
Abstract
In situ x-ray diffraction measurements and inverse Monte Carlo simulations of pair distribution functions were used to characterize the local structure of molten AuGa2 up to 16 GPa and 940 K. Our results document systematic changes in liquid structure due to a combination of bond compression and coordination increase. Empirical potential structure refinement shows the first-neighbor coordination of Ga around Au and of Au around Ga to increase from about 8 to 10 and 4 to 5, respectively between 0 and 16 GPa, and the inferred changes in liquid structure can explain the observed melting-point depression of AuGa2 up to 5 GPa. As intermetallic AuGa2 is an analogue for metallic SiO2 at much higher pressures, our results imply that structural changes documented for non-metallic silicate melts below 100 GPa are followed by additional coordination changes in the metallic state at pressures in the 0.2–1 TPa range achieved inside large planets.
Similar content being viewed by others

Introduction
The study of densified liquid structures provides fundamental information for understanding thermodynamic phase diagrams, including the effects of distinct crystalline phases on melting at various pressures and temperatures. The intermetallic compound AuGa2 exhibits rich polymorphism in the solid state1,2,3, transforming from 8- to 10-coordinated crystal structures (Ga around Au) at pressures below 30 GPa. We present high-pressure x-ray diffraction measurements in order to determine whether analogous structural changes take place in the liquid. The melting temperature of AuGa2 is known to go through a minimum around 5.5 GPa3, presumably because of competing structural changes in the crystalline and liquid phases, but this interpretation needs verification through measurements on the melt under pressure. Developments in experimental techniques over recent years are providing rich information about liquid structures over a much wider range of pressures than heretofore possible, complementing the vast amount of data available for crystal-structural transformations under pressure4,5,6. Moreover, there is only limited understanding of the local structure of solid amorphous AuGa2, with the available information amounting to the total structure factor obtained from electron diffraction7, which does not provide such detail as pair distribution functions from experiment or modelling (e.g., Monte-Carlo modelling).
Results from Experiments and Modeling
Data were obtained by heating each of the crystalline phases of AuGa2 to temperatures slightly above melting (Table 1), the two-dimensional diffraction patterns recorded by image plate confirming the absence of any crystalline phase after melting3. In spite of our limited Q range, which constrains the simulation of measurements to low magnitudes of the scattering vector Q, we were able to observe distinct features in the diffraction patterns for liquid AuGa2 at pressures at which different sub-solidus crystal structures are stable (Table 2, Fig. 1a).

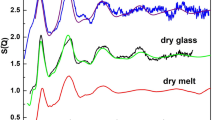
(a) Experimental (thick grey lines) and EPSR simulated (thin black lines) total structure factors, S(Q), for liquid AuGa2 at different pressures. (b) Diffraction patterns for crystalline phases at corresponding pressures. (c) Pressure dependence of the liquid S(Q) peaks for 1.5 < Q < 3.5 Å−1, with Peaks 2 and 3 merging to form a broad peak (BP) at 16 GPa.
The most prominent peak (Peak 3, Fig. 1c) in the structure factor S(Q) at temperatures above the melting temperatures of cubic and orthorhombic phases (P ≤ 11 GPa) has a clear shoulder on the low-Q side (Peak 2, Fig. 1c). This is distinct from the result for the monoclinic-to-melt pattern (P = 16 GPa, Fig. 1a), which shows a broad peak at Q = 2.62(5) Å−1 attributable to overlapping of the prominent peak and shoulder. Peak 3 for the liquid S(Q) is in the range of 2.87–2.95 Å−1 at 11 GPa and below, matching the highest peaks in the diffraction patterns of the three crystalline phases (Fig. 1b). Partial structure factors calculated from our simulations show that Au-Au correlations give the major contribution to the first sharp diffraction peak (Peak 1) in S(Q); Peak 2 arises from Ga-Ga correlations, along with smaller contributions from Au-Ga correlations; and Peak 3 can be attributed to Au-Ga and Au-Au correlations (Fig. 2). Qualitatively, the increases in Q values for Peaks 1–3 to 11 GPa are consistent with compaction of intermediate-range order in the liquid, whereas evolution of the main peak into a broad maximum at Q ~ 2.6 Å−1, similar to the value at zero pressure, suggests an increase in first-neighbor coordination that is causing an increased bond length as the sample is compressed.

Calculated partial structure factors for amorphous (P = 0, T = 300 K) and liquid AuGa2 under pressure. Data are vertically offset for clarity.
For purposes of model validation, we have also considered the amorphous (glass) phase of AuGa2 at P = 0 GPa and T = 300 K based on data of Bohórquez7. In general, the oscillations in S(Q) for liquid AuGa2 under pressure have a lower amplitude and die away more rapidly with Q than do those for the amorphous phase at ambient conditions, while the peak splitting in the 2.0–3.3 Å−1 range for molten AuGa2 becomes indistinct from the corresponding S(Q) range for amorphous AuGa2 (Supplementary Figure S8). The prominent Peak 3 is located at 3.08(5) Å−1 and 2.95(5) Å−1 (~4% change) for amorphous and orthorhombic-to-melt phases, respectively. The position of the shoulder on the low-Q side (Peak 2) is at 2.44(7) Å−1 in the S(Q) data obtained from the amorphous state, and changes to 2.51(8) Å−1 in the liquid at 11 GPa and 863 K, a ~3% change. The ratio of positions for Peak 3 and Peak 2 changing from ~1.26 at ambient conditions to ~1.17 at 11 GPa suggests increasing complexity in the structure of liquid AuGa2 under pressure. The first sharp diffraction peak (Peak 1) in S(Q) of amorphous AuGa2, at ~1.88 Å−1, increases steadily to ~2.01 Å−1 with pressure, corresponding to intermediate-range structural order at a length scale of ~ 3.2 Å (Fig. 1c and Supplementary Figure S11).
We test the idea of increased liquid-structure complexity under pressure by deriving the reduced pair distribution function (PDF) G(r) for the liquid: it has a strong peak at 2.73 to 2.91 Å with increasing pressure and a secondary peak at 4.82 to 5.19 Å over the 0–16 GPa pressure range (Figs 3 and 4), providing a good fit to the experimental data (accurate representation of short range structure) up to ∼6 Å at each pressure point. The differences between calculated and experimental G(r) above ∼6 Å indicate that intermediate range order in liquid AuGa2 is not well represented, however. This is a subject for more detailed PDF analysis using diffraction data collected with higher-energy x-rays, but does not affect the present interpretations.

Calculated (lines) and experimental (grey dots) reduced pair distribution functions, G(r), for liquid AuGa2 under pressure.

Positions of the first peak in the reduced pair distribution functions, G(r), for liquid AuGa2 (squares) and ratio of the second to first peak positions (circles). The dashed lines indicate the ideal ratio determined for a simple close-packed hard sphere-like structures (r2/r1 ≈ 1.91) and for an ideal tetrahedral structural unit (r2/r1 ≈ 1.63)8. The errors in peak positions are in the range of ±0.002–0.018, smaller than the corresponding symbols.
The ratio of second to first neighbor distances shifts from 1.67 to 1.81 with increasing pressure, neither value being close to that expected for simple hard sphere-like structures (for which r2/r1 ≈ 1.91). The Au−Au, Au−Ga and Ga−Ga pair distribution functions, obtained from Fourier transforming the corresponding partial structure factors, show that the main peak in G(r) (1.8 < r < 4.0 Å) has contributions from first-neighbor Au-Ga for r < 2.8 Å and second-neighbor Ga-Ga and Au-Au correlations for r > 2.8 Å (Fig. 5). The Au-Ga and Ga-Ga nearest-neighbor distances are calculated to be in the range 2.50–2.66 Å and 2.88–3.03 Å, respectively, decreasing ∼4.5% and ∼4.8% over our pressure range (Fig. 6a). The pressure-induced broadening observed for the first Au-Ga peak can be associated with a change in coordination number around Au, and the pressure dependence of the Ga-Ga pair distribution function is similar to that of Au-Ga; the Au-Au pair distribution functions have larger statistical uncertainty than for Au-Ga and Ga-Ga, due to the smaller relative fraction of Au atoms in the AuGa2 stoichiometry.

Au-Au, Au-Ga, and Ga-Ga partial pair distribution functions calculated from the EPSR simulations for (a) amorphous AuGa2 at P = 0 and T = 300 K, and (b,c,d) liquid AuGa2 under pressure.

(a) Pressure dependence of Au-Ga (nearest-neighbor) and Ga-Ga bond distances determined from partial pair distribution functions (Fig. 5). (b) Coordination numbers around Au- and Ga-atoms, n AuGa , n GaAu and n AuAu , and experimental and calculated average coordination numbers.
Coordination Changes and Discussion
Nearest-neighbor coordination numbers around Au- and Ga-atoms, n AuGa , n GaAu and n AuAu , obtained by integrating the areas of the corresponding peaks in the g AuAu (r) and g AuGa (r) pair distribution functions (Table 3), show systematic increases with pressure (Fig. 6b). The results at zero pressure are consistent with eight-fold coordination of Ga around Au in the liquid, the coordination in the cubic CaF2 crystal structure that is stable at ambient conditions. However, our model indicates that the first-neighbor coordination increases rapidly in the liquid, toward nine-fold by 1–5 GPa, implying a denser melt relative to the coexisting crystal structure, as documented by the negative Clapeyron slope for the melting temperature3. That is, the pressure-induced melting-point depression for AuGa2 at low pressures, with a turnaround at 5 GPa, can be explained in terms of changes in liquid and crystal structures.
With rising pressure and temperature, we find n AuGa gradually increasing to 9.4(7) at 16.1 GPa, implying nine- and ten-fold coordination around Au in the melt above 15 GPa. Given the stoichiometry of AuGa2, we expect the coordination of Au around Ga to correspondingly rise from about 8/2 = 4 at zero pressure to 10/2 = 5 at 15 GPa, which is consistent with the calculated average value of n GaAu that increases from 3.9(4) to 4.9(5) at 16.2 GPa (Fig. 6b). The presence of GaAu4 structural units in the melt at low pressure is also in line with the ratio of second to first neighbor distances observed in G(r), r2/r1 ≈ 1.63 for tetrahedra (Fig. 4)8,9. Integration of the broad peak in the Au-Au pair distribution function (3 < r < 6 Å) gives a coordination number n AuAu = 12.1(8) at ambient conditions and rising to 14.5(9) by 16 GPa, confirming that liquid AuGa2 densifies via collapse of the second- and higher-coordination shells under pressure. We find that the average coordination number calculated over the range 1.0 < r < 3.8 Å, rises from n average = 10.8(6) at ambient conditions to 13.3(7) 16 GPa based on the fits to our experimental data, and this compares well with n average calculated by integrating the intensity under the peak in g(r), ρ0 4πr2g(r) (Fig. 6b and see Supplementary Figure S12).
Ga-Au-Ga and Au-Ga-Au bond-angle distributions were calculated for neighbors separated by less than 3.3 Å. The bond-angle distributions obtained from amorphous-AuGa2 at ambient conditions are characterized by peaks at ∼68° and 108° for Ga-Au-Ga and Au-Ga-Au, close to the ideal values for 8-fold and tetrahedral (109°) coordination and consistent with our estimates for n AuGa and n GaAu , respectively (Fig. 7). With increasing pressure, the Au-Ga-Au angle distribution forms a broad peak centered near 86° that accords with fivefold coordination. The main Ga-Au-Ga distribution peak broadens and becomes asymmetric with pressure, indicating coordination change toward a distribution of first-neighbor configurations ranging from 7.9 to 9.4 (Fig. 8).

Au-Ga-Au (a) and Ga-Au-Ga (b) bond-angle distributions as functions of pressure, and structural motifs at zero pressure (c,d) captured from EPSR.

Schematic representation of densification mechanism and corresponding structural moieties in liquid AuGa2. Amorphous solid (a) includes GaAu4 tetrahedral units connected to each other via Au-Au bonding and to AuGa8 polyhedral units via Au-Ga bonding. Densification is dominated by large inward shifting of the second- and higher coordination shells under pressure (b). Melt-AuGa2 obtained at highest pressure shows higher order polyhedral units, 5 and 9 coordination around Ga and Au atoms, respectively (c).
Silica is the archetypal rock-forming compound making up terrestrial planets, and it is expected to take on the cotunnite structures and to become metallic at multi-Mbar (~200–500 GPa) pressures1,10,11,12,13,14,15,16. Therefore, we expect that fluid AuGa2 may serve as an analog of the liquid metallic silicates present at the high pressures relevant to the interiors of large planets, such as super-Earths and other extrasolar planets17. Moreover, it has been found that silicate melts (magmas) undergo structural transformations, with increasing first-neighbor coordination reducing the volume difference between crystal and melt, such that magmas can sink at depth rather than being buoyant as observed at the surface (e.g., causing volcanic eruptions)17,18. These conclusions have been documented through high-pressure spectroscopy and x-ray diffraction on silicate (and analogue) melts and glasses, documenting Si-O coordination increasing from 4 to 6 by about 35 GPa19,20,21,22,23,24; recent diffraction results show no significant coordination increase beyond 6–6.8 to nearly 200 GPa25, however. Along with flattening of the melting curve, T m (P), atomistic models predict that the adiabatic temperature gradient (or Grüneisen parameter, γ = (∂lnT/∂lnρ) S ) of the melt exhibits the unusual tendency of increasing over a range of depths due to increasing coordination26,27 (T, P and ρ are temperature, pressure and density; subscripts m and S refer to melting and constant entropy).
Our results provide evidence for pressure-induced coordination change in intermetallic AuGa2, a potential analogue for the liquid metallic state of SiO2 that is stable at pressure relevant to large planetary interiors. Thus, based on our measurements we expect that the Si–O coordination in silicate melts increases beyond the 4- to 6-fold range documented by experiments to date up to 100–200 GPa, and toward 10-fold at pressures above 500 GPa where the cotunnite phase of SiO2 is predicted to be stable. Such changes in liquid structure are expected to affect planetary evolution by influencing the buoyancy of magmas relative to coexisting crystals over specific depth ranges within a planet, with element partitioning between liquid and crystals potentially determining whether melts sink or rise, and the Grüneisen parameter exhibiting anomalous pressure dependencies as at lower pressures.
Methods
Experimental study
AuGa2 was prepared by arc melting the elements under an Ar atmosphere, and x-ray diffraction confirms the expected cubic fluorite-type structure (Fm3m) with lattice parameter, a = 6.079(3) Å28. We used a resistively heated diamond-anvil cell (DAC) driven by a gas membrane for the present experiments29. A 10 × 10 µm beam of 25 keV energy (0.4959 Å wavelength) x-rays, as selected by a Si(111) double-crystal monochromator at the Advanced Light Source beamline 12.2.2, was used to collect diffraction patterns; the beam was focused at the sample position30. We used BN backing plates with different opening angles on the detector side, which allowed diffraction data to be collected up to a maximum scattering vector-magnitude of Q = 7 Å−1, corresponding to a resolution in radial distribution functions Δr ~ 0.740 (±0.045) Å. Diffraction patterns were obtained using a Mar345 image plate located 290.0 (±0.1) mm from the sample, and processed with fit2d and Celref31,32. On-line ruby (Al2O3:Cr3+) and samarium-doped strontium tetra-borate (SrB4O7:Sm2+) fluorescence were used to determine pressure of the hot sample (stimulated with an in-line 200 mW blue diode laser to increase the photon counts at high temperatures). Temperature was measured to an accuracy of ±5 K up to 900 K, using a K-type thermocouple next to the diamond culet3,29. Rhenium was the gasket material, and liquefied argon served as the pressure-transmitting medium3.
For each of our experiments, the sample was taken to the desired pressure and then put through a heating and cooling cycle; we observed consistent pressure drifts during each cycle, caused by thermal expansion of the diamond-cell components (pressure-temperature paths for five runs are given in Table 1). Complete melting of AuGa2 is identified by loss of long-range order, indicated by disappearance of x-ray diffraction peaks of the crystalline phase and a simultaneous increase in diffuse scattering over the entire 2θ range (raw melt data collected by heating various AuGa2 phases are shown in Supplementary Figs S1–S5)3. A new sample was reloaded after each thermal cycle, and diffraction patterns from melting various AuGa2 phases are illustrated in Fig. 1a. The experimental melt patterns shown in Fig. 1a were collected separately on a region free of gasket peaks, with longer exposure time in order to get good peak intensity relative to background.
EPSR method to refine 3D structural model
McGreevy33 reviews a wide range of issues relating to the inversion of diffraction data to derive real-space functions. Although emphasizing reverse Monte Carlo methods, McGreevy also discusses empirical structure refinement (EPSR), and Soper34 provides further details. We use the EPSR method to fit and extrapolate to high Q the total structure factor S(Q) derived from each diffraction pattern (Fig. 1a), using 1000 Au and 2000 Ga atoms in the simulation box at conditions summarized in Table 2. The S(Q) data are then Fourier transformed to get radial distribution functions (see Supplementary Figure S6a). Application of EPSR requires that the sample density is known, which makes characterizing the liquid structures challenging. The density of molten AuGa2 was determined using two different methods: (i) from the slope of the reduced pair distribution functions, G(r), below 2.2 Å (see Supplementary Figure S6b); and (ii) from experimental measurements of the unit-cell volumes of the crystalline phases just below the melting temperature (Supplementary Figure S6c). The former includes truncation errors, so here we present results based on using the latter method (Table 2). Storm et al.35 have estimated the volume changes occurring on melting of AuGa2 by making use of low-pressure dT/dP data, and evaluated entropies of fusion based on the assumption that the addition of the entropies of fusion for the elements along with the entropy of mixing gives a reasonable estimate for the entropy of fusion of the compound. The decrease in volume on melting of AuGa2 is estimated to be 0.5 cc/mole. This results in approximately ≤1% change in volume on melting up to about 10 GPa, and justifies the use of crystalline-phase density just before melting to estimate the liquid density. Atomic number densities calculated using the two methods are compared in Supplementary Table S1. The Lennard-Jones potential-well depth and range for Au (Ga) were set to 0.1632 (0.1750) kJ/mol and 2.93 (1.60) Å, with initial parameters perturbed until we obtained a satisfactory fit to the diffraction data. Minimum approach distances of 2.77, 2.30 and 2.70 Å for, respectively, Au-Au, Au-Ga and Ga-Ga pairs are calculated using atomic radii of Au and Ga, and served as constraints in carrying out Monte Carlo simulations of the experimental S(Q). Corrections for background and Compton scattering, as well as normalization by the atomic form factors (Supplementary Figure S7) are used to obtain the structure factor from the diffraction data36. The EPSR simulations show good agreement with zero-pressure diffraction data for amorphous AuGa2 (Supplementary Figure S8).
Our fits to experimental S(Q) yield chi-square values in the range χ2 = 0.002–0.016 (Supplementary Figure S9), with convergence in ~2600 iterations and best fits achieved after ~8000 iterations of the EPSR steps (Supplementary Figure S10). There remain issues about the reliability of the simulations, but EPSR uses reasonable assumptions to produce close fits to the experimental diffraction data, and the results can be Fourier transformed to obtain the radial distribution functions. The derived structural information (e.g., coordination numbers and bond angle distributions) provides one means of assessing the quality of the results.
References
Leger, J. M., Haines, J. & Atouf, A. The Post-Cotunnite Phase in BaCl2, BaBr2 and BaI2 under High Pressure. J. Appl. Cryst. 28, 416–423 (1995).
Godwal, B. K. et al. Codetermination of crystal structures at high pressure: Combined application of theory and experiment to the intermetallic compound AuGa2. Phys. Rev. B 87, 100101R (2013).
Geballe, Z. M., Raju, S. V., Godwal, B. K. & Jeanloz, R. Clapeyron slope reversal in the melting curve of AuGa2 at 5.5 GPa. J. Phys.: Condens. Matter. 25, 415401 (2013).
Li, R. et al. Local structure of liquid gallium under pressure. Scientific Reports 7, 5666 (2017).
Liu, L. et al. Chain breakage in liquid sulphur at high pressures and high temperatures. Phys. Rev. B 89, 174201 (2014).
Yu, T. et al. Study of liquid gallium at high pressure using synchrotron x-ray. J. Appl. Phys. 111, 112629 (2012).
Bohórquez, A. Difraccion de electrons en peliculas delgadas, amorfas y cristalinas de AuAl2, AuGa2 y AuIn2. Revista Mexicana de Física 37(No. 2), 268–275 (1991).
Petri, I., Salmon, P. S. & Fischer, H. E. Defects in a Disordered World: The Structure of Glassy GeSe2. Phys. Rev. Lett. 84, 2413–2416 (2000).
Wilding, M. C., Wilson, M. & McMillan, P. F. Structural studies and polymorphism in amorphous solids and liquids at high pressure. Chem. Soc. Rev. 35, 964–986 (2006).
Hicks, D. G. et al. Dissociation of liquid Silica at high pressures and temperatures. Phys. Rev. Lett. 97, 025502 (2006).
Spaulding, D. K. et al. Evidence for a Phase Transition in Silicate Melt at Extreme Pressure and Temperature Conditions. Phys. Rev. Lett. 108, 065701 (2012).
Millot, M. et al. Shock compression of stishovite and melting of silica at planetary interior conditions. Science 347, 418–420 (2015).
Scipioni, R., Stixrude, L. & Desjarlais, M. P. Electrical conductivity of SiO2 at extreme conditions and planetary dynamos. Proc. Natl. Acad. Sci. 114, 9009–9013 (2017).
Lyle, M. J., Pickard, C. J. & Needs, R. J. Prediction of 10-fold coordinated TiO2 and SiO2 structures at multimegabar pressures. Proc. Natl. Acad. Sci. 112, 6898 (2015).
Denoed, A. et al. High-pressure structural changes in liquid silica. Phys. Rev. E 94, 031201 (2016).
Gonzalez-Cataldo, F., Davis, S. & Gurierrez, G. Melting curve of SiO2 at multimegabar pressures: implications for gas giants and super-Earths. Scientific Reports 6, 26537 (2016).
Petigura, E. A., Howard, A. W. & Marcy, G. W. Prevalence of Earth-size planets orbiting Sun-like stars. Proc. Natl. Acad. Sci. 110(48), 19273 (2013).
Stolper, E. M. & Ahrens, T. J. On the nature of pressure-induced coordination changes in silicate melts and glasses. Geophys. Res. Lett. 14, 1231–1233 (1987).
Williams, Q. & Jeanloz, R. Spectroscopic evidence for pressure-induced coordination changes in silicate glasses and melts. Science 239, 902–905 (1988).
Meade, C., Hemley, R. J. & Mao, H. K. High-pressure x-ray diffraction of SiO2 glass. Phys. Rev. Lett. 69, 1387–1390 (1992).
Farber, D. L. & Williams, Q. Pressure induced coordination changes in alkali germante melts: An in situ spectroscopic investigation. Science 256, 1427–1430 (1992).
Guthrie, M. et al. Formation and structure of a dense octahedral glass. Phys. Rev. Lett. 93, 115502–1 (2004).
Sato, T. & Funamori, N. High-pressure structural transformations of SiO2 glass up to 100 GPa. Phys. Rev. B 82, 184102 (2010).
Sanloup, C. et al. Structural change in molten basalt at deep mantle conditions. Nature 503, 104–107 (2013).
Prescher, C. et al. Beyond sixfold coordinated Si in SiO2 glass at ultrahigh pressures. Proc. Natl. Acad. Sci. 114, 10041 (2017).
Stixrude, L. & Karki, B. Structure and freezing of MgSiO3 liquid in Earth’s lower mantle. Science 310, 297–299 (2005).
Jeanloz, R. & Roufosse, M. Anharmonic properties: Ionic model of the effects of compression and coordination change. J. Geophys. Res. 87, 10763–10772 (1982).
Garg, A. B., Verma, A. K., Vijayakumar, V., Rao, R. S. & Godwal, B. K. Electronic topological and structural transitions in AuGa2 under pressure. J. Phys.: Condens. Matter. 18, 8523 (2006).
Raju, S. V. et al. Determination of the variation of the fluorescence line positions of ruby, strontium tetraborate, alexandrite, and samarium-doped yttrium aluminum garnet with pressure and temperature. J. Appl. Phys. 110, 023521 (2011).
Kunz, M. et al. A beamline for high-pressure studies at the Advanced Light Source with a superconducting bending magnet as the source. J. Synchrotron Rad. 12, 650 (2005).
Hammersley, A. P., Svensson, S. O., Hanfland, M., Fitch, A. N. & Hausermann, D. Two-dimensional detector software: From real detector to idealised image or two-theta scan. High Press. Res. 14, 235 (1996).
Laugier, J. & Bochu, B. Computer code CELREF. Version 3. Cell parameter refinement program from powder diffraction diagram (Laboratoire des Matériaux et du Génie Physique, Ecole Nationale Supérieure de Physique de Grenoble [INPG], France) (2002).
McGreevy, R. L. R. M. Carlo modelling. J. Phys.: Condens. Matter. 13, R877 (2001).
Soper, A. K. Partial structure factors from disordered materials diffraction data: An approach using empirical potential structure refinement. Phys. Rev. B 72, 104204/1–104204/12 (2005).
Storm, A. B., Wernick, J. H. & Jayaraman, A. Fusion behavior and phase changes at high pressures in some intermetallic compounds with fluorite structure. J. Phys. Chem. Solids 27, 1227–1232 (1966).
Kalkan, B., Dias, R. P., Yoo, C.-S., Clark, S. M. & Sen, S. Polymorphism and pressure-induced metallization at the rigidity percolation threshold in densified GeSe4 glass. J. Phys. Chem. C 118, 10 (2014).
Acknowledgements
B.K. acknowledges financial support from Scientific Research Projects Coordination Unit, Hacettepe University on project number FKG-2016–12828. S.V.R. thanks COMPRES and ALS for the financial support to carry out the above research work. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
B.K.G., S.V.R. and R.J. devised the project, B.K.G. and S.V.R. performed the experiments. B.K. and S.V.R. analyzed the experimental data. B.K. carried out EPSR analysis of diffraction data. All authors contributed in wiring the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kalkan, B., Godwal, B., Raju, S.V. et al. Local structure of molten AuGa2 under pressure: Evidence for coordination change and planetary implications. Sci Rep 8, 6844 (2018). https://doi.org/10.1038/s41598-018-25297-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25297-9
- Springer Nature Limited