CytcO, 20 mM HEPES at pH 7.5, 0.05% DDM, 100 μM EDTA, 2 mM ascorbate, 1 μM hexa-ammine-ruthenium(II) chloride, 1 mM O2 at ~22 °C (black trace). The red traces are averages of three traces obtained under the following conditions: [cyt. c]/[CytcO] in μM (i) 2.1/1.6, (ii) 1.5/1.3, (iii) 1.7/1.3. The mixing ratio was 1:5 with an oxygen-saturated buffer solution (20 mM HEPES at pH 7.5). All traces have been scaled to 1 μM reacting CytcO based on the rapid change in absorbance at 445 nm at t = 0. A laser artifact at t = 0 has been truncated for clarity.
At 445 nm and 605 nm the main contribution to the absorbance changes is from both hemes a and a3. At 830 nm and 550 nm the main contribution is from CuA and cyt. c, respectively. The A → P R and F → O reactions are seen as a decrease in absorbance with time constants of ~40 μs and ~1.5 ms (Fig. 3a,b), respectively, at both 445 nm and 605 nm. The P R → F reaction is not seen at these wavelengths, but fractional oxidation of CuA that is linked in time to the P R → F reaction31 is seen at 830 nm where we observed an increase in absorbance with a time constant of ~200 μs (Fig. 3c). Also the remaining oxidation of CuA, linked in time to the F → O H reaction, is seen as a further increase in absorbance at 830 nm. We did not observe any absorbance changes at 550 nm in the absence of cyt. c, which shows that at this wavelength the contribution from redox changes of CytcO is negligible (Fig. 3d, black trace).
Over a longer time scale we observed a slight re-reduction of hemes a and a3, seen as a small increase in absorbance at 445 nm and 605 nm (see right-hand side boxes of panels a and b).
Reaction of the cyt. c-CytcO complex with O2
A mixture of cyt. c and CytcO, in a low-ionic strength solution (see Materials and Methods), was reduced and incubated under an atmosphere of CO. The cyt. c was added at a slight excess to CytcO (1.2–1.3 cyt. c/CytcO) to assure formation of the cyt. c-CytcO complex in the major fraction of the population (see below). We first discuss the absorbance changes over a time scale of 8 ms (black traces in Fig. 3). Over this time scale four electrons are delivered from CytcO to O2, as discussed above.
When cyt. c is bound to CytcO an electron is transferred from cyt. c to CuA upon the first, fractional oxidation of CuA, i.e. during the P R → F reaction (τ ≅ 200 μs), and then during the F → O H reaction (τ ≅ 1.5 ms) while CuA/heme a are oxidized further (red traces in Fig. 3)26,27,28. These oxidation reactions are seen as a decrease in absorbance at 550 nm with time constants of ~200 μs and ~1.5 ms, respectively. All other absorbance changes over a time scale of 8 ms were diminished in the presence of cyt. c, reflecting re-reduction of the CytcO redox sites by cyt. c (compare the red and black traces in Fig. 3a–c).
The absorption coefficients of the redox sites at the measured wavelengths are listed in Table 1. All data in Fig. 3 were scaled to 1 μM reacting CytcO, based on the absorption coefficient for the CO-dissociation absorbance change at 445 nm. This normalization was only done to simplify comparison of the data at different wavelengths and it does not influence the analysis below.
Further oxidation of the cyt. c-CytcO complex after reaction with O2
In the five-electron cyt. c-CytcO system the single remaining electron after oxidation of the CytcO is not rapidly transferred to the excess oxygen (i.e. to a second O2) because O2 does not bind to the catalytic site when it is reduced by only one electron. Instead, oxidation of the hemes occurred over a longer time scale of ~500 ms, see right-hand side boxes in Fig. 3. This process may involve either (i) transfer of the electron to the optically silent CuB, or (ii) electron exchange between the CytcO molecules, via cyt. c, eventually yielding a thermodynamically stable state. The oxidation state of the CytcO catalytic site in this stable state would depend on the number of electrons transferred to the specific enzyme molecule. The most likely scenario is that an electron is exchanged between two CytcOs such that one molecule becomes oxidized and the other forms state P M .
To discriminate between these two scenarios we reasoned that in case (i) the time constant of the slowest oxidation reaction would be independent of the CytcO concentration while in case (ii) this time constant would decrease upon dilution because the reaction involves electron exchange between CytcO molecules mediated via cyt. c. As seen in Fig. 4, the time constant of the slowest component increased from ~75 ms to ~300 ms upon increasing the dilution of the CytcO-cyt. c sample with the O2-containing buffer from 1:1 to 1:5. This observation indicates that the slowest component is due to electron exchange between CytcO molecules according to scenario (ii).

Kinetics of absorbance changes upon reaction with O2 upon dilution. The experiment is the same as that shown in Fig. 3, but the CytcO-containing sample was mixed 1:1 (black) or 1:5 (red) with the O2-containing buffer. Note the different absorbance scales in the left and right parts of the graph, respectively. The concentrations of CytcO and cyt. c were 5 µM and 7.5 µM, respectively.
At 550 nm we observed a decrease in absorbance in the time range 8-500 ms (see Fig. 3d) that was added on top of the oxidation of the fraction reduced cyt. c in the cyt. c-CytcO complex. This signal was observed because there was an excess of 1.2–1.3 cyt. c compared to CytcO. Over this longer time scale this excess cyt. c in solution, became fully oxidized.
For the CuA absorbance changes at 830 nm (Fig. 3c) the absorbance reached a higher level with than without cyt. c at 500 ms, which is discussed below.
Analysis of the data
We define the absorbance difference, at wavelength λ, after x ms and immediately after the flash, i.e. at time = 0+: ΔAλ(x) = Aλ(t = x ms) - Aλ(t = 0+). As already indicated above, in the absence of cyt. c, ΔAλ(8) reflects oxidation of the reduced CytcO. We note that a comparison of this absorbance change at e.g. 445 nm and 605 nm, and that associated with CO-dissociation typically deviates from the equivalent differences seen in static spectra (Table 1). Therefore, we found that the most reliable approach to analyze the absorbance changes associated with oxidation of hemes a and a3 is to compare ΔA445(8) and ΔA605(8) with and without cyt. c added. This approach does not require the knowledge of absorption coefficients of hemes a and a3, only their relative contribution must be known. As seen in Table 1, at 605 nm hemes a and a3 contribute with 80% and 20%32,33, respectively. At 445 nm we used the relative contributions determined by Vanneste33: 66% and 34% for hemes a3 and a, respectively. The relative contributions determined by Liao et al.32 differed from these values, but the use of these values yields the same conclusions (see below).
The fractions hemes a and a3 (δ(a) and δ(a3), respectively) that become oxidized after 8 ms in the presence of cyt. c are determined from:
which yields δ(a) = 0.40 ± 0.04 and δ(a3) = 0.94 ± 0.06 (SD, 3 measurements). In other words, ~60% of heme a, but only ~6% of heme a3 become re-reduced when cyt. c is present. When using the relative contributions of hemes a and a3, respectively, based on the absorption coefficients determined by Liao et al.32 (see Table 1), the corresponding values would be δ(a) ≅ 0.5 and δ(a3) ≅ 1, i.e. ~50% re-reduced heme a and no heme a3 re-reduced.
Next, we calculate the fraction CuA that becomes re-reduced in the presence of cyt. c from the absorbance changes at 830 nm (Fig. 3c). At this wavelength oxidation of CuA leads to an increase in absorbance.
We compare the absorbance levels after 8 ms obtained in the absence and presence of cyt. c: \(\frac{{\rm{\Delta }}{A}_{+{\rm{cyt}}.c}^{830}(8)}{{\rm{\Delta }}{A}^{830}(8)}=\) \(0.85\pm 0.08\) (SD, three measurements), which yields ~15% re-reduced CuA when cyt. c is present.
We note that the absorbance levels at 830 nm after 500 ms in the presence and absence of cyt. c differ. This difference is most likely due to contribution of state P M , at 830 nm34, formed at the catalytic site after electron exchange between the CytcOs in the presence of cyt. c (see explanation above).
The fraction oxidized cyt. c is calculated from the absorbance changes at 550 nm (Fig. 3d). We calculated the fraction from the ratio of absorbance changes at 500 ms (full oxidation of the cyt. c pool) and 8 ms, respectively, assuming that the fraction oxidized cyt. c over the time scale of 8 ms reflects cyt. c that was bound in complex with CytcO.
The fraction oxidized cyt. c after 8 ms, \(\frac{{\rm{\Delta }}{A}_{+{\rm{cyt}}.c}^{550}(8)}{{\rm{\Delta }}{A}_{+{\rm{cyt}}.c}^{550}(500)}\) is 0.68. This number must be scaled for the molar excess cyt. c as compared to CytcO, which is 1.2 (in the experiment shown in Fig. 3d). Thus, assuming a 1:1 cyt. c-CytcO complex, the absorbance changes at 8 ms yield oxidation of 84 ± 7% (SD, 3 measurements) of the bound cyt. c. The occupancy of the cyt. c-binding site is not known, but because the degree of cyt. c oxidation is close to unity, the assumption of a 1:1 cyt. c:CytcO complex is at least approximately correct.
To test whether or not the presence of detergent could alter the midpoint potentials of CytcO in state O H , we also repeated the experiments with native membranes from R. sphaeroides (Fig. 5). Due to light scattering of the membrane fragment the signal-to-noise ratio in these measurements was much smaller than that seen with detergent-solubilized CytcO. Therefore, we had to exclude data at 830 nm where the absorbance changes associated with redox changes at CuA are very small (see Table 1). We observed a smaller degree of cyt. c oxidation over ~8 ms, presumably because part of cyt. c binds to the membrane fragments, being inaccessible to CytcO. Nevertheless, we could estimate the degree of cyt. c oxidation and reduction of hemes a and a3. The fraction oxidized cyt. c per CytcO was ~0.4 (Fig. 5c), based on a comparison with the data in Fig. 3d. The fraction reduced hemes a and a3 was estimated using Eqs (1 and 2) above and the absorbance differences ΔA445(8) and ΔA605(8) in Fig. 5a,b. These data indicate that at 8 ms after initiation of the reaction, heme a3 is not reduced while heme a is essentially fully reduced, which is qualitatively consistent with the data obtained with CytcO in detergent solution.

Reaction of membrane fragments with O2. The experiment is the same as that shown in Fig. 3. Absorbance changes were monitored at 445 nm (a) 605 nm (b) and 550 nm (c) Experimental conditions after mixing: R. sphaeroides membranes (∼2 µM CytcO, calculated from the reduced minus oxidized difference spectrum) in 20 mM HEPES (pH 7.5), 100 µM EDTA and 1 mM O2 at ∼22 °C. The mixing ratio was 1:1 with an oxygen-saturated buffer (20 mM HEPES pH 7.5). The traces have been scaled to 1 µM reacting CytcO. A laser artifact at t = 0 has been truncated for clarity.
Interpretation of the data obtained with detergent-solubilized CytcO is summarized in the illustration in Fig. 6. We found that at the end of the initial oxidation of the CytcO, i.e. after 8 ms, the fraction reduced cyt. c is 0.16. In other words, we assume that 0.84 electrons reside among the four redox sites of CytcO. The reduction levels of hemes a and a3 were 0.60 and 0.06, respectively, while that of CuA was 0.15. These numbers are obtained from the measured absorbance changes. When adding these fractions reduced hemes a and a3 and CuA we obtain 0.81. Because 0.84 electrons reside at CytcO (based on the degree of oxidation of cyt. c), the fraction reduced CuB is 0.03. It should be noted that this number is not measured directly because CuB is optically silent. It is calculated based on the assumption that the cyt. c-CytcO complex comprises five redox-active centers and that each of these sites can harbor one electron.

Summary of the data. The redox state of the co-factors is indicated with color. The fraction color in the small circles reflects the degree of reduction of each redox site. The apparent redox potentials were estimated from the fractions reduced redox sites assuming a midpoint potential of 250 mV for CuA. The numbers are based on the assumption that the cyt. c-CytcO complex accommodates five redox sites and that each site can accommodate one electron. No interactions between the redox sites are considered (see text).
On the basis of this distribution of the fifth electron among the five redox sites cyt. c, CuA, heme a, heme a3 and CuB, and assuming a midpoint potential of 250 mV for CuA35, we estimate the apparent redox potentials of the redox sites in CytcO in state O H (Fig. 6). We refer to “apparent” redox potentials because the analysis reflects only the distribution of a single electron after 500 ms in the five-electron system (among five sites). Because interactions between the redox sites are not considered (see35,36), the values differ from those obtained in redox titrations, which typically yield a higher midpoint potential for the catalytic site heme (here heme a3) than for the intermediate electron acceptor (here heme a)35,36, although in many bacterial oxidases the difference between the midpoint potentials is reversed37. Furthermore, because a single electron equilibrates among the five redox sites, the situation differs from a redox titration38. The purpose of the exercise to calculate the apparent redox potentials is not to provide the values, only to indicate that the apparent redox potential of CuB is not higher than that of any other site after reaction of the CytcO with O2. The main conclusion is that the fifth electron is not found at the catalytic site.
Belevich and colleagues21 concluded that upon injection of a single electron to state O H less than 2% heme a was reduced and that the midpoint potential of CuB was raised to a value at least 100 mV higher than that of heme a. This contrasts the 60% reduced heme a observed here and we did not find an increased midpoint potential of CuB. One difference between our study and that of Belevich et al. is the different experimental approach; we used cyt. c as the electron donor while Belevich et al. used a Ru-complex excited by light. Even though the binding geometry and overall charge of these two electron donors differ, the differences are not likely to explain the discrepancy in the CuB midpoint potentials because of the significant distance between the site where the electron donor binds and CuB. Belevich and colleagues21 discussed the discrepancy between their results and those obtained in other laboratories prior to the present study, and suggested that they may originate from exposure of the CytcO to detergent or removal of phospholipids during enzyme isolation. As discussed above, we did not find any additional reduction of the catalytic site with CytcO in its native lipid environment.
In earlier studies using an identical experimental approach to that used here39,40 we did observe differences between CytcO that was reconstituted in liposomes and in detergent. However, these differences were mainly observed for the kinetics of reactions that are associated with proton uptake from solution, presumably due to interaction of lipids with groups near the entry point of the CytcO proton pathways40.
The experimental approach used here reflects the conditions in the native system where cyt. c is the electron donor. Yet, one may argue that in our experiments the electron from cyt. c was transferred over a time scale that is shorter than that of O H formation, i.e., an alternative explanation is that the state formed is not O H . State O is defined as the “relaxed form” of the oxidized CytcO, reached after long time without turnover, i.e. not the transient state visited in the experiments described here. Consequently, if we assume that O H is not formed here, we would have to assume a third oxidized state. Furthermore, we would have to postulate that formation of O H requires a delay between oxidation and re-reduction of the CytcO. However, the concentration of O2 in mammalian tissues is only a few μM41, which suggests that at steady state heme a/CuA would be at least partly reduced, resembling the states visited in the current study, as also indicated in42. We also note that in the experiments described by Belevich and colleagues21 the delay between oxidation and injection of the fifth electron was ~5 ms and over this time scale we did not observe any additional heme oxidation.
Summary
An activated form of the oxidized CytcO, O H , was postulated. The difference between states O and O H was suggested to be an elevated midpoint potential of CuB in the latter. In the present study, we used an alternative experimental technique to approach the problem and found that in the O H state the electron resides mainly at heme a and CuA. In other words the CuB midpoint potential was not elevated in O H . A similar behavior was observed in native membrane, which excludes the possibility that the behavior is due to removal of lipids from the protein.
Materials and Methods
Preparation of R. sphaeroides CytcO
The R. sphaeroides bacteria were grown aerobically in the dark at 30 °C in a Sistrom medium. After harvesting, the cells were re-suspended in 50 mM Tris-buffer, pH 8.0 (0.2 g/ml) and stirred for one hour at 4 °C in the presence of DNase I (0.05 mg/ml final concentration). The suspension was subsequently passed twice through a continuous-flow cell disruptor (Constant Systems LTD) operating at 170 MPa. Unbroken bacteria and cell debris were removed from the suspension by low-speed ultra-centrifuging (7800 g for 15 min at 4 °C) followed by high-speed ultra-centrifugation (138 000 g for 90 minutes at 4 °C) to collect the membrane fraction. The membrane pellet was re-suspended (homogenized) in 50 mM Tris-buffer.
For purification of the CytcO the membrane fraction of the cells was solubilized in 1.5% n-dodecyl β -D-maltoside (DDM). The histidine-tagged CytcO was purified using Ni2+-NTA affinity chromatography as described previously43,44.
Measurement of the oxidation kinetics
For studies of CytcO in the native membrane environment the R. sphaeroides membranes were diluted to ~5 μM CytcO in 20 mM HEPES (pH 7.5) and 100 μM EDTA.
For studies of the purified CytcO, the buffer-solution of the enzyme was exchanged for 20 mM HEPES (pH 7.5), 0.05% DDM and 100 μM EDTA using a gel filtration column (PD-10, GE Healthcare), resulting in a final CytcO concentration of ~10 μM. The same method was used to prepare the cyt. c-CytcO complex where equine heart cyt. c (Sigma-Aldrich) was added to the CytcO prior application to the PD-10 column. For the R. sphaeroides membrane fraction, the equine heart cyt. c was added directly to the sample.
The cyt. c-CytcO ratio was 1.2–1.3 (see text and figure legends). The sample was transferred to a locally modified Thunberg cuvette after which air in the cuvette was exchanged for N2 using a vacuum line. The sample was reduced upon anaerobic addition of ascorbate (2 mM) and the redox mediator hexa-ammine-ruthenium(II) chloride (1 μM). After full reduction, the N2 atmosphere in the cuvette was replaced by carbon monoxide, which binds to the reduced heme a3. The redox state of the enzyme as well as formation of the CytcO-CO complex were verified spectrophotometrically.
The CytcO-CO complex (with or without cyt. c) was mixed rapidly with an oxygen-saturated buffer (20 mM HEPES (pH 7.5), 0.05% DDM, 0.1 mM EDTA) at a CytcO:O2-saturated solution ratio of 1:5) in a flow-flash apparatus (Applied Photophysics). The CO ligand was photo-dissociated from heme a3, about 200 ms after mixing, by means of a 10-ns laser flash (Quantel Brilliant B, Nd-YAG, 532 nm), enabling oxygen to bind to heme a3. The subsequent reaction of the reduced CytcO with oxygen was monitored as absorbance changes over time at different single wavelengths45.
References
Kaila, V. R. I., Verkhovsky, M. I. & Wikström, M. Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 110, 7062–7081 (2010).
Hosler, J. P., Ferguson-Miller, S. & Mills, D. A. Energy transduction: Proton transfer through the respiratory complexes. Annual Review of Biochemistry 75, 165–187 (2006).
Yoshikawa, S. et al. Proton pumping mechanism of bovine heart cytochrome c oxidase. Biochimica et Biophysica Acta - Bioenergetics 1757, 1110–1116 (2006).
Namslauer, A. & Brzezinski, P. Structural elements involved in electron-coupled proton transfer in cytochrome c oxidase. FEBS Lett 567, 103–110 (2004).
Brzezinski, P. & Gennis, R. B. Cytochrome c oxidase: exciting progress and remaining mysteries. J. Bioenerg. Biomembr. 40, 521–531 (2008).
Brzezinski, P. & Ädelroth, P. Design principles of proton-pumping haem-copper oxidases. Curr Opin Struct Biol 16, 465–472 (2006).
Richter, O. M. H. & Ludwig, B. Electron transfer and energy transduction in the terminal part of the respiratory chain - Lessons from bacterial model systems. Biochimica et Biophysica Acta - Bioenergetics 1787, 626–634 (2009).
Ferguson-Miller, S., Hiser, C. & Liu, J. Gating and regulation of the cytochrome c oxidase proton pump. Biochimica et Biophysica Acta - Bioenergetics 1817, 489–494 (2012).
Rich, P. R. & Maréchal, A. Functions of the hydrophilic channels in protonmotive cytochrome c oxidase. Journal of the Royal Society Interface 10, 183–196 (2013).
Konstantinov, A. A. Cytochrome c oxidase: Intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett. 586, 630–639 (2012).
Blomberg, M. R. A. & Siegbahn, P. E. M. Proton pumping in Cytochrome c oxidase: Energetic requirements and the role of two proton channels. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1837, 1165–1177, https://doi.org/10.1016/j.bbabio.2014.01.002 (2014).
Popović, D. M., Leontyev, I. V., Beech, D. G. & Stuchebrukhov, A. A. Similarity of cytochrome c oxidases in different organisms. Proteins: Structure, Function and Bioinformatics 78, 2691–2698 (2010).
Von Ballmoos, C., Ädelroth, P., Gennis, R. B. & Brzezinski, P. Proton transfer in ba 3 cytochrome c oxidase from Thermus thermophilus. Biochim. Biophys. Acta 1817, 650–657 (2012).
Wikström, M., Sharma, V., Kaila, V. R. I., Hosler, J. P. & Hummer, G. New perspectives on proton pumping in cellular respiration. Chem. Rev. 115, 2196–2221, https://doi.org/10.1021/cr500448t (2015).
Wikström, M. & Morgan, J. E. The dioxygen cycle. Spectral, kinetic, and thermodynamic characteristics of ferryl and peroxy intermediates observed by reversal of the cytochrome oxidase reaction. J Biol Chem 267, 10266–10273 (1992).
Wikström, M. Identification of the electron transfers in cytochrome oxidase that are coupled to proton-pumping. Nature 338, 776–778 (1989).
Wikström, M. et al. Mechanism of Proton Translocation by the Respiratory Oxidases - the Histidine Cycle. Biochim. Biophys. Acta-Bioenerg. 1187, 106–111 (1994).
Verkhovsky, M. I., Jasaitis, A., Verkhovskaya, M. L., Morgan, J. E. & Wikström, M. Proton translocation by cytochrome c oxidase. Nature 400, 480–483 (1999).
Bloch, D. et al. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc. Natl. Acad. Sci. USA 101, 529–533 (2004).
Wikström, M. & Verkhovsky, M. I. Towards the mechanism of proton pumping by the haem-copper oxidases. Biochimica et Biophysica Acta - Bioenergetics 1757, 1047–1051 (2006).
Belevich, I., Bloch, D. A., Belevich, N., Wikström, M. & Verkhovsky, M. I. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. USA 104, 2685–2690 (2007).
Sharma, V., Karlin, K. D. & Wikström, M. Computational study of the activated OH state in the catalytic mechanism of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 110, 16844–16849, https://doi.org/10.1073/pnas.1220379110 (2013).
Blomberg, M. R. A. & Siegbahn, P. E. M. Protonation of the binuclear active site in cytochrome c oxidase decreases the reduction potential of Cu B. Biochimica et Biophysica Acta - Bioenergetics 1847, 1173–1180, https://doi.org/10.1016/j.bbabio.2015.06.008 (2015).
Jancura, D. et al. Spectral and kinetic equivalence of oxidized cytochrome c oxidase as isolated and “activated” by reoxidation. J. Biol. Chem. 281, 30319–30325, https://doi.org/10.1074/jbc.M605955200 (2006).
Brand, S. E. et al. A new ruthenium complex to study single-electron reduction of the pulsed OH state of detergent-solubilized cytochrome oxidase. Biochemistry 46, 14610–14618, https://doi.org/10.1021/bi701424d (2007).
Hill, B. C. Modeling the sequence of electron transfer reactions in the single turnover of reduced, mammalian cytochrome c oxidase with oxygen. J Biol Chem 269, 2419–2425 (1994).
Hill, B. C. The reaction of the electrostatic cytochrome c-cytochrome oxidase complex with oxygen. J. Biol. Chem. 266, 2219–2226 (1991).
Hirota, S. et al. A flash-photolysis study of the reactions of a caa3-type cytochrome oxidase with dioxygen and carbon monoxide. J Bioenerg Biomembr 28, 495–501 (1996).
Brzezinski, P. & Larsson, G. Redox-driven proton pumping by heme-copper oxidases. Biochim. Biophys. Acta 1605, 1–13 (2003).
Einarsdóttir, Ó. Fast Reactions of Cytochrome-Oxidase. Biochim. Biophys. Acta 1229, 129–147 (1995).
Karpefors, M., Ädelroth, P., Zhen, Y., Ferguson-Miller, S. & Brzezinski, P. Proton uptake controls electron transfer in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 95, 13606–13611 (1998).
Liao, G. L. & Palmer, G. The reduced minus oxidized difference spectra of cytochromes a and a3. Biochim. Biophys. Acta 1274, 109–111 (1996).
Vanneste, W. H. The stoichiometry and absorption spectra of components a and a-3 in cytochrome c oxidase. Biochemistry 5, 838–848 (1966).
Szundi, I., Liao, G. L. & Einarsdóttir, O. Near-infrared time-resolved optical absorption studies of the reaction of fully reduced cytochrome c oxidase with dioxygen. Biochemistry 40, 2332–2339, https://doi.org/10.1021/bi002220v (2001).
Gorbikova, E. A., Vuorilehto, K., Wikström, M. & Verkhovsky, M. I. Redox titration of all electron carriers of cytochrome c oxidase by Fourier transform infrared spectroscopy. Biochemistry 45, 5641–5649, https://doi.org/10.1021/bi060257v (2006).
Sousa, F. L. et al. Redox properties of thermus thermophilusba3: Different electron-proton coupling in oxygen reductases? Biophys. J. 94, 2434–2441 (2008).
Melin, F. et al. The unusual redox properties of C-type oxidases. Biochimica et Biophysica Acta - Bioenergetics 1857, 1892–1899, https://doi.org/10.1016/j.bbabio.2016.09.009 (2016).
Alric, J. et al. Electrostatic interaction between redox cofactors in photosynthetic reaction centers. J. Biol. Chem. 279, 47849–47855, https://doi.org/10.1074/jbc.M408888200 (2004).
Öjemyr, L. N., Von Ballmoos, C., Faxén, K., Svahn, E. & Brzezinski, P. The membrane modulates internal proton transfer in cytochrome c oxidase. Biochemistry 51, 1092–1100 (2012).
Näsvik Öjemyr, L., Lee, H. J., Gennis, R. B. & Brzezinski, P. Functional interactions between membrane-bound transporters and membranes. Proc Natl Acad Sci USA 107, 15763–15767 (2010).
Wittenberg, B. A. & Wittenberg, J. B. Transport of oxygen in muscle. Annual Review of Physiology 51, 857–878 (1989).
Wikström, M. Active site intermediates in the reduction of O 2 by cytochrome oxidase, and their derivatives. Biochimica et Biophysica Acta - Bioenergetics 1817, 468–475 (2012).
Mitchell, D. M. & Gennis, R. B. Rapid purification of wildtype and mutant cytochrome c oxidase from Rhodobacter sphaeroides by Ni(2+)-NTA affinity chromatography. FEBS Lett. 368, 148–150 (1995).
Zhen, Y. et al. Overexpression and purification of cytochrome coxidase from Rhodobacter sphaeroides. Protein Expression and Purification. Protein Expr. Purif. 13, 326–336 (1998).
Brändén, M. et al. On the role of the K-proton transfer pathway in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 98, 5013–5018 (2001).
Van Gelder, B. F. & Slater, E. C. The extinction coefficient of cytochrome c. BBA - Biochimica et Biophysica Acta 58, 593–595 (1962).
Boelens, R. & Wever, R. Redox reactions in mixed-valence cytochrome c oxidase. FEBS Lett. 116, 223–226, https://doi.org/10.1016/0014-5793(80)80649-1 (1980).
Acknowledgements
We would like to thank Margareta Blomberg and Pia Ädelroth for valuable discussions. The study was supported by grants from the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
Author information
Authors and Affiliations
Contributions
J.V. performed experiments. P.B. and J.V. wrote the manuscript and prepared figures. R.B.G., P.B. and J.V. planned research. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vilhjálmsdóttir, J., Gennis, R.B. & Brzezinski, P. The electron distribution in the “activated” state of cytochrome c oxidase. Sci Rep 8, 7502 (2018). https://doi.org/10.1038/s41598-018-25779-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25779-w
- Springer Nature Limited