
Abstract
The electronic and magnetic properties of Nd2Fe17 and Nd2Fe17X3 (X = C or N) compounds have been calculated using the first-principles density functional calculations. Among these, the nitrogen and carbon interstitial compounds exhibit all of the required properties such as a saturation moment of 1.6 T, Curie temperature of 700–750 K, however easy magnetic axis lies in the planar direction making them less attractive for permanent magnet applications. The calculated magnetocrystalline anisotropy energy is found to be −2.7 MJ/m3 for Nd2Fe17C3 and −4.7 MJ/m3 for Nd2Fe17N3. We further explored the possibility of changing the easy axis direction through La/Ce alloying at Nd site. Although the MAE is found to be smaller in magnitude for all the La/Ce alloys it still maintains planar direction.
Similar content being viewed by others

Introduction
The Magnetocrystalline anisotropy energy (MAE), the energy required to switch crystal magnetization from the favorable direction to other spatial directions is the most important property of a ferromagnet. As formulated by Van Vleck1, in general MAE is an intrinsic property which mainly originates from spin-orbit coupling (SOC) interaction. A large value of MAE is essential for wide range of applications, from permanent magnets to magnetic storage devices2,3,4,5,6,7,8. The SOC is strong in heavy elements such as rare-earth compounds which consequently exhibit large MAE. Together with large MAE a good permanent magnet requires large value of magnetization, large coercivity, high temperature stability and high Curie temperature6,7. For example, one of the best magnetic materials, Nd2Fe14B, has T c 588 K, M s 1.28 MA/m, K1 4.9 MJ/m3 and an easy magnetic axis along the c-axis9.
Nd2Fe17 is another Nd based interesting magnetic material. Nd2Fe17 is known to crystallize in rhombohedral Th2Zn17 structure type with the space group R−3 m. This compound has gained back interest since it was shown that insertion of interstitial atoms could raise the usually low Curie temperature (T c ) of this ferromagnetic compound. In the case of Nd2Fe17, carbon and nitrogen insertion increases T c from 325 K to 658 K for carbon interstitial (Nd2Fe17C2) and to 732 K for nitrogen interstitial (Nd2Fe17N2.3) compound10. Although both carbon and nitrogen interstitial compounds exhibit high magnetic moments and Curie point11,12,13,14,15, unlike Sm2Fe17C3(N3)8,11,12,13,14,16,17,18,19 they suffer from planar MAE. One possibility for changing the easy axis direction is through alloying. Previous theoretical calculations20,21 for these compounds treat Nd f electrons as core electrons (also known as open core approximation) which do not take part in hybridization. However for the rare earth elements the most of the contribution to MAE is shown to be from f electrons, hence a proper treatment of f electrons is crucial for accurate description of MAE22,23,24. To this end, here we have applied a Hubbard U correction on the Nd-f orbitals which splits the f bands into lower and upper Hubbard bands. Our results are in better agreement with experiments than previous theoretical studies where f orbitals were treated under open core approximation rather than as valence electrons. We also investigate the effect of elemental substitutions of La and Ce on Nd site on MAE. Our calculations show these substitutions do not switch the easy axis direction.
Results
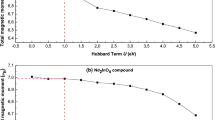
We begin our study by determining the ground state for Nd2Fe17 and its carbides and nitrides. To estimate the ground state, calculations were performed by assuming, non magnetic, ferromagnetic (parallel alignment of spins between Fe and Nd atoms), and anti-ferromagnetic (anti-parallel alignment of spins between Fe and Nd atoms) configurations. For all three systems Nd2Fe17, Nd2Fe17C3, and Nd2Fe17N3 we find the ground state to have the Nd spin moment opposite to that of the Fe and energy difference between ground state and non magnetic was found to be as 360, 460, and 510 meV on per Fe atom basis. The calculated magnetic moments are summarized in Table 1. The variation in Nd orbital and spin magnetic moments for Nd2Fe17N3 and Nd2Fe17C3 is presented in Fig. 1(a) and (b) respectively. As can be seen from Fig. 1(a) the orbital magnetic moments (M L ) is positive for rare earth atom, indicating that the direction of the orbital magnetic moment is opposite to the spin magnetic moment. This is consistent with the Hund’s rule for half filled rare earth ions. The calculated orbital moment of Nd atoms without U is 1.49 μ B for both Nd2Fe17N3 and Nd2Fe17C3. On varying U in GGA + SOC + U calculations, although the orbital moment of Nd atoms increases, overall the orbital moment displays a weak dependence on U. The rate of increase of orbital moments with U is higher for carbon interstitial compound than in nitrogen interstitial compound. The variation of Nd spin moments with U parameter is also shown in the Fig. 1(b). The Nd spin moments are almost independent of U values used.

U dependence of calculated (a) Nd orbital and (b) spin magnetic moments for Nd2Fe17N3, and Nd2Fe17C3.
The calculated Fe orbital moments for Nd2Fe17 lie between 0.04 to 0.05 μ B . On introducing interstitial nitrogen/carbon atom the Fe orbital moments are increased, and the enhancement is higher for Nd2Fe17C3. The total magnetic moments for the crystallographically nonequivalent Fe sites are plotted in Fig. 2. The calculated total magnetic moments agree reasonably with experiments. Furthermore, on introducing nitrogen/carbon interstitial atoms, while the spin moment of Fe-18f and Fe-18h sites, which are close to N/C atoms decreases, the moments on the distant Fe-9d site are enhanced as shown in Fig. 2 and Table 1. This trend of Fe moments on nitrogenation/carbonation is in good qualitative agreement with previous studies and has been attributed to hybridization between N(C) and Fe atoms21. Similar variation in magnetic moments upon nitrogenation and carbonization has also been observed for Sm2Fe17. Regardless of this slight variation the average Fe-spin moment of these systems remains in the range of 2.3–2.5 μ B , which is significantly higher than the value for BCC Fe 2.2 μ B . The experimental values of Fe moments for Nd2Fe17N3 by neutron powder diffraction measurements show large variation depending on the stoichiometry of the nitrogen in the system. For example the total magnetic moments of Nd2Fe17N2.85 and Nd2Fe17N2.91 are 32.5 μ B and 40.6 μ B , respectively19, indicating the sensitivity of magnetic properties on nitrogen stoichiometry. This large variation is mainly due to the difference in Fe-Fe distance depending upon on nitrogen stoichiometry. As explained above due to the sensitivity of magnetic properties on nitrogen/carbon content, a good quantitative agreement between experiments and theory is unlikely. Nonetheless our calculated total magnetic moment of 36.3 μ B for Nd2Fe17N3 lies well within the range proposed by experimental measurements as shown in Table 1. The calculated Nd total magnetic moments are also listed in Table 1. We see that due to the presence of 4f electrons, Nd atom has a large (~1.47 μ B ) orbital moment. For both carbon and nitrogen interstitial compounds the orbital moment of Nd is slightly increased from 1.47 μ B to 1.50 μ B . The magnetic moment for La/Ce substituted compounds are also listed in Table 1. For the substituted compounds the magnetic moment on Fe and Nd sites mostly remain unchanged. On the other hand the magnetic moment on the substituted RE atomic sites (La/Ce) is greatly reduced as La has no f electrons and Ce has only one outermost f electron.

Calculated local magnetic moments for the crystallographically nonequivalent Fe sites in Nd2Fe17 (blue cross), Nd2Fe17C3 (red squares), and Nd2Fe17N3 (black circles). The calculated magnetic moments for BCC-Fe is also shown for comparison.
Next, we calculate the magnetocrystalline anisotropy energy (MAE) constant K1, which is shown in Table 1. MAE is an important parameter for generating high coercivity in a permanent magnets. For accurate estimate of MAE values a proper treatment of strongly correlated f electrons is essential by applying a onsite Hubbard U parameter. Here a U value of 3 and 5 eV is used for Ce, and Nd, respectively. The convergence of MAE with respect to U parameter is shown in Fig. 3. For Nd2Fe17N3 where the experimental data of MAE is available, our calculated MAE with UNd as 5 eV is in reasonable agreement with the experimental data. We found the parent compound Nd2Fe17 to be planar with K1 value as −2.1 MJ/m3, which is in fair agreement with the experimentally measured value of −3.7 MJ/m312. Unlike Sm2Fe17 where carbon and nitrogen interstitial atoms switch the direction of MAE from planar to axial, Nd2Fe17N3 and Nd2Fe17C3 does not exhibit this change in MAE sign and still maintain planar anisotropy. This planar anisotropy is less desirable for permanent magnet applications. However the magnitude of K1 is quite significant and a value of −2.7 MJ/m3 and −4.7 MJ/m3 was obtained for Nd2Fe17C3 and Nd2Fe17N3, respectively. The measured anisotropy values for Nd2Fe17N3 are also listed in Table 1, which is in fair agreement with our calculated value. Given the high magnetization along with large magnitude of K1 one might obtain a coercivity higher than 1.5 Tesla in these compounds if the MAE was uniaxial.

The Hubbard U dependence of calculated magneto-crystalline anisotropy energy (MAE) for Nd2Fe17N3, and Nd2Fe17C3. Experimental value of MAE for Nd2Fe17N3 is also marked for comparison.
To explore the possibility of switching the sign of MAE and reducing the rare earth content in these magnets next we study the effect of La/Ce substitution on MAE. As the MAE is sensitive to the nature of electronic structure around the Fermi energy, it can be controlled by tuning the band structure around the Fermi energy. In practice this can be done, for example, by doping/alloying. Theoretical calculations were done in order to find a possible alloy based on NdLaFe17X3 and NdCeFe17X3 (X = C or N) with uniaxial anisotropy. We thus began with substituting one out of two Nd atoms in the primitive cell by La/Ce. Results of these calculations are summarized the in Table 1. As seen, for all the La/Ce substituted systems the MAE still remains planar. While for Ce substituted compounds the MAE is reduced by 25–31% (on going from nitrogen interstitial to carbon interstitial), a higher 40–60% reduction was observed for La substituted compounds.
Figure 4 presents the density of states (DOS) for Nd2Fe17N3. The total DOS illustrates behavior of a ferromagnetic system. In the Fig. 4(b) and (c) the DOS for RE-4f and Fe-3d states are shown. On comparing the total DOS with partial DOS in the lower panels, we can see that the states in the vicinity of the Fermi level are mainly composed of Fe-3d states. The RE-4f states are not occupied in the spin-up channel and are partially occupied in the spin-down channel, confirming that the Nd spin-moments align in the opposite direction compared to the Fe moments. Due to Hubbard U parameter the Nd spin down band split into lower and upper Hubbard bands separated by 6 eV (marked by red arrow in Fig. 4(b)). To understand the effect of La/Ce substitution at the Nd site on the magnetic properties of these compounds, we next analyze their DOS. The total and partial DOS for the these compounds are shown in Figs. 4(a)–(c). The zoomed version of RE-f DOS and Fe-d DOS are shown in Figs. 4(d) and (e). As shown in Figs. 4(c) and (e) Ce/La substitution at Nd site does not affect Fe-d DOS which overlaps with the Fe-d DOS for Nd3Fe17N3. Due to this reason the magnetic moments on Fe sites are largely unaffected by La/Ce substitution. The only significant change is in the RE-f states, which are shifted to higher energies. As shown in Fig. 4(d) upon La/Ce substitution the Nd DOS around Fermi level is reduced. This reduction in RE DOS is responsible for the reduction in magnitude of MAE on La/Ce substitution (Table 1).

The total and partial density of states for Nd2Fe17N3 (red), NdLaFe17N3 (blue), NdCeFe17N3 (green), La in NdLaFe17N3 (cyan), and Ce in NdCeFe17N3 (orange). In panel (a) the Total DOS (b) the RE atom DOS, and (c) the averaged Fe atoms DOS are compared. Panel (d) and (f) show the zoomed version of panel (b) and (c) in the vicinity of Fermi level. The positive and negative DOS values correspond to spin-up and spin-down channels respectively.
The important insights into the effect of SOC on magnetic properties can be obtained from perturbation theory25,26,27. According to this theory the MAE can be described by the electronic structure near the Fermi energy, in-terms of coupling between occupied and unoccupied levels. To obtain information about which regions are particularly important to the MAE, the band structures after applying SOC with magnetization along either in plane or out of plane directions are plotted in Fig. 5(a) and (b)). From these bands the MAE contribution per k-point can be evaluated using the magnetic force theorem28,29,30, by taking the difference of the sum over occupied energy eigenvalues for different magnetization directions, which is also plotted (blue line, right y-axis) in Fig. 5(a) and (b) for Sm2Fe17N3 and Nd2Fe17N3 respectively. The force theorem calculations produce a MAE of 12.1 MJ/m3 for Sm2Fe17N3, and −4.1 MJ/m3 for Nd2Fe17N3, which is in nice agreement with the values obtained from total energy calculations. Since the total MAE is positive for Sm2Fe17N3, as can be seen in Fig. 5(a) overall the MAE remains positive throughout the Brillouin zone (except along Γ to \(\frac{1}{2}-Z\) direction where it takes a negative value). On the other hand for Nd2Fe17N3 although the overall profile of MAE with respect to k-points remains the similar as Sm2Fe17N3, MAE takes negative values in the majority of brillouin zone.

Band structure including SOC with magnetization along inplane [11-2] direction (marked as M100 by black dash-dotted line) and out of plane [111] direction (marked as M001 by red dashed line) as well as the MAE contribution per k-point (blue solid line), calculated via the magnetic force theorem (a) Sm2Fe17N3 and (b) Nd2Fe17N3. The band energies are marked on the left y-axis and MAE contribution per k-points is marked on the right y-axis. In order to capture the MAE behavior at various band crossing, band structure was plotted by taking 3000 k-points along the entire high symmetric path. (c) The Brillouin along with the high symmetry directions.
Figure 6 compares the DOS for Nd2Fe17N3, and Sm2Fe17N3. As shown in the Fig. 6(a) in close vicinity of Fermi level (EF), the majority spin DOS is similar for the two compounds. However, as Sm is exchanged for Nd more electrons are added into the system and the minority spin states become occupied, whereby these 4f levels are shifted more towards the left in the middle panel of Fig. 6(b) and, as a result, Sm-4f level in the minority spin DOS of Sm2Fe17N3 shifts towards Fermi level. The Fe-DOS for both majority and minority spin in both Nd2Fe17N3, and Sm2Fe17N3 is quite similar. The important observation is the presence of minority 4f spin channels at the the Fermi level of large uniaxial anisotropy in Sm2Fe17N3, although other differences in the electronic structure are also important.

Comparison of density of states for Nd2Fe17N3 and Sm2Fe17N3. The DOS is plotted within GGA + SOC + U calculations with a U value of 5 eV for Nd and Sm sites. In panel (a) the Total DOS (b) the RE atom DOS, and (c) the averaged Fe atoms DOS are compared. Panel (d) and (f) show the zoomed version of panel (b) and (c) in the vicinity of Fermi level.
The MAE of a permanent magnet is dependent on SOC, and can be further quantified by considering its dependence on orbital moments. As shown by Bruno et al.26 by using perturbation theory that if deformations of the Fermi surface can be neglected and the MAE is dominated by spin-diagonal coupling, the MAE and orbital magnetic-moment anisotropy are proportional. The relation between MAE and orbital magnetic anisotropy is presented in Fig. 7. As can be seen from Fig. 7 the MAE is directly proportional to the orbital magnetic anisotropy. For Sm2Fe17C3, and Sm2Fe17N3 where a uniaxial anisotropy is obtained the orbital magnetic moments is also maximum along easy axis. On the other for Nd2Fe17C3, and Nd2Fe17N3 where the anisotropy is in hexagonal plane, orbital magnetic moment is maximum along the hard magnetization axis.

Calculated MAE vs orbital magnetic anisotropy for Nd2Fe17C3, Nd2Fe17N3, Sm2Fe17C3, and Sm2Fe17N3. The orbital magnetic anisotropy is averaged over both Nd/Sm and Fe atoms.
Discussions
We present a computational study of intrinsic magnetic properties such as saturation magnetization and MAE for Nd2Fe17, Nd2Fe17X3 and related compounds. For all compounds the rare earth and Fe atoms spin moments are anti-aligned in the ground state. The calculated magnetic properties of the base compounds agree well with available experimental data. The treatment of the Nd-f electrons in our calculations is different from the previously reported first principles calculations. For example the calculation of Lai et al.15 which were performed within orthogonalized linear combination of atomic orbitals (OLCAO) method, reported a decrease in total magnetic moments from 37.3 μ B /unit-cell (for to Nd2Fe17) to 36.3 μ B /unit-cell (for Nd2Fe17N3) which is contradictory to the experimental observation19, where on nitrogenation an increase in total magnetic moment has been reported. As shown in Table 1 the observed enhancement of magnetic moments in Nd2Fe17 upon nitrogenation (for Nd2Fe17N3) is nicely captured by our GGA + SOC + U calculations. Similarity Drebov et al.20 used open-core method for treatment of f-electrons by assuming RE3+ configuration. Such treatment of f electrons cannot reproduce some of the observed magnetic properties. For instance within open core method a moment of 2.5 μ B has been reported on Nd site, which is overestimated on comparing with the available experimental data19 (1.6 μ B ). On the other hand, within our calculations the f-electrons are treated as valence electrons and, as a result we could correctly reproduce the reported moment for Nd site. Overall our all-electron calculations can reproduce a number of experimentally observed properties. For example, experimentally19 for Nd2Fe17 Fe-9d site has the lowest magnetic moment of 1.6 μ B . Upon introducing the interstitial nitrogen the moment of Fe-9d increases significantly and for Nd2Fe17N3 the Fe-9d site has the maximum moment. This is nicely produced by our calculations as shown in Fig. 2. Furthermore the calculated MAE for Nd2Fe17 and Nd2Fe17N3 is in fair agreement with the available experimental data as shown in Table 1.
Although Nd2Fe17X3 compounds has many of the desirable properties for a permanent magnet such as high Curie point, large saturation magnetization, the easy axis of magnetization lies in the hexagonal plane hindering it practical application. To investigate the possibility of switching the MAE direction from hexagonal plane to c-axis we also investigated the effect of La/Ce substitution at Nd site. These calculations indicate that although the MAE on La/Ce substitution is reduced by 30–40%, its still lies in the hexagonal plane, which is not very promising for technological applications. The MAE has been also analyzed in terms of the electronic structure and by using the magnetic force theorem to compute k-point resolved contributions to the MAE. For Nd2Fe17N3 the density of states at the Fermi energy is dominated by 3d states, 4f states only contribute notably to the MAE in small regions of the Brillouin zone, however for Sm2Fe17N3 the DOS around the Fermi level is dominated by both Fe-3d, Sm-4f states. As the Fe-d states exhibit similar features for Nd2Fe17N3 and Sm2Fe17N3, the origin of uniaxial MAE could lie in the 4f-3d hybridization.
Methods
Density functional theory (DFT) calculations in the generalized gradient approximation31 (GGA) were performed with the full-potential linearized augmented plane waves (FP-LAPW) method as implemented in WIEN2k32,33,34. The sphere radii were set to 2.50, 1.88, 1.61, and 1.61 Bohr for Nd, Fe, N and C. All calculations were performed with the experimental lattice parameters8,35,36. The structure relaxations were performed within spin-polarized calculations without spin orbit coupling and all internal coordinates were relaxed until internal forces on atoms were less than 1 mRyd/Bohr. For structural relaxation 500 k-points were in the full Brillouin zone. SOC was included in the MAE calculations within a second variational approach37. For all the calculations a RKmax = 7 was used. RKmax is typically defined as the product of the smallest muffin-tin sphere and the largest reciprocal lattice vector, and describes the basis set size for a calculation. For La/Ce substitution at Nd site, one out of two Nd atoms in the primitive cell was replace by La/Ce. These substituted structure were subsequently relaxed to their ground state by minimizing the forces on all the atoms.
All the calculations are performed in the collinear spin alignment. The magnetic anisotropy energy (MAE) is obtained by calculating the total energies of the system with spin obit coupling (SOC) as K = E a − E c , where E a and E c are the total energies for the magnetization oriented along the a and c directions, respectively. Positive (negative) K corresponds to uniaxial (planar) anisotropy. For MAE calculations the convergence with respect to K-points was carefully checked all the MAE results reported in this paper correspond to 2000 reducible K-points in full Brillouin zone. To correctly treat the strong interactions between the Nd/Ce-f electrons, the Hubbard “U” correction was applied with U Nd = 5.0 eV, and U Ce = 3.0 eV with the Hund’s coupling parameter J as zero. For DFT + U calculations, the standard self interaction correction (SIC) method38,39 was used where onsite Coulomb interaction for localized orbitals is parametrized by Ueffective = U − J. The values of U parameter were obtained by optimized various magnetic properties with respect to available experimental data, and lies in the typical range that has been used successfully to describe various properties of Nd/Ce compounds before22,40,41,42,43.
References
Van Vleck, J. H. On the anisotropy of cubic ferromagnetic crystals. Phys. Rev. 52, 1178 (1937).
Buschow, K. New developments in hard magnetic materials. Rep. Prog. Phys. 54, 1123 (1991).
Buschow, K. H. J. & Boer, F. R. Physics of magnetism and magnetic materials, vol. 92 (Springer, 2003).
Kramer, M., McCallum, R., Anderson, I. & Constantinides, S. Prospects for non-rare earth permanent magnets for traction motors and generators. Jom 64, 752–763 (2012).
Coey, J. M. Magnetism and magnetic materials (Cambridge University Press 2010).
Coey, J. Hard magnetic materials: A perspective. IEEE Trans. Magn. 47, 4671–4681 (2011).
Coey, J. Permanent magnets: Plugging the gap. Scr. Mater. 67, 524–529 (2012).
Katter, M., Wecker, J., Kuhrt, C., Schultz, L. & Grössinger, R. Structural and intrinsic magnetic properties of Sm2(Fe1−xCo x )17N y . J. Magn. Magn. Mater. 114, 35–44 (1992).
Givord, D., Li, H. & Moreau, J. Magnetic properties and crystal structure of Nd2Fe14B. Solid State Commun. 50, 497–499 (1984).
Oyama, S. T. Introduction to the chemistry of transition metal carbides and nitrides. In The chemistry of transition metal carbides and nitrides, 1–27 (Springer, 1996).
Fa-Shen, L., Fu-Sheng, W., Dong, Z., Liang, Q. & Wen-Liang, Z. Microwave magnetic properties of Nd2Fe17N3−δ with planar anisotropy. Chin. Phys. Lett. 25, 1068 (2008).
Isnard, O. & Guillot, M. Investigation of the magnetic properties of Nd2Fe17 and Nd2Fe17H x (x = 3, 4.9) in high magnetic field. J. Appl. Phys. 87, 5326–5328 (2000).
Xiu-Feng, H., Han-Min, J., Zi-Jun, W., Zhao, T. & Sun, C. Analysis of the magnetic properties of R2Co17 (R = Pr, Nd, Sm, Gd, Tb, Dy, Ho, and Er). Phys. Rev. B 47, 3248 (1993).
Helmholdt, R. & Buschow, K. Crystallographic and magnetic structure of ternary carbides of the type Nd2Fe17C x . Journal of the Less Common Metals 155, 15–21 (1989).
Gu, Z. & Lai, W. Calculated electronic and magnetic structures of the new ternary rare-earth–iron nitride Nd2Fe17N3. J. Appl. Phys. 71, 3911–3916 (1992).
Coey, J. & Sun, H. Improved magnetic properties by treatment of iron-based rare earth intermetallic compounds in anmonia. J. Magn. Magn. Mater. 87, L251–L254 (1990).
Sun, H., Otani, Y. & Coey, J. Gas-phase carbonation of R2Fe17. J. Magn. Magn. Mater. 104, 1439–1440 (1992).
Sun, H., Coey, J., Otani, Y. & Hurley, D. Magnetic properties of a new series of rare-earth iron nitrides: R2Fe17N y (y approximately 2.6). J. Phys. Condens. Matter 2, 6465 (1990).
Kajitani, T. et al. High-resolution neutron powder diffraction study on nitrogenated Nd2Fe17. J. Appl. Phys. 73, 6032–6034 (1993).
Drebov, N. et al. Ab initio screening methodology applied to the search for new permanent magnetic materials. New J. Phys. 15, 125023 (2013).
Steinbeck, L., Richter, M., Nitzsche, U. & Eschrig, H. Ab initio calculation of electronic structure, crystal field, and intrinsic magnetic properties of Sm2Fe17, Sm2Fe17N3, Sm2Fe17C3, and Sm2Co17. Phys. Rev. B 53, 7111 (1996).
Larson, P., Mazin, I. & Papaconstantopoulos, D. Calculation of magnetic anisotropy energy in SmCo5. Phys. Rev. B 67, 214405 (2003).
Larson, P., Mazin, I. & Papaconstantopoulos, D. Effects of doping on the magnetic anisotropy energy in SmCo5−xFe x and YCo5−xFe x . Phys. Rev. B 69, 134408 (2004).
Larson, P. & Mazin, I. I. Magnetic properties of SmCo5 and YCo5. J. Appl. Phys. 93, 6888–6890 (2003).
Andersson, C. et al. Influence of ligand states on the relationship between orbital moment and magnetocrystalline anisotropy. Phys. Rev. Lett. 99, 177207 (2007).
Bruno, P. Tight-binding approach to the orbital magnetic moment and magnetocrystalline anisotropy of transition-metal monolayers. Phys. Rev. B 39, 865 (1989).
Wang, D.-S., Wu, R. & Freeman, A. First-principles theory of surface magnetocrystalline anisotropy and the diatomic-pair model. Phys. Rev. B 47, 14932 (1993).
Wu, R. & Freeman, A. Spin-orbit induced magnetic phenomena in bulk metals and their surfaces and interfaces. J. Magn. Magn. Mater. 200, 498–514 (1999).
Liechtenstein, A. I., Katsnelson, M., Antropov, V. & Gubanov, V. Local spin density functional approach to the theory of exchange interactions in ferromagnetic metals and alloys. J. Magn. Magn. Mater. 67, 65–74 (1987).
Daalderop, G., Kelly, P. & Schuurmans, M. First-principles calculation of the magnetocrystalline anisotropy energy of iron, cobalt, and nickel. Phys. Rev. B 41, 11919 (1990).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple [phys. rev. lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396–1396 (1997).
Blaha, P., Schwarz, K., Madsen, G. K. H., Kvasnicka, D. & Luitz, J. WIEN2K, An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties. (Karlheinz Schwarz, Techn. Universität Wien, Austria, 2001).
Sjostedt, E., Nordstrom, L. & Singh, D. J. An alternative way of linearizing the augmented plane-wave method. Solid State Commun. 114, 15–20 (2000).
Singh, D. J. & Nordstrom, L. Planewaves Pseudopotentials and the LAPW Method. (Springer, Berlin, 2006).
Christodoulou, C. N. & Takeshita, T. Interstitial carbonation of the Sm2Fe17 phase by reaction with hydrocarbons. J. Alloys Compd. 190, 41–48 (1992).
Djega-Mariadassou, C., Bessais, L., Nandra, A., Grenèche, J. & Burzo, E. Structure and hyperfine properties of Sm2(Fe,Si)17. Phys. Rev. B 65, 014419 (2001).
Koelling, D. & Harmon, B. A technique for relativistic spin-polarised calculations. J. Phys. C 10, 3107 (1977).
Anisimov, V. I., Solovyev, I., Korotin, M., Czyżk, M. & Sawatzky, G. Density-functional theory and NiO photoemission spectra. Phys. Rev. B 48, 16929 (1993).
Liechtenstein, A., Anisimov, V. & Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467 (1995).
Yan, X.-W., Wang, Y., Gao, M., Ma, D. & Huang, Z. Magnetic and electronic properties of samarium-doped phenanthrene from first-principles study. J. Phys. Chem. C 120, 22565–22570 (2016).
Morari, C. et al. Magnetism and electronic structure calculation of SmN. J. Phys. Condens. Matter 27, 115503 (2015).
Aryasetiawan, F., Karlsson, K., Jepsen, O. & Schönberger, U. Calculations of Hubbard U from first-principles. Phys. Rev. B 74, 125106 (2006).
Shi, L., Vathonne, E., Oison, V., Freyss, M. & Hayn, R. First-principles DFT + U investigation of charged states of defects and fission gas atoms in CeO2. Phys. Rev. B 94, 115132 (2016).
Acknowledgements
This research was supported by the Critical Materials Institute, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Advanced Manufacturing Office.
Author information
Authors and Affiliations
Contributions
D.S.P. conceived the idea. T.P. conducted the calculations. T.P. and D.S.P. analyzed the results and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pandey, T., Parker, D.S. Magnetic properties and magnetocrystalline anisotropy of Nd2Fe17, Nd2Fe17X3, and related compounds. Sci Rep 8, 3601 (2018). https://doi.org/10.1038/s41598-018-21969-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21969-8
- Springer Nature Limited
This article is cited by
-
Inter-element miscibility driven stabilization of ordered pseudo-binary alloy
Nature Communications (2022)
-
An ab initio study of the magnetic properties of strontium hexaferrite
Scientific Reports (2021)