
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) is a virus susceptible to antibody dependent enhancement, causing reproductive failures in sows and preweaning mortality of piglets. Modified-live virus (MLV) vaccines are used to control PRRS in swine herds. However, immunized sows and piglets often generate variable antibody levels. This study aimed to detect significant genes and pathways involved in antibody responsiveness of pregnant sows and their offspring post-PRRSV vaccination. RNA sequencing was conducted on peripheral blood-mononuclear cells (PBMCs), which were isolated from pregnant sows and their piglets with high (HA), median (MA), and low (LA) PRRS antibody levels following vaccination. 401 differentially expressed genes (DEGs) were identified in three comparisons (HA versus MA, HA versus LA, and MA versus LA) of sow PBMCs. Two novel pathways (complement and coagulation cascade pathway; and epithelial cell signaling in H. pylori infection pathway) revealed by DEGs in HA versus LA and MA versus LA were involved in chemotactic and proinflammatory responses. TNF-α, CCL4, and NFKBIA genes displayed the same expression trends in subsequent generation post-PRRS-MLV vaccination. Findings of the study suggest that two pathways and TNF-α, CCL4, and NFKBIA could be considered as key pathways and potential candidate genes for PRRSV vaccine responsiveness, respectively.
Similar content being viewed by others


Introduction
The use of vaccines for controlling and eliminating virus-induced diseases is an important and complex process that highly depends on both antigen properties and host responses. Porcine reproductive and respiratory syndrome (PRRS), which is caused by the PRRS virus (PRRSV), is characterized by acute reproductive failure in sows and respiratory disorders in piglets, resulting in significant economic losses to the swine industry worldwide1,2,3. Thus, PRRS control strategies via vaccine prevention are commonly used to reduce porcine production losses4.
The commercially modified live-attenuated PRRSV vaccine (PRRS-MLV) is widely used to induce protective immunity in pigs. However, host immune responses against PRRSV infection induced by PRRSV vaccine cannot be maintained in the long term5,6. Thus, to solve this problem, sows, including gestating sows, are vaccinated several times per year with PRRS-MLV in some pig farms. PRRSV is also an antibody-dependent enhancement (ADE) virus, which may induce the susceptibility of pigs to PRRSV infection with decreasing levels of PRRSV-specific antibodies of maternal origin or with antibodies induced by exposure to PRRSV vaccination7,8. Numerous studies have also described that vaccination with PRRS-MLV vaccine during sow gestation may exhibit negative effects on reproductive performance, such as reduced pigs born alive and increased pigs born dead9. Therefore, the interaction basis and genetic fundamentals of immunogenetics between sow immune responses and PRRSV vaccines remain to be elucidated. Identification of affected genes and pathways involved in immune responses allows researchers to understand variable antibody responses of PRRSV vaccination in sows.
The transcriptome of peripheral blood mononuclear cells (PBMCs) not only indicates primary immune response of leukocytes but also shows the extent and dynamics of differentially expressed genes (DEGs) induced by antigens in these cells10,11,12. We first conducted genome-wide transcriptional analyses of PBMCs, which were isolated from gestating sows with high (HA), median (MA), and low (LA) levels of PRRS-antibody post-vaccination of highly pathogenic PRRSV vaccine strains TJM, to investigate the key pathways and genes involved in immune responses to PRRSV vaccination of sows. Then, transcriptomes of offspring after PRRSV vaccination were analyzed to compare common genes and immune-related pathways between two generations of pigs. Figure 1 shows our experimental design. Finally, key genes and pathways involved in porcine PBMCs post-vaccination of PRRSV-MLV were identified.

Experimental design. Pregnant sows were vaccinated with PRRS-MLV vaccine (day 0). At 21 and 35 days after vaccination, blood samples were collected. Antibody levels in sample serum at days 21 and 35 were determined using enzyme-linked immunosorbent assay (ELISA). PBMCs were isolated from blood samples at 35 days post-vaccination and used for DEG identification. Piglets were initially vaccinated with PRRS-MLV vaccine at 28 days old. Second vaccination of PRRS-MLV was performed at 58 days old. Heparinized blood samples of piglets were collected directly at 21 days after the second vaccination (79 days old). Antibody levels of piglets were determined using ELISA. PBMCs were isolated from blood samples and used for DEG identification. HA: High antibody level; MA: Median antibody level; LA: Low antibody level.
Results
Different immune responses of pregnant sows to PRRSV vaccination
Serum samples were isolated from the jugular vein of sows at days 21 and 35 post-vaccination of PRRSV-MLV (21 and 35 dpi) and were screened by enzyme-linked immunosorbent assay (ELISA) to evaluate antibody responses of pregnant sows induced by PRRSV vaccine.
At 21 dpi, we rated 94 pregnant sows from high to low sample-to-positive (S/P) ratio (3.004 to 0.305). Then, 27 samples were selected and divided into three groups based on S/P ratio: HA group with the highest antibody level (n = 10, S/P = 2.911 ± 0.094), MA group with median antibody level (n = 7, S/P = 1.857 ± 0.059), and LA with the lowest antibody level (n = 10, S/P = 0.666 ± 0.190). At 35 dpi, PRRSV-specific antibody levels of these 27 pregnant sows were measured again. We observed that the antibody levels of the three groups were consistent with those at 21 dpi. The findings were as follows: the HA group incurred high S/P ratio (n = 10, S/P = 3.714 ± 0.461), the MA group with median antibody level (MA, n = 7, S/P = 2.335 ± 0.519), and the LA group with low antibody level (LA, n = 10, S/P = 1.177 ± 0.277) (Fig. 2A). Four sows in each group at 35 dpi were randomly selected for RNA sequencing (RNA-seq), and their specific antibody levels differed significantly among the three groups (P < 0.05) (Fig. 2B). Quantitative reverse transcription polymerase chain reaction (RT-qPCR) was conducted in PBMCs to assess responses of known host-molecular signature genes (IFN-α and TLR3) to viral vaccine13,14 between the HA and LA groups. Results indicated significantly higher transcriptional expression levels of IFN-α and TLR3 in the HA group than in the LA group (Fig. 2C,D, respectively; P < 0.05).

Immune responses of pregnant sows to PRRSV-MLV vaccination. (A) Sample-to-positive (S/P) ratio varied among the three groups at days 21 and 35 post-PRRSV-MLV vaccination (21 and 35 dpi). HA: high antibody group (n = 10); MA: median antibody group (n = 7); LA: low antibody group (n = 10). (B) S/P ratio of four individuals in each group which were selected for RNA-seq. S/P values differed among the three groups at 35 dpi (n = 4). (C) Expression levels of IFN-α gene in HA and LA groups with quantitative reverse transcription-polymerase chain reaction (RT-qPCR) (n = 4). (D) Expression levels of TLR3 in HA and LA groups with RT-qPCR (n = 4). * and ** mean significant levels of P < 0.05 and P < 0.01 with Student’s t-test, respectively.
Differentially expressed genes (DEGs) in pregnant sows with different antibody levels post-PRRSV vaccination
Subsequently, four individuals in each group were used for identifying DEGs of sow PBMCs via RNA-seq to analyze genome-wide transcriptional expression differences among the three groups. Under the criteria of |fold change| (|FC|) > 1.5 and q < 0.05, 401 DEGs, including 91 up-regulated and 310 down-regulated DEGs, were identified by comparing the three groups. During comparison of HA versus MA, 89 significantly DEGs were detected (Table 1). Among these 89 genes, 45% were up-regulated (9/20) with |FC| > 2, whereas 36% were down-regulated (25/69) with |-FC| > 2. Genes MYL4 and TMP-CH242-74M17.6 showed 10 times upregulation and downregulation in HA sows compared with MA sows, respectively. Table 1 lists the numbers of DEGs during comparison of HA versus LA and MA versus LA. Results revealed the highest number of DEGs in HA versus LA, whereas the least DEG number was detected in HA versus MA. MYL4 was also 10 times up-regulated in HA compared with LA. IL-1α, IL-1β, and ENSSSCG00000009469 displayed 10 times downregulation in HA compared with that in LA. These data indicate strong inducement of proinflammatory cytokines interleukin (IL)-1α and IL-1β in LA pigs.
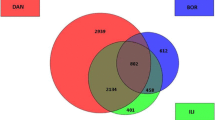
Figure 3 shows the commonly up-regulated and down-regulated DEGs observed among the three comparisons. Data indicated that down-regulated DEGs between HA versus LA and MA versus LA (68 DEGs) were 11 times higher than up-regulated DEGs (6 DEGs). Only one common gene (TNF-α) was detected in three comparisons in down-regulated DEGs (Fig. 3B).

DEGs in different comparisons in sow PBMCs. (A,B) Venn diagrams of up-regulated and down-regulated DEGs after vaccination against PRRS.
According to biological pathways of involved DEGs, we proposed that general immune responses of pigs are susceptible to modulation by PRRSV vaccine. Supplementary Figure S1 describes the transcriptional responses and interprets vaccine–host interaction of sow PBMCs after PRRS-MLV vaccination. As shown in the figure, we observed significant downregulation of various important inflammatory mediators (tumor necrosis factor (TNF)-α, IL-1α, and IL-1β) and chemokines (chemokine (C-C motif) ligand (CCL) 2, CCL4, CCL8, chemokine (C-X-C motif) ligand 2 (CXCL2), and CXCL8) in the HA group compared with that in the LA group. However, seven genes (SLA-DOB, C-JUN, EGR1, CD69, MAL, CDH2, and FOS) were regulated in opposite directions when comparing HA versus LA and HA versus MA (Supplementary Figure S1).
Validation of RNA sequencing by RT-qPCR
Seven genes, including three up-regulated (IFN-γ, CXCR4, and CCR1) and four down-regulated ones (CCL8, CCL4, IL1B, and CXCL8), were selected for RT-qPCR confirmation to verify the DEGs obtained by RNA-seq. Results were presented as |FC|s of gene expression levels normalized to GAPDH gene. Expression profiles of genes determined by RT-qPCR were consistent with those of RNA-seq results (Supplementary Figure S2), confirming the reliability of our RNA-seq data.
Gene ontology (GO) terms and Kyoto Encyclopedia of genes and genomes (KEGG) pathways of DEGs in sows after PRRSV vaccination
GO annotates genes to biological, cellular, and molecular terms in a hierarchically structured way, whereas KEGG divides DEGs according to functional pathways. We therefore further analyzed GO terms and KEGG pathways of 401 DEGs using Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resources (version 6.7).
With regard to the 91 up-regulated DEGs, we found no GO terms after three comparisons. However, two pathways (ErbB signaling pathway and T cell receptor signaling pathway) were detected during comparison of MA versus LA, and related DEGs (C-JUN, HBEGF, IFN-γ, and FOS) were up-regulated in the MA group compared with LA (Table 2).
For the down-regulated DEGs, 30 enriched GO terms (Fig. 4A) and six pathways (Table 2) were obtained in comparison of HA versus LA. Significantly enriched biological processes were related to immune response functions, including inflammatory responses, response to wounding, cytokine production, defense response, cell proliferation, leukocyte chemotaxis and migration, stimulus response, and immune system processes (Fig. 4A). With regard to the six pathways detected in comparison of HA versus LA (Table 2), epithelial cell signaling in H. pylori infection serves as a novel pathway related to PRRSV vaccination, whereas the other five pathways were reportedly involved in immune responses to PRRSV infection15 and PRRSV vaccination16.

Gene Ontology (GO) term analysis of the DEGs. Biological process enriched by down-regulated DEGs in HA versus LA (A) and MA versus LA (B) induced by PRRSV-MLV vaccine in sows.
During comparison of MA versus LA, 15 enriched GO terms were obtained from down-regulated DEGs (Fig. 4B). Such biological processes involved in immune responses were similar to those in comparison of HA versus LA, in which six pathways were obtained (Table 2). Among these pathways, a unique hematopoietic cell lineage was observed during comparison. The remaining five pathways were consistent with those in comparison of HA versus LA. Results showed the identical key pathways enriched in innate and cell-mediated immune responses between comparisons of HA versus LA and MA versus LA.
Protein–Protein interaction (PPI) of DEGs in sow and piglet PBMCs induced by PRRSV vaccination
PPI analysis was performed, and we observed significant interplay in a subset of 73 proteins encoded by DEGs in sows (Fig. 5A). In the network, the proteins encoded by C-JUN, FOS, CCL2, CCL4, CCL8, CLU, CXCR4, ID2, IL1A, IL1B, IL8, IRG6, MX1, MX2, MYD88, NFKBIA, TNF-α, VEGFA, IRF1, PF4, and TLR4 were highly interconnected (Fig. 5A). Considering significant differences in the two extreme sow groups (HA and LA) for antibody levels, we followed up on the offspring of these groups and performed further PPI analysis. Results showed that the proteins encoded by C-JUN, FOS, CCL4, CCL8, CXCR4, NFKBIA, and TNF-α were also present as pivotal nodes in the HA and LA groups in the next generation (Fig. 5B). These data suggest that the common node genes (C-JUN, FOS, NFKBIA, and TNF-α), which are encircled in Fig. 5A,B, play the most significant roles in pregnant sows and piglets induced by PRRSV vaccine.

Protein–protein interaction network of DEGs produced by STRING analysis. STRING analysis was used to analyze DEGs in PBMCs of sows (A) and piglets (B). Differently colored lines represent seven types of evidence used in predicting associations. Red line: fusion evidence; blue line: co-occurrence evidence; yellow line: text mining evidence; green line: neighborhood evidence; purple line: experimental evidence; light blue line: database evidence; and black line: co-expression evidence.
Transcriptomic homogeneity in pregnant sows and their offspring
Upon follow-up of piglets of pregnant sows, we compared antibody levels and DEGs in the two generation of pigs for comparison of HA versus LA (Fig. 6A,B). Results showed that during comparison of HA versus LA, 17 out of 159 DEGs (10.69%) in sows were also detected in their offspring (Fig. 6B). Signaling pathways involving toll-like receptor (TLR) signaling pathway were also affected in both sows and their offspring. Out of 17 common genes between sows and their piglets, 15 genes (88.24%) displayed the same expression tendency in comparison of HA versus LA. As shown in Fig. 6C, TNF-α, CCL8, and CCL4 gene expression tendency in the HA versus LA groups in offspring agreed with that in sows. Supplementary Figure S3A summarizes the antibody levels of vaccinated animals compared with naive piglet serum responses. Findings indicated the lower PRRSV antibody levels in newborn pre-immune piglets (mean of their S/P ratio is 6.07E-18) in comparison with vaccinated animals. Investigation by RT-qPCR on biological variations in TNF-α, CCL4, and CXCL8 genes in naive piglets showed increased sensitivity compared with PRRSV antibody levels (Supplementary Figure S3B). Data indicated that common, sensitive, and specific gene-expression patterns in two successive generations can provide further insights into host–vaccine interaction post-PRRSV vaccination.

Unique and mutual transcriptomic changes between sows and offspring in comparison of HA versus LA. (A) S/P ratio between sows (n = 4) and piglets (n = 3). (B) Venn diagram showing the number of DEGs overlapping in sows and their offspring. (C) Expression trend of three common DEGs (TNFα, CCL8, and CCL4) between sows (n = 4) and piglets (n = 3). * and ** mean P < 0.05 and P < 0.01 with Student’s t-test, respectively.
Remarkably, we observed negative regressions between expression levels of TNF-α, CCL4, and NFKBIA with PRRS-MLV-specific antibody levels in HA, MA, and LA sows (upper panel in Fig. 7). Similar expression trends of the three genes were observed in piglets in comparison of HA versus LA (lower panel in Fig. 7). Thus, sows with low PRRSV antibody levels presented high transcript abundances of cellular immune-related genes.

Regression analysis of divergent PRRSV-MLV-specific antibody levels (x-axis) with gene expression levels (y-axis) in sows and piglets.
Discussion
PRRS is an economically important and panzootic disease leading to reproductive failure in breeding pigs and respiratory tract sickness in piglets. Simultaneously, the use of vaccines to control PRRS is a tough choice for pig farms as PRRSV vaccination poses safety risks on virulent reversion of modified live vaccines (MLV) and depends on immune responses of pigs to the vaccine17,18. This study investigated genome-wide transcriptional changes in PBMCs associated with high, medium, and low antibody responses over two generations of Landrace pigs after PRRSV vaccination. Finally, our results revealed several significant genes and pathways involved in variable antibody responsiveness of pregnant sows and their offspring post-PRRSV vaccination via next-generation sequencing technology.
Our results demonstrated that more than 400 DEGs and 12 KEGG pathways were involved in gestating sows of HA, MA, and LA PRRSV-antibody levels. A total of 17 DEGs and two pathways recurred in piglets during comparison of HA versus LA. To integrate DEGs in sows with healthy and PRRSV susceptibility-related quantitative trait loci (QTLs), we matched positions of DEGs to QTL regions for health traits (including PRRSV susceptibility) in the pig QTL database (http://www.animalgenome.org/cgi-bin/QTLdb/SS/index). Our search discovered that 19 DEGs were distributed in 17 different QTL regions associated with pig healthy and PRRSV susceptibility-related traits (Table 3). Out of 19 DEGs, MEGF9, NDRG1, SLA-8, and C2 were located within QTL regions, which were validated to display remarkable genetic effects on PRRSV susceptibility19, whereas STX11 was located in the QTL region of CD4-positive leukocyte percentage. Thus, these five genes were suggested to be powerful candidate genes for PRRSV vaccine responsiveness in porcine. Transcriptional responses and underlying mechanisms associated with high, medium, and low level of neutralizing antibody must be measured to improve the protection of PRRSV vaccination.
In the current study, two immune response-related pathways (i.e., TLR signaling pathway and T cell receptor signaling pathway) were exposed to PRRSV vaccination responsiveness in the two generations of pig. Especially, TLR signaling pathway plays a significant role in adaptive immunity16,20. This pathway is primarily represented by CD1421, which was significantly up-regulated in LA pigs and moderately up-regulated in MA pigs compared with that in the HA group after PRRSV-MLV vaccination (Table 2 and Supplementary Figure S1). On the other hand, in PPI analysis, CD14 was closely interconnected with TLR2 and TLR4 (Fig. 5), which are members of the TLR family and may play roles in virus recognition22,23. T cell receptor pathway performs a central role in regulation of thymocyte development and T cell differentiation24. The present study observed that DEGs (C-JUN, IFN-γ, and FOS) in the pathway were up-regulated in MA pigs compared with that in LA animals. TLR signaling pathway and T cell receptor pathway are known to be involved in host–vaccine interaction14,22; however, further research at more time points is needed to dissect whether these genes in the two pathways actually affect the responsiveness of PRRSV vaccination.
The present study reported for the first time the association of two pathways (i.e., complement and coagulation cascade pathway and epithelial cell signaling in H. pylori infection pathway) with PRRSV vaccination responsiveness. The complement and coagulation cascade pathway exhibits a first line of defense against pathogens and other invaders that may enter circulation and affect inflammatory responses25. In this study, epithelial cell signaling in H. pylori infection known as infectious disease-related pathway was detected for the first time to be associated with PRRSV vaccination, indicating that animal immune responses to pathogens can share certain evolutionary conservation26. DEGs (NFKBIA and CXCL8) in the two pathways showed the potential to distinguish antibody levels in PRRSV-vaccinated pigs; these DEGs were down-regulated in HA pigs compared with that in the LA group.
Cytokines and chemokines in cellular immune response play a pivotal role in the initial stage of post-vaccination responsiveness27,28. We observed that cytokine TNF-α and chemokine CCL4 were down-regulated in pigs with high antibody levels. Similar downregulation of CCL4 was reported in piglets with high humoral immune responses (ELISA) after primary PRRSV vaccination29. These negative regressions (Fig. 7) were validated by adding more sows (Supplementary Figure S4). TNF-α is a primary mediator of systemic inflammation and is one of the cytokines participating in acute phase reaction30. TNF-α plays a significant role in efficient innate immunity, antiviral effect initiation, activation, maturation and differentiation of monocyte/macrophage lineages, and stimulation of natural killer cells to produce IFN-γ31,32. Chemokine CCL4 (MIP-1β) is the most potent chemoattractant and mediator of virus-induced inflammation in vivo33. These findings imply that cellular immune response could be improved in sows and piglets with low antibody levels after PRRSV vaccination to compensate for inferior humoral immune responses.
Encoded proteins of FOS, C-JUN, and NFKBIA were identified as central nodes of vaccine-induced transcriptional network in PBMCs of PPI networks (Fig. 5A). The proto-oncogenes FOS and C-JUN work mutually as derivable transcription factors in signal transduction. FOS is a nuclear phosphoprotein involved in important cellular events, including cell proliferation, differentiation, and survival, and controlled replication of hepatitis C virus34,35. C-JUN is known as a regulator of primary biological processes35,36 and downregulates p53, which is a suppressor of apoptosis and cell differentiation37,38 and encodes an avian sarcoma virus-like protein39. We observed that expression trends of key node genes fluctuated between the two generations (Fig. 7). Therefore, further study is warranted to dissect whether the variably expressed genes are involved in variable PRRSV vaccination responsiveness in pigs.
The third node gene NFKBIA (Iκ-Bα) encodes NFKB inhibitor alpha (NFKBIA) protein, which is a repressor of nuclear factor (NF)-κB transcription by covering nuclear localization signals of NF-κB proteins and maintaining their inactivity in cytoplasm40. NFKBIA also inhibits NF-κB from binding to DNA; thus, NF-κB cannot function as a transcription factor41,42. NF-κB is a universal transcription factor that induces several relevant downstream signal transductions, resulting in upregulation of proinflammatory cytokines, type-I interferon, and chemokines, which promote inflammatory processes, apoptosis, and phagocytosis40,43. Our results suggest that NFKBIA is the node with the most potential to prevent inflammatory process following PRRS-MLV vaccination in PBMCs of sows (Fig. 5A). With NFKBIA showing a similar trend in the next generation of sows post-PRRS-MLV vaccination (Fig. 7, Supplementary Figure S4), these data suggest that NFKBIA could be a candidate gene relevant with pig PRRSV vaccine responsiveness.
In conclusion, we performed genome-wide transcriptional profiles to investigate genes, pathways, and networks related with immune responses in PBMCs of Landrace sows and their offspring following PRRS-MLV vaccination. This study revealed 401 DEGs in three comparisons (HA versus MA, HA versus LA, and Ma versus LA) and identified NFKBIA, TNF-α, CXCL8, and CCL4 as the most promising candidate genes that can contribute to the interaction between sows and PRRS-MLV vaccine. TNF-α, CCL4, and NFKBIA are more possibly related with variable antibody responses in PRRSV vaccination in both sows and offspring. On the other hand, two pathways were detected to be associated with sow PRRSV vaccine responsiveness: complement and coagulation cascade pathway and epithelial cell signaling in H. pylori infection pathway. These DEGs and novel pathways provide new insights into the molecular mechanism of immune response in sow and piglet PRRS vaccination. Future studies will be needed to identify these novel gene functions in PRRSV vaccine responsiveness.
Materials and Methods
Animals, vaccination, and PRRS antibody detection
All the following procedures involving animals were approved by the Animal Welfare Committee of China Agricultural University in Beijing, China. Animal experiments were also conducted in strict accordance with regulations and guidelines established by this committee (permit number: DK996).
Sows
Landrace pregnant sows (1–3 months of pregnancy) were obtained from Tianjin Ninghe Primary Pig Breeding Farm (Tianjin, China). Sows were reared in the same normal conditions with natural room temperature and light, fed twice daily, and provided ad libitum access to water. Pedigree information was available for all animals. In this study, sows were free of serum antibody of PRRSV, classical swine fever virus, and pseudorabies virus, as confirmed by ELISA detection.
Figure 1 outlines the experimental design. A total of 94 Landrace pregnant sows in the middle stages of gestation (30–80 days of pregnancy, 2nd–3rd parity, and age of approximately 2.5 years old) were inoculated (day 0) intramuscularly with one dose (1 mL) of PRRS-MLV and named highly pathogenic porcine PRRS vaccine (live strain TJM-F92) (Pfizer, USA). Sow blood samples were collected for analysis at days 21 and 35 post-inoculation of PRRS-MLV vaccine. PBMCs at day 35 were isolated from heparinized blood collected by Ficoll–Hypaque (TBD, Tianjin, China) gradient centrifugation and stored at −80 °C. Sera at two time points were isolated from blood samples by centrifuging at 2000 g for 10 min at room temperature.
Serum PRRSV antibody levels were determined using an ELISA kit (HerdCheck PRRS 3X, IDEXX Laboratories Inc., USA) according to manufacturer’s protocol. Optical density (OD) of each well was determined at 630 nm using a microplate reader (Multiskan MK3, Thermo Scientific, USA). Presence or absence of PRRSV-specific antibodies in samples was determined by calculating S/P ratio. S/P ratio was calculated according to the following equation: S/P ratio (%) = 100 × [(OD of test sample—mean OD of negative controls)/(mean OD of positive controls—mean OD of negative controls)]. Samples were considered positive for PRRSV antibody when S/P ratio reached 0.4 or higher44. Three groups (HA, MA, and LA) with different PRRSV antibody levels were classified according to S/P ratio at 21 dpi and confirmed by S/P ratio at 35 dpi. Four sows in each group (HA, MA, and LA) were randomly selected to perform RNA-seq according to S/P ratio at 35 dpi.
Piglets
Sow piglets in the same farm were earmarked by small cuts with a sharp pincer to record PRRSV antibody levels of their offspring piglets from the HA and LA groups. All piglets were raised under the same standard indoor conditions according to their growth stages. In detail, the piglets were raised in farrowing house from birth to 28 days old. Then, the piglets were moved to nursery pens until their weight reached approximately 30 kg. Subsequently, the animals were moved to growing–finishing pens to be sold out as breeders (~150 days old). According to routine vaccination program of the farm, the piglets were initially vaccinated (28 days old) intramuscularly with one dose (1 mL) of PRRS-MLV vaccine. The second vaccination was administered after 30 days (58 days old). Heparinized blood samples were collected directly 21 days after the second vaccination (79 days old). Levels of serum PRRSV antibody at 79 days old were measured by an ELISA kit (HerdCheck PRRS 3X, IDEXX Laboratories Inc., USA). Finally, three animals from each group were selected as follows: HA piglet group from the HA sow group (S/P = 2.499 ± 0.111) and LA piglet group from the LA sow group (S/P = 1.227 ± 0.542). The selected animals were used to conduct transcriptome examination with PBMCs at 79 days old. Newborn piglets (n = 4) in a naive population were not vaccinated but served as controls.
RNA isolation, cDNA library preparation, and RNA-seq
Total RNA of pig PBMCs was extracted directly from each sample using TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA) according to manufacturer’s protocol. RNA quality and integrity were measured with the Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). All RNA integrity numbers of samples were above nine. The 28S:18S rRNA ratios of all samples ranged from 1.7 to 2.4.
Equivalent amount (4 µg) of total RNA from PBMCs of each pig was used to construct RNA-seq libraries. Poly-A-containing mRNA was purified from total RNA using Oligo (dT) magnetic beads (NEB, Ipswich, MA, USA) to create cDNA sequencing libraries. Fragmentation reagent was then added to break the purified mRNA into short fragments at 94 °C for 15 min. First-strand cDNA synthesis was conducted using ProtoScript II reverse transcriptase (NEB, Ipswich, MA, USA) and random primers (NEB). A second strand cDNA was generated using Second Strand Synthesis Enzyme Mix (NEB) and then purified using AMPure XP Beads (Beckman Coulter, Beverly, USA). End repair, dA tailing, and sequencing adapter ligation were performed, followed by cDNA purification with AMPure XP beads (Beckman Coulter) and 12 cycles of polymerase chain reaction (PCR) amplification. Then, the libraries were purified by AMPure XP beads (Beckman Coulter). Subsequently, PCR products with lengths of 350–450 bp were selected in 2% agarose gel and quantified with QUBIT DNA HS Assay Kit (Invitrogen Corporation, Carlsbad, CA). Quality of cDNA library was evaluated by 2100 Bioanalyzer chip (Agilent). Libraries were then denatured using sodium hydroxide (NaOH) and diluted to a final concentration of 20 pM. Finally, clusters for RNA-seq were generated on flow cell using TruSeq Rapid PE Cluster Kit (Illumina). After cluster generation, cDNA libraries were sequenced by 2 × 125 paired-end sequencing on an Illumina HiSeq. 2500 platform (Illumina, San Diego, CA, USA). Raw data were stored in FASTQ format. Complete sequencing data obtained in this study have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive with accession number SRP110797.
Sequencing quality control and data analysis
Quality of raw sequences was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adaptor sequences, unknown sequences (N), low-quality reads (Q < 20), and their paired sequences measuring less than 50 bases were eliminated from raw reads using Next Generation Sequencing Quality Control Toolkit version 2.3.3.
Reference genome and gene annotation files were downloaded from Ensembl (http://www.ensembl.org/info/data/ftp/index.html). A set of genomic index files of reference genome was built using Bowtie version 2.2.5, and clean reads were mapped to the Sus scrofa reference genome (Sscrofa10.2) from NCBI using Tophat version 2.1.045. Supplementary Table 1 presents the detailed alignment information, including total numbers of reads (37.28–42.34 million) and mapped rate (80.7–85.7%). Then, the transcripts were assembled using Cufflinks version 2.2.1. Transcript files generated by Cufflinks were added to a single-merged transcriptome annotation using Cuffmerge version 2.2.1. DEGs and transcripts were identified between different sample groups using Cuffdiff version 2.2.146. Gene expression values were calculated by counting the number of fragments per kilobase of transcript per million mapped fragments. Cuffdiff applies a corrected p-value, known as q-value, to measure significant differences between two sample groups (q < 0.05).
Quantitative reverse transcription PCR (RT-qPCR) validation for RNA-seq
DEGs were validated by RT-qPCR for the following transcripts: IFN-γ, CXCR4, CCR1, CCL8, CCL4, IL1B, and CXCL8. One microgram of total RNA per sample was used to perform first-strand cDNA synthesis using a PrimeScript™ RT (Perfect Real Time) reagent kit with gDNA Eraser (Takara, Tokyo, Japan). RT reaction was performed at 37 °C for 15 min and 85 °C for 5 s. Gene-specific primers were designed according to their gene sequences (Supplementary Table 2). cDNA samples were then quantified with RT-qPCR using LightCycler® 480 Real-Time PCR System (Roche, Hercules, CA, USA). RT-qPCR was carried out in a reaction volume of 20 µL with Roche SYBR Green PCR Kit (Roche, Hercules, CA, USA) as follows: 95 °C for 5 min, followed by 40 cycles of 95 °C for 5 s, 60 °C for 15 s, and 72 °C for 15 s. GAPDH gene was selected as internal control to normalize cDNA input. Three technical replicates were conducted on each cDNA, and average Ct value was used for further analysis. Amplification efficiencies were close to 100%. Relative expression values were calculated using the 2−ΔΔCt method.
Gene ontology (GO) terms and Kyoto Encyclopedia of genes and genome (KEGG) pathways
DEGs with criteria of q < 0.05 and |fold change| (|FC|) > 1.5 were selected to annotate biological functions. GO term and KEGG pathway analyses of DEGs were performed on an online bioinformatics system (DAVID) (https://david.ncifcrf.gov/)47. False discovery rate (FDR) was determined to correct p-values. Significant GO categories were enriched (P < 0.01, FDR < 0.05), whereas only pathway categories with P < 0.05 were selected.
Protein–Protein interaction (PPI) network analysis
DEGs were inputted into STRING v10 to generate a PPI network with a reliability score higher than 0.4. STRING database was used to predict physical/functional PPIs from four sources, namely, text mining, high-throughput experiments, coexpression, and databases48.
Statistical analyses
Student’s t-test was conducted to analyze antibody levels (ELISA) and mRNA expression levels (RT-qPCR) between the two groups. A p-value less than 0.05 was considered statistically significant for all analyses.
References
Neumann, E. J. et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. Journal of the American Veterinary Medical Association 227, 385–392 (2005).
Holtkamp, D. J. et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod 21, 72–84 (2013).
Nieuwenhuis, N., Duinhof, T. F. & van Nes, A. Economic analysis of outbreaks of porcine reproductive and respiratory syndrome virus in nine sow herds. Vet Rec 170, 225−+, https://doi.org/10.1136/vr.100101 (2012).
Straw, B. E., Zimmerman, J. J., D’Allaire, S. and Taylor, D. J. Diseases of Swine. 9th edn, (Blackwell Publishing Professional, 2006).
Labarque, G., Van Gucht, S., Van Reeth, K., Nauwynck, H. & Pensaert, M. Respiratory tract protection upon challenge of pigs vaccinated with attenuated porcine reproductive and respiratory syndrome virus vaccines. Vet Microbiol 95, 187–197, https://doi.org/10.1016/S0378-1135(03)00157-3 (2003).
van Woensel, P. A., Liefkens, K. & Demaret, S. European serotype PRRSV vaccine protects against European serotype challenge whereas an American serotype vaccine does not. Adv Exp Med Biol 440, 713–718 (1998).
Shibata, I., Mori, M. & Uruno, K. Experimental infection of maternally immune pigs with porcine reproductive and respiratory syndrome (PRRS) virus. The Journal of veterinary medical science 60, 1285–1291 (1998).
Qiao, S. et al. Porcine FcgammaRIIb mediates enhancement of porcine reproductive and respiratory syndrome virus (PRRSV) infection. PloS one 6, e28721, https://doi.org/10.1371/journal.pone.0028721 (2011).
Dewey, C. E., Wilson, S., Buck, P. & Leyenaar, J. K. The reproductive performance of sows after PRRS vaccination depends on stage of gestation. Preventive veterinary medicine 40, 233–241 (1999).
Liew, C. C., Ma, J., Tang, H. C., Zheng, R. & Dempsey, A. A. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med 147, 126–132, https://doi.org/10.1016/j.lab.2005.10.005 (2006).
Kohane, I. S. & Valtchinov, V. I. Quantifying the white blood cell transcriptome as an accessible window to the multiorgan transcriptome. Bioinformatics 28, 538–545, https://doi.org/10.1093/bioinformatics/btr713 (2012).
Moller-Levet, C. S. et al. Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. P Natl Acad Sci USA 110, E1132–E1141, https://doi.org/10.1073/pnas.1217154110 (2013).
Gitlin, L. et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 103, 8459–8464, https://doi.org/10.1073/pnas.0603082103 (2006).
Perry, A. K., Chen, G., Zheng, D., Tang, H. & Cheng, G. The host type I interferon response to viral and bacterial infections. Cell research 15, 407–422, https://doi.org/10.1038/sj.cr.7290309 (2005).
Badaoui, B. et al. Pig immune response to general stimulus and to porcine reproductive and respiratory syndrome virus infection: a meta-analysis approach. BMC genomics 14, 220, https://doi.org/10.1186/1471-2164-14-220 (2013).
Islam, M. A. et al. Deciphering transcriptome profiles of peripheral blood mononuclear cells in response to PRRSV vaccination in pigs. BMC genomics 17, 641, https://doi.org/10.1186/s12864-016-2849-1 (2016).
Botner, A. et al. Appearance of acute PRRS-like symptoms in sow herds after vaccination with a modified live PRRS vaccine. Vet Rec 141, 497–499 (1997).
Key, K. F. et al. Development of a heteroduplex mobility assay to identify field isolates of porcine reproductive and respiratory syndrome virus with nucleotide sequences closely related to those of modified live-attenuated vaccines. J Clin Microbiol 41, 2433–2439, https://doi.org/10.1128/Jcm.41.6.2433-2439.2003 (2003).
Boddicker, N. J. et al. Genome-wide association and genomic prediction for host response to porcine reproductive and respiratory syndrome virus infection. Genetics, selection, evolution: GSE 46, 18, https://doi.org/10.1186/1297-9686-46-18 (2014).
Sharma, N., Akhade, A. S. & Qadri, A. Sphingosine-1-phosphate suppresses TLR-induced CXCL8 secretion from human T cells. Journal of leukocyte biology 93, 521–528, https://doi.org/10.1189/jlb.0712328 (2013).
Barton, G. M. & Medzhitov, R. Toll-like receptor signaling pathways. Science 300, 1524–1525, https://doi.org/10.1126/science.1085536 (2003).
Shinya, K. et al. The TLR4-TRIF Pathway Protects against H5N1 Influenza Virus Infection. Journal of virology 86, 19–24, https://doi.org/10.1128/Jvi.06168-11 (2012).
Delaloye, J. et al. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS pathogens 5, e1000480, https://doi.org/10.1371/journal.ppat.1000480 (2009).
Li, M. O. & Rudensky, A. Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol 16, 220–233, https://doi.org/10.1038/nri.2016.26 (2016).
Oikonomopoulou, K., Ricklin, D., Ward, P. A. & Lambris, J. D. Interactions between coagulation and complement-their role in inflammation. Semin Immunopathol 34, 151–165, https://doi.org/10.1007/s00281-011-0280-x (2012).
Fang, L. et al. Genome-Wide Transcriptional and Post-transcriptional Regulation of Innate Immune and Defense Responses of Bovine Mammary Gland to Staphylococcus aureus. Frontiers in cellular and infection microbiology 6, 193, https://doi.org/10.3389/fcimb.2016.00193 (2016).
Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nature immunology 5, 987–995, https://doi.org/10.1038/ni1112 (2004).
Pashine, A., Valiante, N. M. & Ulmer, J. B. Targeting the innate immune response with improved vaccine adjuvants. Nature medicine 11, S63–68, https://doi.org/10.1038/nm1210 (2005).
Ait-Ali, T. et al. Distinct functional enrichment of transcriptional signatures in pigs with high and low IFN-gamma responses after vaccination with a porcine reproductive and respiratory syndrome virus (PRRSV). Veterinary research 47, 104, https://doi.org/10.1186/s13567-016-0392-3 (2016).
Strieter, R. M., Kunkel, S. L. & Bone, R. C. Role of tumor necrosis factor-alpha in disease states and inflammation. Critical care medicine 21, S447–463 (1993).
Corradi, A., Ferrari, L. & Borghetti, P. Parameters for evaluating the cell-mediated immune response during viral infection: diagnostic and prognostic applications. Veterinary research communications 31(Suppl 1), 103–107, https://doi.org/10.1007/s11259-007-0075-7 (2007).
Murtaugh, M. P. & Foss, D. L. Inflammatory cytokines and antigen presenting cell activation. Veterinary immunology and immunopathology 87, 109–121 (2002).
Salazar-Mather, T. P. & Hokeness, K. L. Cytokine and chemokine networks: pathways to antiviral defense. Curr Top Microbiol Immunol 303, 29–46 (2006).
Kang, S. M. et al. c-Fos regulates hepatitis C virus propagation. FEBS letters 585, 3236–3244, https://doi.org/10.1016/j.febslet.2011.08.041 (2011).
Kovary, K. & Bravo, R. The Jun and Fos Protein Families Are Both Required for Cell-Cycle Progression in Fibroblasts. Molecular and cellular biology 11, 4466–4472 (1991).
Fan, F. J. et al. The AP-1 Transcription Factor JunB Promotes Multiple Myeloma (MM) Cell Proliferation, Survival and Drug Resistance in the Bone Marrow Microenvironment. Blood 124 (2014).
Schreiber, M. et al. Control of cell cycle progression by c-Jun is p53 dependent. Gene Dev 13, 607–619, https://doi.org/10.1101/Gad.13.5.607 (1999).
Mariani, O. et al. JUN oncogene amplification and overexpression block adipocytic differentiation in highly aggressive sarcomas. Cancer cell 11, 361–374, https://doi.org/10.1016/j.ccr.2007.02.007 (2007).
Adiseshaiah, P., Li, J., Vaz, M., Kalvakolanu, D. V. & Reddy, S. P. ERK signaling regulates tumor promoter induced c-Jun recruitment at the Fra-1 promoter. Biochemical and biophysical research communications 371, 304–308, https://doi.org/10.1016/j.bbrc.2008.04.063 (2008).
Niyazoglu, M. et al. Association of PARP-1, NF-kappaB, NF-kappaBIA and IL-6, IL-1beta and TNF-alpha with Graves Disease and Graves Ophthalmopathy. Gene 547, 226–232, https://doi.org/10.1016/j.gene.2014.06.038 (2014).
Verma, I. M., Stevenson, J. K., Schwarz, E. M., Vanantwerp, D. & Miyamoto, S. Rel/Nf-Kappa-B/I-Kappa-B Family-Intimate Tales of Association and Dissociation. Gene Dev 9, 2723–2735, https://doi.org/10.1101/gad.9.22.2723 (1995).
Schimke, L. F. et al. A Novel Gain-of-Function IKBA Mutation Underlies Ectodermal Dysplasia with Immunodeficiency and Polyendocrinopathy. J Clin Immunol 33, 1088–1099, https://doi.org/10.1007/s10875-013-9906-1 (2013).
Hansen, J. D., Vojtech, L. N. & Laing, K. J. Sensing disease and danger: A survey of vertebrate PRRs and their origins. Developmental and comparative immunology 35, 886–897, https://doi.org/10.1016/j.dci.2011.01.008 (2011).
Kittawornrat, A. et al. Detection of Porcine reproductive and respiratory syndrome virus (PRRSV) antibodies in oral fluid specimens using a commercial PRRSV serum antibody enzyme-linked immunosorbent assay. J Vet Diagn Invest 24, 262–269, https://doi.org/10.1177/1040638711435679 (2012).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111, https://doi.org/10.1093/bioinformatics/btp120 (2009).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57, https://doi.org/10.1038/nprot.2008.211 (2009).
Franceschini, A. et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research 41, D808–815, https://doi.org/10.1093/nar/gks1094 (2013).
Acknowledgements
We are very grateful to Prof. Junjie Xu, Associate Prof. Xin Wang, and two anonymous reviewers for their constructive comments and suggestion on this manuscript. This work was supported by the Beijing Innovation Consortium of Agriculture Research System (BAIC02-2016), the National Natural Science Foundation of China (31572361), the Twelfth Five-Year Plan for National Science and Technology Projects in Rural Areas (2011BAD28B01), the Project for Science and Technological Innovation of Yunnan Province (2010AB001), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1191).
Author information
Authors and Affiliations
Contributions
Y.Y. and W.C. conceived the experiment. Y.T., Z.F., Z.L., T.Z., S.Y., W.Y., Y.Y. and L.L. collected samples. Y.T. performed experiments. Y.Y., Y.T., H.W. and W.C. designed the study. Y.T., H.W., N.C., Y.Y., A.H. and W.C. analyzed the results and wrote the manuscript. Z.W. collaborated in the experiments. All authors reviewed and contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, T., Zhang, F., Zhai, L. et al. Transcriptome of Porcine PBMCs over Two Generations Reveals Key Genes and Pathways Associated with Variable Antibody Responses post PRRSV Vaccination. Sci Rep 8, 2460 (2018). https://doi.org/10.1038/s41598-018-20701-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20701-w
- Springer Nature Limited