
Abstract
Psoriasis is a common inflammatory disease. It affects 1–3% of the population worldwide and is associated with increasing medical costs every year. Typical psoriatic skin lesions are reddish, thick, scaly plaques that can occur on multiple skin sites all over the body. Topical application of imiquimod (IMQ), a toll-like receptor (TLR)7 agonist and potent immune system activator, can induce and exacerbate psoriasis. Previous studies have demonstrated that isoflavone extract has an antioxidant effect which may help decrease inflammation and inflammatory pain. Through in vivo studies in mice, we found that the topical application to the shaved back and right ear of mice of isoflavone extract prior to IMQ treatment significantly decreased trans-epidermal water loss (TEWL), erythema, blood flow speed, and ear thickness, while it increased surface skin hydration, and attenuated epidermal hyperplasia and inflammatory cell infiltration. Through in vitro experiments, we found that isoflavone extract can reduce IL-22, IL-17A, and TNF-α-induced MAPK, NF-κB, and JAK-STAT activation in normal human epidermal keratinocytes. At the mRNA level, we determined that isoflavone extract attenuated the increased response of the TNF-α-, IL-17A-, and IL-22- related pathways. These results indicate that isoflavone extract has great potential as an anti-psoriatic agent and in the treatment of other inflammatory skin diseases.
Similar content being viewed by others

Introduction
Psoriasis is a common chronic immune-mediated inflammatory disease with a prevalence of 1–3% worldwide1,2,3, and is characterized by red, scaly, raised plaques4,5. The pathologic features of psoriatic skin lesions are due to hyperproliferation of epidermal keratinocytes, aberrant differentiation of epidermal keratinocytes, infiltration of distinct types of inflammatory and immune cells, and over-activation of angiogenesis5. Genetic and immunological studies have noted the critical role of cytokines in the pathogenesis of psoriasis6,7. Psoriatic inflammation was initially thought to be mediated by T-helper 1 (TH1) cells; however, more recent studies have revealed that TH17 cells play an indispensable role in the progression of the disease by producing IL-17 or IL-225. The involvement of these cytokines in psoriasis has also been revealed through the therapeutic silencing of key immune system components, such as TNF-α, IL-17, IFN-γ, IL-22, and the Th17/IL-23 axis. This approach has been highly successful and has demonstrated the importance of cytokine networking in psoriasis. Further, it strongly supports the proposed immunopathogenic mechanisms of psoriasis6,7,8,9,10,11.
Soybean is a common agricultural crop often used in Asian cuisine. It contains many nutrients including plant protein, saponins, phytosterol, oligosaccharides, and isoflavones12,13, making it beneficial to the human body. There are 12 different soybean isoflavone isomers. Genistein and daidzein are both major isoflavones but genistein is the most widely investigated14,15. Isoflavones (Fig. 1) are a type of phytoestrogen, and have similar molecular structures to animal oestrogen. This leads to weak oestrogenic effects15 including the treatment of menopausal symptoms16 and prostate cancer17, bone strengthening18, and reduction of breast cancer risk19. Some immune cells, in turn, such as thymocytes, lymphocytes, and macrophages have been reported to contain oestrogen receptors. These receptors bind oestrogen that can inhibit over-activation of immune cells along with delayed hypersensitivity reactions20. Isoflavones have been reported to bind to oestrogen receptors and to increase eicosanoid function, and play effectiveness of oestrogen. They also possess antioxidant properties and can scavenge reactive oxygen species, leading to a strong anti-inflammatory effect21,22,23. In this study, we applied the rodent model of psoriasis to evaluate the in vivo protective effects of isoflavone extract in alleviating imiquimod-induced inflammatory effects on the development and physiological parameters of mice. Further, we evaluated the efficacy of isoflavone extract in preventing epidermal keratinocytes from psoriatic activity.

The structure of isoflavone.
Results
Isoflavone extract attenuates skin inflammation and hyperplasia in a murine IMQ–induced model
We studied the potential beneficial effect of isoflavone extract in a murine IMQ-induced psoriasis-like skin inflammation model in which IMQ, a TLR7/8 agonist, was applied to the whole back of the mice daily over 6 days. This repetitive application of IMQ onto mouse skin led to psoriasis-like inflammation with significant thickening, redness, and scaling caused by keratinocyte hyperproliferation and leukocyte infiltration into the skin (Fig. 2A,B).

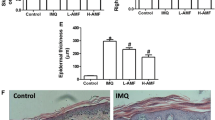
Isoflavone extract ameliorates imiquimod (IMQ)-induced skin inflammation. Isoflavone extract was topically applied 1 hour before IMQ or vehicle cream administration for 6 consecutive days. (A) Phenotypical presentation of mouse skin after 6 days of treatment. (B) Macroscopic appearance of the ear during 6 days of the experiment. (C) Histological analyses of mouse skin are after haematoxylin and eosin (H&E) staining of skin biopsies. In each group, mice were applied with vaseline or IMQ after pretreatment with isoflavone extract.
Histological analysis of lesion skin biopsies using H&E staining showed a decrease of inflammatory cell infiltrate in isoflavone extract-pretreated mice compared with IMQ-treated mice. In addition, isoflavone extract treatment prevented the characteristic epidermal hyperplasia induced by IMQ (Fig. 2C).
Topical pre-treatment with isoflavone extract prior to IMQ application ameliorated lesion formation in a dose-dependent manner and significantly reduced the development of erythema at the site of application (Fig. 3B). There were no significant changes after treatment with isoflavone extract alone. Moreover, our data showed that the treatment with isoflavone extract inhibited the TEWL, erythema, blood flow, ear thickness, and increased a corneometer in a dose-dependent manner. There were no significant differences in melanin or pH value between the IMQ treatment alone and mice treated with the isoflavone extract before IMQ treatment (Fig. 3A–E).

Change in physiological parameters on mice skin surface after treatment with isoflavone extract. Analysis of the change in ear thickness, trans-epidermal water loss (TEWL), erythema, and blood flow values in different treatments. A gradual decrease in the value of the parameters was observed after pre-treatment with isoflavone extract before stimulation with IMQ (A–D). Corneometer a tool used to determine the hydration level of the skin surface and the value of the corneometer increases with isoflavone extract treatment after IMQ stimulation (E). (A) TEWL (B) erythema (C) blood flow (D) ear thickness (E) corneometer measurement. Data are the mean ± SEM from at least six independent experiments.
Isoflavone extract is not cytotoxic to NHEKs
The cytotoxic effect of isoflavone extract on NHEKs was determined via MTT, trypan blue, and crystal violet assays. Treatment with isoflavone extract in a concentration range of 1–10 μg·mL−1 for 24 hours did not reduce cell viability (Fig. 4). However, the trial of 30 μg·mL−1 isoflavone extract showed slight cytotoxicity. We, therefore, used isoflavone extract at concentrations of 1–10 μg·mL−1 in our experiments.

Normal human epidermal keratinocyte cells show viability under different concentrations of isoflavone extract treatment. (A) Cell viability in normal human keratinocyte cells was determined via MTT assay after incubation with 1, 3, 10, and 30 μg/mL isoflavone extract, respectively, for 24 h. (B) Cell viability in NHEKs was determined via crystal violet assay and (C) trypan blue assay. Values represent means of optical density values measured at 550 nm ± standard error of mean (SEM) from at least three independent experiments.
Isoflavone extract inhibits TNF-α-, IL-17A-, and IL-22-induced MAPK pathway phosphorylation in NHEKs
MAP kinase signalling is critically involved in TNF-α-activated pathways and regulates various downstream effects in skin cells. This study, therefore, investigated possible signalling pathways modulated by isoflavone extract. A western blot analysis of NHEKs performed after they were pre-incubated with isoflavone extract for 24 h and stimulated with TNF-α (50 ng·ml−1) showed that the changes in phosphorylated ERK, p38, and c-Jun N-terminal kinase (JNK) expression were induced by TNF-α alone. Isoflavone extract-pretreatment suppressed the production of TNF-α-induced ERK, p38, and JNK phosphorylation in a dose-dependent manner (Fig. 5A).

Isoflavone extract inhibits TNF-α-, IL-17A-, and IL-22-induced MAPK and JAK/STAT pathway activation in human primary keratinocytes. Primary keratinocytes were pretreated with different doses of isoflavone extract for 24 hours and then treated with TNF-α, IL-17A, or IL-22. After treatment of TNF-α (A), IL-17A (C), or IL-22 (E), with the quantification data shown in the right panel (B,D,F), the total protein was extracted from the cells and associated protein expression were determined via western blotting. Data are the mean ± SEM from at least three independent experiments. *P < 0.05, indicating significant effects of isoflavone extract as compared to control.
To assess the activation of MAPK following exposure to IL-17A (50·ng mL−1), we examined phosphorylation levels of p38, ERK, and JNK as the relevant downstream molecules via western blot. Our results showed a significant increase in the phosphorylation of p38, ERK, and JNK after treatment with IL-17A alone. These increases could be significantly lowered with isoflavone extract in a dose-dependent manner (Fig. 5B). Our results demonstrate that isoflavone extract can effectively reduce the activation of MAPK.
The JAK-STAT pathway is a classical signal transduction pathway for numerous cytokines and growth factors24. IL-22 activates the JAK/STAT, ERK, JNK, and p38 MAPK pathways25. Therefore, we assessed how IL-22-induced activation is affected by the presence of isoflavone extract in NHEKs using western blot analysis. The analysis of IL-22-induced molecular cascades demonstrated that JAK/STAT and MAPK pathways were rapidly induced in NHEKs and that the phosphorylations of STAT3, JAK2, ERK, JNK, and p38 were inhibited by treatment with isoflavone (Fig. 5C).
Isoflavone extract inhibits TNF-α- and IL-17A-induced NF-κB pathway activation in NHEK
Since phosphorylation and degradation of the IκB protein, especially the α isoform (IκBα) are critical steps in the activation of the NF-κB signalling pathway26, we measured the effect of isoflavone extract on the TNFα- and IL-17A-induced phosphorylation and degradation of IκBα. Once NHEKs are stimulated by TNF-α or IL-17A, IκBαs are phosphorylated and degraded rapidly. As shown in Fig. 6, treatment of NHEKs with TNF-α or IL-17A increased the phosphorylation and degradation of IκBα, and this stimulation was prevented significantly by pre-treatment with isoflavone extract (Fig. 6). These results indicate that isoflavone extract influences NF-κB activation by regulating the phosphorylation and degradation of IκBα.

Isoflavone extract inhibited TNF-α- and IL-17A-induced NF-κB phosphorylation in human primary keratinocytes. Primary keratinocytes were pretreated with different doses of isoflavone extract for 24 hours and then treated with TNF-α or IL-17A. After treatment with TNFα (A) or IL-17A (C), with the quantification data shown in the right panel (B,D), the total protein was extracted from the cells and IκBα expression was determined via western blotting. Data were the mean ± SEM from at least three independent experiments. *P < 0.05, indicating significant effects of isoflavone extract as compared to control.
Isoflavone extract inhibits TNF-α-, IL-17A-, and IL-22-induced mRNA expression in NHEKs
Antimicrobial peptides and proteins (AMPs) are a diverse group of small molecules (12–50 amino acids), which are ubiquitously distributed in mammals, insects, frogs, and which can even be found in plants and invertebrates27. The main function of AMPs is to resist and kill pathogenic microorganisms such as bacteria, fungi, and viruses. In general, AMPs are concentrated in animal body surfaces, such as the skin and gastrointestinal tract28,29. In patients with psoriatic lesions, there are three subclasses of AMPs, including cathelicidin, S100 proteins, and defensins, which are highly expressed, and which are considered to play an important role in the pathogenesis of psoriasis30,31. RT-qPCR analysis of NHEKs revealed that during the development of psoriatic inflammation induced by TNF-α, IL-17A, or IL-22, increased levels of the AMPs CCL20, S100A7, S100A8, S100A9, hBD2, and LL-37 modulated and triggered host immune response. Moreover, we found that expression of CCL20, S100A7, S100A8, S100A9, hBD2, and LL-37 significant decreased after pretreatment with isoflavone extract, compared with the group treated with TNF-α, IL-17A, or IL-22 alone (Fig. 7). These data indicate that isoflavone extract attenuated the TNF-α-, IL-17A-, or IL-22-induced pathway at the mRNA level.

Real-time polymerase chain reaction (PCR) quantification of fold changes in CCL20, S100A7, S100A8, S100A9, hBD2, and LL-37 transcripts in human primary keratinocytes after the addition of TNFα, IL-17A, and IL-22 recombination proteins. Primary keratinocytes were pretreated with different doses of isoflavone extract for 24 hours and then incubated with TNFα (A), IL-17A (B), and IL-22 (C) for 6 or 8 h. Total RNA was then isolated and mRNA analysis performed via real-time RT-PCR. Data are expressed as fold induction of relevant mRNAs sequences as compared to untreated controls. Data are mean ± SEM from at least three independent experiments performed in triplicate; *P < 0.05 determined by Student’s t-test.
Discussion
The topical application of isoflavone extract prior to IMQ treatment significantly decreased the levels of TEWL and erythema, the speed of blood flow, and the thickness of the ear. The in vitro study demonstrated that, in human keratinocytes, cytokines such as TNF-α and IL-17A trigger the phosphorylation of the MAPK and NF-κB pathways and that IL-22 induces JAK/STAT and MAPK pathways, all of which induces inflammation. In the MAPK pathway, we found that the phosphorylation of signal transduction molecules such as ERK, p38 kinase, and JNK is upregulated by TNF-α, IL-17A, and IL-22. IκBα expression in the NF-κB pathway was also significantly upregulated after stimulation with TNF-α and IL-17A, and STAT3 and JAK2 expression in the JAK/STAT pathway was rapidly induced by IL-22. In contrast, pre-treatment with isoflavone extract downregulated the phosphorylation levels of p38 kinase, ERK, JNK, IκBα, STAT3, and JAK2.
Furthermore, the levels of CCL20 and the AMPs, includind S100A7, S100A8, S100A9, hBD2, and LL-37 alternate mRNA were analysed via qRT-PCR after treatment with the recombination proteins TNF-α, IL-17A, and IL-22 with or without pre-treatment with isoflavone extract. The results show that pre-treatment with isoflavone extract downregulated the levels of the AMPs.
The current study found that human Th17 pro-inflammatory cells produce IL-17A, IL-17F, TNF-α, IL-6, IL-21, IL-22, and IL-2332. Recent reports have discovered six isoform members of the IL-17 family, including IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F. Several isoforms of the IL-17 receptor family have also been discovered, consisting of IL-17R, IL-17RH1, IL-17RL, IL-17RD, and IL-17RE33. Thus far, a number of autoimmune diseases are considered to be mediated by Th17, and the biological function of IL-17 is consistent with chronic inflammation and destructive inflammation responses. Other cell types are also involved in inflammation, including γδ T cells, natural killer T cells, natural killer cells, neutrophils, and eosinophils, all of which produce IL-17A and IL-17F. Both IL-17A and IL-17F are pro-inflammatory and interact with various cell types to induce the release of cytokines, such as transforming growth factor beta (TGF-β), tumour necrosis factor (TNF), interleukins (IL-1β, IL-6, IL-21, IL-23), granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), chemokines (CXCL1, CXCL8, CXCL10), and metalloproteinase34. In addition, IL-17A and IL-17F are critical in neutrophils recruitment, activation, and migration35.
Interleukin-22 (IL-22) is a member of the IL-10 family, which consists of 179 residues and has 25% identity with human IL-1036. Human IL-22 is characterized by six alpha-helices that help maintain its secondary structure37. IL-22 was originally found to be stimulated by IL-19 in mice with T lymphocytes using the cDNA subtraction method, and is commonly considered an immunosuppressive cytokine, even though it can induce an immune response, as well. IL-22 has been shown to induce rheumatoid arthritis, along with an increase in IL-17 in patients’ plasma and synovial fluid, resulting in serious joint damage38. IL-22 also prevents inflammatory bowel disease (IBD) by producing a pancreatitis-associated protein in pancreatic acinar cells39,40, and can prevent autoimmune liver diseases, such as primary biliary cirrhosis (PBC). IL-22 also shows protective effects that ameliorate liver inflammation by decreasing T cell and B cell infiltration in PBC mice41. After IL-22 binds to the receptor complex on the cell membrane, which consists of two receptor chains, the IL-22R1 chain and the IL-10R2 chain42, it activates the JAK-STAT pathway43,44. Further, IL-22 regulates the expression of genes responsible for antimicrobial defence, cellular differentiation, mobility in keratinocytes45, and inhibition of epidermal differentiation46. These bioactivities suggest that IL-22 plays an important role in inflammatory skin processes and wound healing, but may be harmful to patients with psoriasis. The level of IL-22 was positively correlated with the production of IL-22 in plasma and the severity of psoriasis, indicating that IL-22 has a significant effect on psoriasis47. Besides that, high IL-22 levels in psoriatic skin were associated with strongly upregulated cutaneous S100A7, S100A8, S100A9, and MMP1 expression45.
TNF-α is the primary mediator of inflammatory skin disease pathology. It is a multifunctional cytokine that plays an important role in inflammation, immune response, and apoptosis48. Patients with psoriasis have higher levels of TNF-α and translation factors such as NF-κB in their skin lesions49,50. This is due to TNF-α’s ability to trigger NF-κB activation, which leads to the activation of other cytokines and upregulation of TNF-α itself, in a positive feedback loop49,51. In addition, TNF-α causes mTOR activation and ROS generation, which leads to IκB degradation, NF-κB translocation, and, finally, the production and secretion of inflammatory cytokines52. Moreover, TNF-α was shown to activate the Ras/Raf/MEK/ERK and protein kinase B/Akt pathways. In a human epidermal cell line, phosphatidylinositol (PI)3-kinase/Akt transduction was activated by TNF-α, which subsequently triggered the activation of NF-κB, which, in turn, regulated gene expression, and induced innate and adaptive immune responses and inflammation53. TNF-α also induces phosphorylation of JNK and reduces the expression of filaggrin and loricrin–two major proteins expressed by terminally differentiated epidermal keratinocytes54. Therefore, the use of a TNF-α antagonist may increase the expression of the skin barrier and ameliorate the symptoms of psoriasis by improving the barrier protein expression55. Antioxidants, also, have been shown to reduce the TNF-α-induced production of cytokines52, and may, therefore, play an important role in inflammatory skin diseases, especially psoriasis52. It is reasonable to assume that antioxidants have the potential to be developed into a new drug which would have both anti-inflammatory and barrier repair properties.
Skin is the body’s largest organ56 and serves as the main barrier to the body’s internal and external environment in order to maintain body homeostasis57, and to protect the human body from external damage. In addition, the skin can recognize, discriminate, and integrate various signals from the outside environment58, choose appropriate responses, and trigger a variety of physiological reactions including skin immune, pigmentary, epidermal, adnexal systems, systemic immune, neural, and endocrine systems59,60,61,62. In previous studies, it was reported that many skin-related responses are mediated via the cutaneous neuroendocrine system63. For example, in response to environmental changes, the skin can generate signals to produce neural, humoral, or immune responses at the local and systemic levels. In this study, we explored the effect of inflammatory-related molecules on the mechanisms of psoriasis. In the future, we hope to study the cutaneous neuroendocrine system in more detail.
Flavonoids have been widely investigated in the past. One flavonoid, epigallocatechin gallate (EGCG), has been reported to possess anti-inflammatory effects in the skin. EGCG has been found effective against UVB-induced prostaglandin metabolism, and also to reduce UVB-induced erythema, myeloperoxidase activity, hydrogen peroxide generation, and leukocyte infiltration64,65. In addition, EGCG inhibits UVB-induced AP-1 upregulation and the expression of apoptosis-regulatory genes (p53-p21)66, and possesses a protective effect on UVA-induced damage in human keratinocytes67. Similar to EGCG, (−)-epicatechin-3-gallate (ECG) can protect against UVA-induced cell death of human keratinocytes by reducing the generation of H2O2 and hypoxanthine-xanthine oxidase68,69. Our previous research also found that (+)-catechin provides an anti-oxidation effect after UVB exposure, but the mechanism of (+)-catechin differs from that of EGCG, since (+)-catechin protects against UV and hydrogen peroxide damage via inhibition of JNK phosphorylation70. Genistein, found in soybeans, can prevent UVB-induced H2O2 generation, lipid peroxidation, and oxidative DNA damage, and inhibits the progression and promotion of cancer via the inhibition of tyrosine protein kinase71. Further studies have shown that genistein can also suppress the expression of UVB-induced c-fos and c-jun proto-oncogenes, inhibiting the transformation of a normal cell into a tumour cell72.
Previous studies in our laboratory have shown that isoflavone extract works better against photo-aging (UVB) than genistein and is almost non-toxic. These two properties give isoflavones excellent potential for therapeutic applications and development in the future73,74. Theaflavins, the main component of black tea, are also reported to possess excellent antioxidant activity along with the ability to downregulate UV-induced lipid oxidation to reduce skin damage further75. Theaflavins also suppress the activation of transcription factor AP-176, UVB-induced phosphorylation of STAT1 at Ser72777, and arsenite-induced apoptosis78. In another study, 12-o-tetradecanoylphorbol-13-acetate (TPA)-induced inflammation in a rodent model showed that theaflavins reduced ornithine decarboxylase (ODC) levels, which led to an anti-inflammatory effect79, and decreased NF-κB phosphorylation, thereby inhibiting tumour production76.
Another plant, indigo naturalis has been used as a medicine for the treatment of skin diseases in the past. In recent years, more and more reports have shown that indigo naturalis and indirubin can be effectively and safely used for the treatment of psoriasis80, especially nail psoriasis and plaque-type psoriasis81,82,83. Indigo naturalis upregulates claudin-1 expression and tight-junctions in human keratinocytes and also has a strong inhibitory effect on the inflammatory response of human neutrophils84,85. Therefore, an exploration of the relationship between natural products and activity will be of great value to the development of anti-inflammatory and barrier repair skin treatments.
The use of steroids can suppress both the immune system and inflammation, but it does not cure the source of the inflammation. When the use of steroids is discontinued, the inflammatory response will flare up again, unless the allergens causing the skin inflammation have been neutralized. Moreover, long-term and significant steroid use inevitably leads to systemic side effects, since steroids absorbed by the body inhibit the normal secretion of epinephrine. The long-term result is a reduction in pituitary and adrenal gland secretions, which causes dermis layer thinning that does not return to normal even after cessation of steroid use, microvascular proliferation, loss of melanin, and skin that begins to fade86. Thus, the use of steroids involves many limitations, while the use of natural products and Chinese herbal medicines is possible at a low cost, without significant restrictions, or side effects, which is a significant advantage in the treatment of inflammatory skin diseases such as psoriasis.
Materials and Methods
Ethics Statement
The foreskins used in the experiments was provided by the Mackay Memorial Hospital to the Fu Jen Catholic University with a letter providing an exemption from the institutional review board (IRB) (#13MMHIS022), as no interaction occurred with the foreskin donors themselves and no identifiable information was made available to the researchers. All animal tests were approved by the Fu Jen Catholic University’s Institutional Animal Care and Use Committee (IACUC) policy (approval #A10367).
Animals
Mice were obtained from the National Laboratory Animal Center, Taipei, Taiwan. Animals were housed and handled according to institutional guidelines. Briefly, mice were housed one per cage in a controlled environment for 1 week with the temperature set at 21–25 °C, humidity at 60 ± 5%, and light in a 12/12 h light/dark cycle. Alfalfa-free food and water were given ad libitum. All of the animal-experiment protocols were reviewed by the committee and conducted after obtaining an Affidavit of Approval of Animal Use Protocol from Fu Jen Catholic University.
IMQ-induced psoriasis-like skin inflammation in mice
Male BALB/c mice (8–11 weeks) received a daily topical dose of 62.5 mg of commercially available IMQ cream (Aldara 5%; Meda AB, Solna, Sweden) on shaved areas of the dorsal and lumbar back and on the right ear for 6 consecutive days. Similarly, a vehicle cream (Vaselina Pura, Laboratorios Rida, Valencia, Spain) was applied to the IMQ-untreated mice. The trans-epidermal water loss (TEWL), amount of erythema, amount of melanin, skin hydration, pH value, ear thickness, and blood flow were measured before treatment. The surface changes in the dorsal skin were recorded using photography. The TEWL, erythema, melanin level, skin hydration, and blood flow were regularly measured using an MPA-580 cutometer (Courage & Khazaka, Cologne, Germany) and FLO-N1 laser tissue blood flowmeter (Omegawave, Tokyo, Japan). One hour before the cream was applied, isoflavone extract (10 mg/mL) or vehicle were topically administered. At the end of the experiment, the mice were killed, and tissues were collected and stored at −80 °C for subsequent homogenization and fixture in formalin. Animals were removed from the study and euthanized with CO2 when clear suffering negated the need to continue, in accordance with the Fu Jen Catholic University’s IACUC policy.
Histopathological analysis
Mouse tissues were fixed overnight with neutral buffered 4% paraformaldehyde (PFA) at 4 °C and then directly embedded in paraffin. Blocks of skin biopsies in paraffin were prepared using routine methods and sectioned to obtain consecutive levels. Five-micrometre sections were stained either with haematoxylin and eosin (H&E) or processed further. Images from H&E staining were obtained using a ZEISS Axioskop 40 Inverted System Microscope (NY, United States) and SPOT Cam software (Sterling Heights, MI).
Primary human keratinocyte isolation from foreskin and cell culture
Normal human epidermal keratinocytes (NHEKs) were obtained from normal adult human foreskins. All protocols and procedures were approved by the local ethics committee and carried out in accordance with the Declaration of Helsinki. Written consent was obtained from each donor before the experiments were performed. In brief, the skin was divided and incubated in 0.2% protease (Sigma-Aldrich, St Louis, MO) for 2 days at 4 °C. After the epidermis separated from the dermis, the keratinocytes were disaggregated into a single-cell suspension in serum-free Dulbecco’s modified Eagle’s medium (DMEM) and then centrifuged at 1100 × rpm for 5 minutes. The keratinocytes were then cultured in Keratinocyte-SFM (Gibco BRL/Invitrogen, Carlsbad, CA), supplemented with recombinant epidermal growth factor (0.1–0.2 ng·mL−1), bovine pituitary extract (20–30 μg·mL−1), and 1% penicillin/streptomycin in a humidified atmosphere at 37 °C and 5% CO2. The second- to fourth-passage cells were used in the experiments.
Stimulation of primary human keratinocytes
NHEKs were seeded in 12-well plates at a density of 5 × 104 cells/well. After reaching subconfluency, the keratinocyte medium was renewed, and the cells were subjected to a 24 h-pretreatment with isoflavone extract. Control keratinocytes were placed in an equal volume of vehicle. Finally, cells were stimulated with either IL-17A (50 ng·mL−1), IL-22 (50 ng·mL−1), or TNF-α (50 ng·mL−1) from PeproTech (Rocky Hill, NJ).
Cell viability assays (MTT, trypan blue assay, and crystal violet assay)
Cell viability was determined via MTT, trypan blue, and crystal violet assays. The MTT assay was performed according to a previously described protocol68. Briefly, cells were pretreated with double-distilled (dd)H2O or isoflavone extract and incubated for 24 hours. After being washed with the phosphate buffer solution (PBS), MTT (0.5 mg/mL in Keratinocyte-SFM) was used for the quantification of metabolically active cells. Mitochondrial dehydrogenases metabolized MTT to a purple formazan dye, which was analysed photometrically at 550 nm. Cell viability was proportional to the absorbance.
The Trypan Blue exclusion method was used, according to the manufacturer’s protocols, to accurately determine cell viability subsequent to isolation. Briefly, NHEKs were seeded in a 35 mm culture dish. After reaching subconfluency, the Keratinocyte-SFM was renewed, and the cells were subjected to a 24-h pretreatment with isoflavone extract. The cells were then collected into cellular suspension with TrypLE Express (Gibco BRL/Invitrogen, Carlsbad, CA) and stained with equal volumes of 0.4% trypan blue dye for 1 min. The cells were counted using a dual-chamber haemocytometer and a light microscope.
The crystal violet assay was used to determining the viability of cultured cells87. Crystal violet is a triarylmethane dye (purple) that stains DNA and proteins in cells that are grown into a monolayer. The cytotoxicity of a drug is determined by measuring the absorbance of the crystal violet-stained cells via spectrophotometry. Cell treatments were performed as above described. After a 24-h incubation period, the cells were washed with PBS to remove non-adherent cells. Then, methanol (500 μL) was added to each well to fix the living cells to the bottom of the plate, and the cells were incubated for 30 min at 25 °C. The methanol was discarded and 0.1% crystal violet staining solution (300 μL) was added for 1 h. The crystal violet was discarded, and the plate was washed three times with ddH2O. The plates were left to dry and 33% acetic acid (500 μL) was added to dissolve the stained cells. Absorbance optical density (OD) was read using a Tecan Sunrise spectrophotometer (Tecan, Crailsheim, Germany) at a wavelength of 550 nm.
Cell lysate preparation and western blot analysis
NHEKs treated with or without cytokines were washed twice with PBS and lysed with radioimmunoprecipitation assay (RIPA) buffer [17 mM Tris–HCl, pH 7.4, 50 mM NaCl, 5 mM EDTA, 1 mM sodium fluoride, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS), 1 mM sodium orthovanadate, 1 mM phenylmethane sulfonyl fluoride (PMSF), 1 μg/mL aprotinin (freshly prepared), and 1 μg/mL leupeptin (freshly prepared)]. After sonication, the lysate was centrifuged (14,000 × g for 10 min at 4 °C), and the supernatant was removed. The protein content was quantified using a Pierce protein assay kit (Pierce, Rockford, IL). The total protein was separated via electrophoresis on 10% SDS–polyacrylamide gels. The proteins were then electroblotted onto polyvinylidene fluoride (PVDF) membranes and probed using the indicated specific antibodies. Immunoblots were detected via enhanced chemiluminescence (Chemiluminescence Reagent Plus, NEN, Boston, MA). For some of the experiments, the PVDF membrane was stripped at 60°C for 10 minutes with a stripping buffer (62.5 mM Tris-HCl, pH 6.7, 2% SDS, and 100 mM β-mercaptoethanol).
Real-time quantitative reverse transcriptase – polymerase chain reaction (RT-qPCR)
After these treatments, the cells were lysed and the total RNA was extracted using a total RNA isolation kit (GeneDireX®, Vegas, NV). First strand cDNAs were synthesized using the SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. qPCR was performed using an CFX96™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA) and under the following conditions: heat to 95 °C for 3 min, followed by 54 denaturation cycles at 95 °C for 3 s each, primer annealing at 60 °C for 20 s, and primer extension at 95 °C for 10 s. The melting temperature curve ranged from 60 to 95 °C, increasing in increments of 0.3 °C. cDNA was amplified using SYBR green (Kapa Biosystems, Wilmington, MA) and using the following primers: human CCL2088, forward TACTCCACCTCTGCGGCGAATCAGAA, and reverse GTGAAACCTCCAACCCCAGCAAGGTT; human S100A789, forward GCATGATCGACATGTTTCACAAATACAC, and reverse TGGTAGTCTGTGGCTATGTCTCCC; human S100A890, forward TGAAGAAATTGCTAGAGAC, and reverse CTTTATCACCAGAATGAGGA; human S100A946, forward GCTCCTCGGCTTTGACAGAGTGCAAG, and reverse GCATTTGTGTCCAGGTCCTCCATGATGTGT; human BD291, forward CCAGCCATCAGCCATGAGGGT, and reverse GGAGCCCTTTCTGAATCCGCA; human LL-3792, forward GCAGTCACCAGAGGATTGTGAC, and reverse CACCGCTTCACCAGCCC; human β-Actin93,94, forward CGGGGACCTGACTGACTACC, and reverse AGGAAGGCTGGAAGAGTGC. The CCL20, S100A7 to 9, hBD2, and LL-37 amplification signals were normalized relative to β-actin expression and evaluated using the equation: fold change = 2−ΔΔCT.
Statistical analysis
Unless otherwise indicated, data are expressed as mean ± standard error of the mean (SEM) using the GraphPad Prism Program 6 software (GraphPad Software, San Diego, CA). Comparison of the mean survival rates of cells with and without isoflavone extract was performed using a one-way analysis of variance (ANOVA) followed by Dunnett’s t-test for multiple comparisons. We considered P < 0.05 to be statistically significant.
References
Raychaudhuri, S. P. & Farber, E. M. The prevalence of psoriasis in the world. Journal of the European Academy of Dermatology and Venereology: JEADV 15, 16–17 (2001).
Schon, M. P. & Boehncke, W. H. Psoriasis. The New England journal of medicine 352, 1899–1912, https://doi.org/10.1056/NEJMra041320 (2005).
Chandran, V. & Raychaudhuri, S. P. Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. Journal of autoimmunity 34, J314–321, https://doi.org/10.1016/j.jaut.2009.12.001 (2010).
Krueger, J. G. The immunologic basis for the treatment of psoriasis with new biologic agents. Journal of the American Academy of Dermatology 46, 1–23; quiz 23-26 (2002).
Lowes, M. A., Bowcock, A. M. & Krueger, J. G. Pathogenesis and therapy of psoriasis. Nature 445, 866–873, https://doi.org/10.1038/nature05663 (2007).
Elder, J. T. et al. Molecular dissection of psoriasis: integrating genetics and biology. The Journal of investigative dermatology 130, 1213–1226, https://doi.org/10.1038/jid.2009.319 (2010).
Khan, S. Psoriasis. The New England journal of medicine 353, 848–850; author reply 848–850 (2005).
Nickoloff, B. J. Cracking the cytokine code in psoriasis. Nat Med 13, 242–244, https://doi.org/10.1038/nm0307-242 (2007).
Di Cesare, A., Di Meglio, P. & Nestle, F. O. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. The Journal of investigative dermatology 129, 1339–1350, https://doi.org/10.1038/jid.2009.59 (2009).
Nestle, F. O., Kaplan, D. H. & Barker, J. Psoriasis. The New England journal of medicine 361, 496–509, https://doi.org/10.1056/NEJMra0804595 (2009).
American Academy of Dermatology Work, G. et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case-based presentations and evidence-based conclusions. Journal of the American Academy of Dermatology 65, 137–74, https://doi.org/10.1016/j.jaad.2010.11.055 (2011).
Sean O’ Keefe, L. B. a. J. S. Soybean Nutrition. SM Journal of Nutrition and Metabolism 1, 1006 (2015).
Araújo, M. M., Fanaro, G. B. & Villavicencio, A. L. C. H. Soybean and Isoflavones – From Farm to Fork (2013).
Sakai, T. & Kogiso, M. Soy isoflavones and immunity. J Med Invest 55, 167–173 (2008).
Zhang, H. Y. et al. Isoflavones and Prostate Cancer: A Review of Some Critical Issues. Chin Med J (Engl) 129, 341–347, https://doi.org/10.4103/0366-6999.174488 (2016).
Coon, J. T., Pittler, M. H. & Ernst, E. Trifolium pratense isoflavones in the treatment of menopausal hot flushes: a systematic review and meta-analysis. Phytomedicine 14, 153–159, https://doi.org/10.1016/j.phymed.2006.12.009 (2007).
Swami, S. et al. Calcitriol and genistein actions to inhibit the prostaglandin pathway: potential combination therapy to treat prostate cancer. J Nutr 137, 205S–210S (2007).
Yamaguchi, M. Regulatory mechanism of food factors in bone metabolism and prevention of osteoporosis. Yakugaku Zasshi 126, 1117–1137 (2006).
Messina, M., McCaskill-Stevens, W. & Lampe, J. W. Addressing the soy and breast cancer relationship: review, commentary, and workshop proceedings. J Natl Cancer Inst 98, 1275–1284, https://doi.org/10.1093/jnci/djj356 (2006).
Yellayi, S. et al. The phytoestrogen genistein suppresses cell-mediated immunity in mice. J Endocrinol 176, 267–274 (2003).
Sirotkin, A. V. & Harrath, A. H. Phytoestrogens and their effects. European Journal of Pharmacology 741, 230–236, https://doi.org/10.1016/j.ejphar.2014.07.057 (2014).
Ming, L.-G., Chen, K.-M. & Xian, C. J. Functions and action mechanisms of flavonoids genistein and icariin in regulating bone remodeling. Journal of Cellular Physiology 228, 513–521, https://doi.org/10.1002/jcp.24158 (2013).
Kladna, A., Berczynski, P., Kruk, I., Piechowska, T. & Aboul-Enein, H. Y. Studies on the antioxidant properties of some phytoestrogens. Luminescence. https://doi.org/10.1002/bio.3091 (2016).
Bao, L., Zhang, H. & Chan, L. S. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT 2, e24137, https://doi.org/10.4161/jkst.24137 (2013).
Racz, E. & Prens, E. P. Molecular pathophysiology of psoriasis and molecular targets of antipsoriatic therapy. Expert Rev Mol Med 11, e38, https://doi.org/10.1017/S146239940900129X (2009).
Li, Q., Withoff, S. & Verma, I. M. Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol 26, 318–325, https://doi.org/10.1016/j.it.2005.04.003 (2005).
Morizane, S. & Gallo, R. L. Antimicrobial peptides in the pathogenesis of psoriasis. The Journal of dermatology 39, 225–230, https://doi.org/10.1111/j.1346-8138.2011.01483.x (2012).
Ganz, T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3, 710–720, https://doi.org/10.1038/nri1180 (2003).
Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals (Basel) 7, 545–594, https://doi.org/10.3390/ph7050545 (2014).
Batycka-Baran, A., Maj, J., Wolf, R. & Szepietowski, J. C. The new insight into the role of antimicrobial proteins-alarmins in the immunopathogenesis of psoriasis. J Immunol Res 2014, 628289, https://doi.org/10.1155/2014/628289 (2014).
Bierkarre, H. et al. Differential expression of antimicrobial peptides in psoriasis and psoriatic arthritis as a novel contributory mechanism for skin and joint disease heterogeneity. Scand J Rheumatol 45, 188–196, https://doi.org/10.3109/03009742.2015.1091497 (2016).
Fujimura, K., Oyamada, A., Iwamoto, Y., Yoshikai, Y. & Yamada, H. CD4 T cell-intrinsic IL-2 signaling differentially affects Th1 and Th17 development. Journal of leukocyte biology 94, 271–279, https://doi.org/10.1189/jlb.1112581 (2013).
Chen, Z. & O’Shea, J. J. Th17 cells: a new fate for differentiating helper T cells. Immunologic research 41, 87–102, https://doi.org/10.1007/s12026-007-8014-9 (2008).
Maddur, M. S., Miossec, P., Kaveri, S. V. & Bayry, J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. The American journal of pathology 181, 8–18, https://doi.org/10.1016/j.ajpath.2012.03.044 (2012).
Korn, T., Bettelli, E., Oukka, M. & Kuchroo, V. K. IL-17 and Th17 Cells. Annual review of immunology 27, 485–517, https://doi.org/10.1146/annurev.immunol.021908.132710 (2009).
Dumoutier, L., Louahed, J. & Renauld, J. C. Cloning and characterization of IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J Immunol 164, 1814–1819 (2000).
Nagem, R. A. et al. Crystal structure of recombinant human interleukin-22. Structure 10, 1051–1062 (2002).
Evans, H. G. et al. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proceedings of the National Academy of Sciences of the United States of America 106, 6232–6237, https://doi.org/10.1073/pnas.0808144106 (2009).
Aggarwal, S., Xie, M. H., Maruoka, M., Foster, J. & Gurney, A. L. Acinar cells of the pancreas are a target of interleukin-22. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research 21, 1047–1053, https://doi.org/10.1089/107999001317205178 (2001).
Zenewicz, L. A. et al. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29, 947–957, https://doi.org/10.1016/j.immuni.2008.11.003 (2008).
Hsueh, Y. H., Chang, Y. N., Loh, C. E., Gershwin, M. E. & Chuang, Y. H. AAV-IL-22 modifies liver chemokine activity and ameliorates portal inflammation in murine autoimmune cholangitis. J Autoimmun 66, 89–97, https://doi.org/10.1016/j.jaut.2015.10.005 (2016).
Kotenko, S. V. et al. Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10Rbeta) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. The Journal of biological chemistry 276, 2725–2732, https://doi.org/10.1074/jbc.M007837200 (2001).
Kotenko, S. V. et al. Identification, cloning, and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. J Immunol 166, 7096–7103 (2001).
Nagalakshmi, M. L., Rascle, A., Zurawski, S., Menon, S. & de Waal Malefyt, R. Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. Int Immunopharmacol 4, 679–691, https://doi.org/10.1016/j.intimp.2004.01.008 (2004).
Wolk, K. et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 36, 1309–1323, https://doi.org/10.1002/eji.200535503 (2006).
Boniface, K. et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol 174, 3695–3702 (2005).
Lo, Y. H. et al. Serum IL-22 correlates with psoriatic severity and serum IL-6 correlates with susceptibility to phototherapy. Journal of dermatological science 58, 225–227, https://doi.org/10.1016/j.jdermsci.2010.03.018 (2010).
Locksley, R. M., Killeen, N. & Lenardo, M. J. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501 (2001).
Bachelez, H. Immunopathogenesis of psoriasis: recent insights on the role of adaptive and innate immunity. Journal of autoimmunity 25, Suppl, 69–73, https://doi.org/10.1016/j.jaut.2005.09.025 (2005).
Veale, D. J., Ritchlin, C. & FitzGerald, O. Immunopathology of psoriasis and psoriatic arthritis. Annals of the rheumatic diseases 64(Suppl 2), ii26–29, https://doi.org/10.1136/ard.2004.031740 (2005).
Punzi, L. et al. Pathogenetic and clinical rationale for TNF-blocking therapy in psoriatic arthritis. Autoimmunity reviews 6, 524–528, https://doi.org/10.1016/j.autrev.2006.12.003 (2007).
Young, C. N. et al. Reactive oxygen species in tumor necrosis factor-alpha-activated primary human keratinocytes: implications for psoriasis and inflammatory skin disease. J Invest Dermatol 128, 2606–2614, https://doi.org/10.1038/jid.2008.122 (2008).
Ghosh, S. & Hayden, M. S. New regulators of NF-kappaB in inflammation. Nature reviews. Immunology 8, 837–848, https://doi.org/10.1038/nri2423 (2008).
Steven, A. C., Bisher, M. E., Roop, D. R. & Steinert, P. M. Biosynthetic pathways of filaggrin and loricrin–two major proteins expressed by terminally differentiated epidermal keratinocytes. J Struct Biol 104, 150–162 (1990).
Kim, B. E. et al. TNF-alpha downregulates filaggrin and loricrin through c-Jun N-terminal kinase: role for TNF-alpha antagonists to improve skin barrier. J Invest Dermatol 131, 1272–1279, https://doi.org/10.1038/jid.2011.24 (2011).
Swann, G. The skin is the body’s largest organ. J Vis Commun Med 33, 148–149, https://doi.org/10.3109/17453054.2010.525439 (2010).
Zmijewski, M. A. et al. Synthesis and photochemical transformation of 3beta,21-dihydroxypregna-5,7-dien-20-one to novel secosteroids that show anti-melanoma activity. Steroids 76, 193–203, https://doi.org/10.1016/j.steroids.2010.10.009 (2011).
Slominski, A. & Wortsman, J. Neuroendocrinology of the skin. Endocr Rev 21, 457–487, https://doi.org/10.1210/edrv.21.5.0410 (2000).
Arck, P. C., Slominski, A., Theoharides, T. C., Peters, E. M. & Paus, R. Neuroimmunology of stress: skin takes center stage. J Invest Dermatol 126, 1697–1704, https://doi.org/10.1038/sj.jid.5700104 (2006).
Slominski, A. T. et al. Sequential metabolism of 7-dehydrocholesterol to steroidal 5,7-dienes in adrenal glands and its biological implication in the skin. PLoS One 4, e4309, https://doi.org/10.1371/journal.pone.0004309 (2009).
Slominski, A., Tobin, D. J., Shibahara, S. & Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev 84, 1155–1228, https://doi.org/10.1152/physrev.00044.2003 (2004).
Slominski, A., Wortsman, J., Tuckey, R. C. & Paus, R. Differential expression of HPA axis homolog in the skin. Mol Cell Endocrinol 265-266, 143–149, https://doi.org/10.1016/j.mce.2006.12.012 (2007).
Slominski, A. T. et al. Sensing the environment: regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv Anat Embryol Cell Biol 212, v, vii, 1–115 (2012).
Katiyar, S. K., Bergamo, B. M., Vyalil, P. K. & Elmets, C. A. Green tea polyphenols: DNA photodamage and photoimmunology. Journal of photochemistry and photobiology. B, Biology 65, 109–114 (2001).
Katiyar, S. K., Afaq, F., Azizuddin, K. & Mukhtar, H. Inhibition of UVB-induced oxidative stress-mediated phosphorylation of mitogen-activated protein kinase signaling pathways in cultured human epidermal keratinocytes by green tea polyphenol (−)-epigallocatechin-3-gallate. Toxicology and applied pharmacology 176, 110–117, https://doi.org/10.1006/taap.2001.9276 (2001).
Luo, D. et al. Effect of epigallocatechingallate on ultraviolet B-induced photo-damage in keratinocyte cell line. The American journal of Chinese medicine 34, 911–922, https://doi.org/10.1142/S0192415X06004387 (2006).
Tobi, S. E., Gilbert, M., Paul, N. & McMillan, T. J. The green tea polyphenol, epigallocatechin-3-gallate, protects against the oxidative cellular and genotoxic damage of UVA radiation. International journal of cancer. Journal international du cancer 102, 439–444, https://doi.org/10.1002/ijc.10730 (2002).
Huang, C. C. et al. Protective effects of (−)-epicatechin-3-gallate on UVA-induced damage in HaCaT keratinocytes. Archives of dermatological research 296, 473–481, https://doi.org/10.1007/s00403-005-0540-5 (2005).
Bickers, D. R. & Athar, M. Oxidative stress in the pathogenesis of skin disease. The Journal of investigative dermatology 126, 2565–2575, https://doi.org/10.1038/sj.jid.5700340 (2006).
Wu, W. B. et al. (+)-Catechin prevents ultraviolet B-induced human keratinocyte death via inhibition of JNK phosphorylation. Life sciences 79, 801–807, https://doi.org/10.1016/j.lfs.2006.02.028 (2006).
Barnes, S. Effect of genistein on in vitro and in vivo models of cancer. The Journal of nutrition 125, 777S–783S (1995).
Wang, Y., Zhang, X., Lebwohl, M., DeLeo, V. & Wei, H. Inhibition of ultraviolet B (UVB)-induced c-fos and c-jun expression in vivo by a tyrosine kinase inhibitor genistein. Carcinogenesis 19, 649–654 (1998).
Chiu, T. M. et al. In vitro and in vivo anti-photoaging effects of an isoflavone extract from soybean cake. Journal of ethnopharmacology 126, 108–113, https://doi.org/10.1016/j.jep.2009.07.039 (2009).
Huang, C. C. et al. Anti-photoaging effects of soy isoflavone extract (aglycone and acetylglucoside form) from soybean cake. International journal of molecular sciences 11, 4782–4795, https://doi.org/10.3390/ijms11124782 (2010).
Zhang, S., Lu, J., Li, J. & Zheng, Q. [Protective effect of tea pigments on mice skin photoaging induced by UV irradiation]. Zhong yao cai=Zhongyaocai=Journal of Chinese medicinal materials 25, 29–31 (2002).
Nomura, M., Ma, W., Chen, N., Bode, A. M. & Dong, Z. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced NF-kappaB activation by tea polyphenols, (−)-epigallocatechin gallate and theaflavins. Carcinogenesis 21, 1885–1890 (2000).
Zykova, T. A., Zhang, Y., Zhu, F., Bode, A. M. & Dong, Z. The signal transduction networks required for phosphorylation of STAT1 at Ser727 in mouse epidermal JB6 cells in the UVB response and inhibitory mechanisms of tea polyphenols. Carcinogenesis 26, 331–342, https://doi.org/10.1093/carcin/bgh334 (2005).
Chen, N. Y., Ma, W. Y., Yang, C. S. & Dong, Z. Inhibition of arsenite-induced apoptosis and AP-1 activity by epigallocatechin-3-gallate and theaflavins. Journal of environmental pathology, toxicology and oncology: official organ of the International Society for Environmental Toxicology and Cancer 19, 287–295 (2000).
Liang, Y. C. et al. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced inflammatory skin edema and ornithine decarboxylase activity by theaflavin-3,3′-digallate in mouse. Nutrition and cancer 42, 217–223, https://doi.org/10.1207/S15327914NC422_11 (2002).
Lin, Y. K. et al. Anti-psoriatic effects of indigo naturalis on the proliferation and differentiation of keratinocytes with indirubin as the active component. J Dermatol Sci 54, 168–174, https://doi.org/10.1016/j.jdermsci.2009.02.007 (2009).
Liang, C. Y., Lin, T. Y. & Lin, Y. K. Successful treatment of pediatric nail psoriasis with periodic pustular eruption using topical indigo naturalis oil extract. Pediatr Dermatol 30, 117–119, https://doi.org/10.1111/j.1525-1470.2012.01721.x (2013).
Lin, Y. K. Indigo naturalis oil extract drops in the treatment of moderate to severe nail psoriasis: a small case series. Archives of dermatology 147, 627–629, https://doi.org/10.1001/archdermatol.2011.112 (2011).
Lin, Y. K. et al. The efficacy and safety of topically applied indigo naturalis ointment in patients with plaque-type psoriasis. Dermatology 214, 155–161, https://doi.org/10.1159/000098576 (2007).
Lin, Y. K. et al. Indigo naturalis upregulates claudin-1 expression in human keratinocytes and psoriatic lesions. J Ethnopharmacol 145, 614–620, https://doi.org/10.1016/j.jep.2012.11.044 (2013).
Lin, Y. K. et al. Anti-inflammatory effects of the extract of indigo naturalis in human neutrophils. J Ethnopharmacol 125, 51–58, https://doi.org/10.1016/j.jep.2009.06.014 (2009).
Fuchs, E. & Raghavan, S. Getting under the skin of epidermal morphogenesis. Nat Rev Genet 3, 199–209, https://doi.org/10.1038/nrg758 (2002).
Feoktistova, M., Geserick, P. & Leverkus, M. Crystal Violet Assay for Determining Viability of CulturedCells. Cold Spring Harb Protoc 2016, pdbprot087379, https://doi.org/10.1101/pdb.prot087379 (2016).
Harper, E. G. et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol 129, 2175–2183, https://doi.org/10.1038/jid.2009.65 (2009).
Aries, M. F. et al. Anti-inflammatory and immunomodulatory effects of Aquaphilus dolomiae extract on in vitro models. Clin Cosmet Investig Dermatol 9, 421–434, https://doi.org/10.2147/CCID.S113180 (2016).
Altmae, S. et al. Endometrial gene expression analysis at the time of embryo implantation in women with unexplained infertility. Mol Hum Reprod 16, 178–187, https://doi.org/10.1093/molehr/gap102 (2010).
Li, D. et al. A novel lipopeptide from skin commensal activates TLR2/CD36-p38 MAPK signaling to increase antibacterial defense against bacterial infection. PLoS One 8, e58288, https://doi.org/10.1371/journal.pone.0058288 (2013).
Villanueva, E. et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187, 538–552, https://doi.org/10.4049/jimmunol.1100450 (2011).
Wu, C. A., Huang, D. Y. & Lin, W. W. Beclin-1-independent autophagy positively regulates internal ribosomal entry site-dependent translation of hypoxia-inducible factor 1alpha under nutrient deprivation. Oncotarget 5, 7525–7539, https://doi.org/10.18632/oncotarget.2265 (2014).
Tian, H. et al. Ethanol extract of propolis protects macrophages from oxidized low density lipoprotein-induced apoptosis by inhibiting CD36 expression and endoplasmic reticulum stress-C/EBP homologous protein pathway. BMC Complement Altern Med 15, 230, https://doi.org/10.1186/s12906-015-0759-4 (2015).
Acknowledgements
This work is supported by the research grants from Fu-Jen Catholic University, Ministry of Science and Technology (MOST104-2320-B-030-008-MY3), in Taiwan.
Author information
Authors and Affiliations
Contributions
H.-J.L., N.-L.W., and C.-F.H. conceived and designed the experiments; H.-J.L. and C.-F.H. performed the experiments; H.-J.L. and N.-L.W. analysed the data; N.-L.W., G.-A.L., and C.-F.H. contributed reagents, materials, and analysis tools; H.-J.L., N.-L.W., and C.-F.H. wrote and revised the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, HJ., Wu, NL., Lee, GA. et al. The Therapeutic Potential and Molecular Mechanism of Isoflavone Extract against Psoriasis. Sci Rep 8, 6335 (2018). https://doi.org/10.1038/s41598-018-24726-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24726-z
- Springer Nature Limited
This article is cited by
-
Investigating the clinical implication of corneometer and mexameter readings towards objective, efficient evaluation of psoriasis vulgaris severity
Scientific Reports (2022)
-
Impact of isoflavone genistein on psoriasis in in vivo and in vitro investigations
Scientific Reports (2021)
-
Chrysin alleviates imiquimod-induced psoriasis-like skin inflammation and reduces the release of CCL20 and antimicrobial peptides
Scientific Reports (2020)