
Abstract
Globally, downy mildews are among the important foliar diseases of maize that cause significant yield losses. We conducted a genome-wide association study for sorghum downy mildew (SDM; Peronosclerospora sorghi) resistance in a panel of 368 inbred lines adapted to the Asian tropics. High density SNPs from Genotyping-by-sequencing were used in GWAS after controlling for population structure and kinship in the panel using a single locus mixed model. The study identified a set of 26 SNPs that were significantly associated with SDM resistance, with Bonferroni corrected P values ≤ 0.05. Among all the identified SNPs, the minor alleles were found to be favorable to SDM resistance in the mapping panel. Trend regression analysis with 16 independent genetic variants including 12 SNPs and four haplotype blocks identified SNP S2_6154311 on chromosome 2 with P value 2.61E-24 and contributing 26.7% of the phenotypic variation. Six of the SNPs/haplotypes were within the same chromosomal bins as the QTLs for SDM resistance mapped in previous studies. Apart from this, eight novel genomic regions for SDM resistance were identified in this study; they need further validation before being applied in the breeding pipeline. Ten SNPs identified in this study were co-located in reported mildew resistance genes.
Similar content being viewed by others


Introduction
Maize is the world’s leading cereal in terms of production, with 1,016 million metric tons (MMT) produced on 184 million hectares (M ha) globally1. The crop is produced in both temperate and tropical zones across all continents. Tropical maize is largely (about 80%) grown under rainfed conditions in sub-Saharan Africa, South and Southeast Asia, and Latin America, and is particularly vulnerable to an array of abiotic and biotic stresses2. In Asia, eight major maize growing countries (China, India, Indonesia, Nepal, Pakistan, Philippines, Thailand and Vietnam) together produce 98 per cent of Asia’s maize and 28 per cent of the world’s maize3. However, maize grown in the Asian tropics, especially during the monsoon season, is constrained an array of diseases, including downy mildews, leaf blights, rusts, stalk rots and ear rots. Among these diseases, the downy mildews (DMs) are widely prevalent across Asia, significantly affecting maize production and productivity. Globally, DM affected areas, including lowland tropical, subtropical, mid-altitude, transition zone and highland environments, suffer significant economic losses, reported to be as high as 30%4. Heavy losses (as high as 100%) due to different DM pathogens were reported on maize in several countries in Asia, including Indonesia, India, Nepal, Philippines and Thailand5. Disease transmission occurs mainly through oospores which survive in the soil6, but it can also be transmitted from plant to plant by airborne conidia or through infected seed. Because of the systemic nature of the disease, infected susceptible varieties usually die as the plants get infected at a very early stage, i.e., immediately after seedling emergence. When infection occurs during later stages of plant growth, the plants may survive, but without ear development. At least six pathogens that cause downy mildew are reported to infect maize in Asia, including sorghum downy mildew (SDM) caused by Peronosclerospora sorghi (Weston & Uppal).
Sorghum DM has a global distribution and is found at different altitudes and in different agro-ecological environments in the American, African, Australian and Asian subcontinents7,8. It causes considerable yield losses (as high as 30–40%) in several maize growing states of India, particularly Karnataka, Tamil Nadu and Rajasthan9. Despite the introduction of DM resistant cultivars in some countries and the extensive use of the systemic fungicide Metalaxyl, severe incidences of DM diseases are still reported in localized areas10. The pathogen’s resistance to Metalaxyl was reported by Raymundo11, and was also observed at Mandya, Karnataka, which is an SDM hot spot. Moreover, seed treatments with systemic fungicides make the seeds more expensive and beyond the reach of resource-poor farmers. Therefore, developing and deploying DM-resistant maize varieties is considered a cost-effective and environmentally-safer alternative for effectively overcoming the DM problem in maize.
Extensive studies on downy mildew resistance in maize were undertaken earlier through the CIMMYT co-ordinated Asian Maize Biotechnology Network (AMBIONET) in four Asian countries, namely India, Indonesia, Philippines and Thailand12,13,14. Limited variability was observed for resistance to some of the DMs, especially SDM13,14. Considering the quantitative nature of DM resistance, several groups undertook QTL mapping experiments in different countries with diverse mapping populations5,15,16,17,18,19. George et al.5 identified QTLs involved in resistance to all the major DM pathogens in Asia. Six genomic regions on chromosomes 1, 2, 6, 7 and 10 were identified for downy mildew resistance, including a major QTL on chromosome 6 that influenced resistance to five different DMs in the Asian region. Agrama et al.15 identified three QTLs for SDM resistance; two of them were closely mapped on chromosome 1 and the third one on chromosome 9. Sabry et al.17 detected three putative QTLs, one on chromosome 2 with large effect, and two on chromosomes 3 and 9 with minor effects. Studies by Jampatong et al.18 and Lohithaswa et al.19 identified QTLs for SDM resistance on chromosomes 2, 3, 4, 5, 6 and 9. However, the QTL mapping approach has several limitations, for example, it confines QTL to a 10–20 cM interval because of the limited number of recombination events during the development of the mapping population; also, only two alleles from each of the parents of the mapping population at any locus are studied in a biparental mapping population20,21.
A genome-wide association study (GWAS) is a powerful tool that identifies specific allele variants that confer improved resistance to various diseases in maize. It has been used in studying resistance to Fusarium ear rot22, gray leaf spot23, head smut24, northern corn leaf blight25, southern corn leaf blight26, sugarcane mosaic virus27, maize streak virus28 and maize lethal necrosis29. In Thailand, an association study on 60 inbred lines using 48 SSR markers identified the marker umc1033 as being significantly associated with SDM resistance30. As opposed to QTL mapping, which is a powerful method that identifies genomic regions and spans broad chromosome regions in a bi-allelic context, the main advantages of association mapping are increased mapping resolution, reduced research time, and the possibility of identifying a greater number of favorable alleles responsible for the trait of interest31.
CIMMYT has a long history of breeding for downy mildew resistance (DMR); as a result, specific populations like populations 22 (Mezcla Tropical Blanca), 28 (Amarillo Dentado), and 31 (Amarillo Cristalino-2) have been improved for DMR32. Four genetically broad-based populations were developed for DMR by the CIMMYT Asian Regional Maize Program (ARMP) and two sets of CIMMYT maize lines (CMLs) resistant to DM were released for use in DM resistance breeding in Asia. CIMMYT developed a panel of elite Asia-adapted tropical and subtropical maize inbred lines, called CIMMYT Asia Association Mapping (CAAM) panel. This panel enables association mapping studies for an array of major agronomic, biotic and abiotic stress tolerance traits in the region. The panel is particularly important for SDM analysis, considering the lack of adequate genetic variability for the trait in the region’s maize germplasm. DM resistant inbred lines developed in Thailand and Philippines as well as lines developed from DM resistant maize populations, such as early white DMR (EW-DMR), early yellow DMR (EY-DMR), late white DMR (LW-DMR) and late yellow DMR (LY-DMR) are included in the CAAM panel. Improved lines developed by the CIMMYT-Asia program by crossing elite materials tolerant to abiotic stresses like drought, heat and excess moisture with DMR lines were also included in the panel. Considering the CIMMYT ARMP’s history of incorporating resistance to DM, the CAAM panel is expected to have greater frequency of alleles favourable for DMR in the Asian region. The main objective of the present study was to identify genomic regions associated with SDM resistance and to validate previously reported QTL responsible for DM resistance in maize.
Results
Phenotypic evaluation for SDM resistance
The CAAM panel with a set of 368 lines was evaluated against SDM at Mandya, India, during the years 2012 and 2015 under artificial epiphytotic conditions. The disease severity was high in both years, with the standard susceptible check CM500 showing 100 per cent mortality. The mean disease severity of the trials was also high, with 96.28 and 89.21 per cent during 2012 and 2015, respectively. A Box-Cox transformation lambda (λ) value of 0.96 was obtained and used to transform the data. There was significant genotypic variance (p ≤ 0.001) for the trait in the panel and the broad-sense heritability estimated by the combined analysis was high (0.72). Heritability estimates for 2012 and 2015 were 0.92 and 0.74, respectively (Table 1).
Population structure and linkage disequilibrium (LD) decay
Principal component analysis using 63,546 SNPs (CR 0.9, and MAF ≥0.1 with LD pruning) did not reveal a clear population structure with the first three principal components (Fig. 1). All the important tester lines from various maturity groups used in the Asian tropics and mapped within the 3D PCA plot revealed the spread of SDM resistant lines within the studied panel. Two early maturing testers developed in the Asian tropics, CML470 and CML474, clearly grouped into one cluster in the PCA plot. The Scree plot plotted (Supp. Figure 1) with all the eigen values suggested 10 principal components that should be considered in the GWAS to account for the population structure.

Population structure based on the first three eigenvalues of principal component (PC) analysis of the CAAM panel using 63,546 SNPs. Clusters in dark blue represent moderate resistant (MR) lines, florescent green (CL02450) and black (CML474) dots represent tester lines of CIMMYT’s heterotic group A, red dots are moderately susceptible (MS) lines, pink are downy mildew resistant (DMR) lines, and clusters of florescent blue dots are susceptible lines (SUS), while yellow (CML451) and green (CML470) dots represent CIMMYT’s heterotic group B tester lines.
Genome-wide LD across 37,043 SNPs with minor allele frequency (MAF) ≥0.3 was investigated in the CAAM panel. The genome-wide LD decay was plotted as LD (r2) between adjacent pairs of markers versus distance in kb which showed that LD decay was 15.53 Kb at r2 = 0.1 and 5.39 kb at r2 = 0.2, (Fig. 2). Chromosome-wise LD analysis showed fastest LD decay in chromosome 5 (10.35 Kb at r2 = 0.1 and 3.59 Kb at r2 = 0.2), followed by chromosome 2 (11.39 Kb at r2 = 0.1 and 3.95 Kb at r2 = 0.2). However, the slowest LD decay was observed on chromosome 8 (28.8 Kb at r2 = 0.1 and 9.99 Kb at r2 = 0.2), followed by chromosome 4 (22.14 Kb at r2 = 0.1 and 7.68 Kb at r2 = 0.2) in this panel (Supp. Table 1).

Linkage disequilibrium (LD) plot representing the average genome-wide LD decay of the CAAM panel using 37,043 genome-wide SNP markers. The values on the Y-axis represent the squared correlation coefficient r2 and the X-axis represents the genetic distance in kilobases (Kb).
Marker-trait associations for SDM resistance
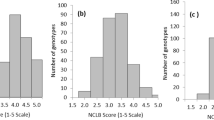
A subset of markers with a call rate ≥0.7 and minor allele frequency ≥0.03 were used for association studies. With the total 333,039 SNPs fulfilling these criteria, a naïve association analysis with only these SNPs was carried out. The Quantile-quantile plot (QQ plot) drawn using the observed against the expected -log10 (P values) showed significant genomic inflation (Fig. 3a). Following this, to control the genomic inflation, a general linear model analysis including the genotypes and 10 PCs (G + Q) was carried out, which also showed high genomic inflation in the QQ plot. Further, a single marker mixed linear model including the genotypes, 10 PCs and the kinship matrix (G + Q + K) was able to keep the genomic inflation within acceptable limits. Based on this analysis, 26 significant SNP hits for disease response were identified that had Bonferroni-corrected P values ≤ 0.05 on chromosomes 1, 2, 3, 5, 6, 8 and 9 (Fig. 3b; Supp. Table 2). SNP S2_6154311 on chromosome 2 showed the strongest association with the lowest P value (1.0E-012) and Bonferroni-corrected P value (1.0E-06). The phenotypic variance explained by significant SNPs ranged from 5.50 to 13.07%; SNP S2_6154311 and S1_88004654 explained the highest and lowest proportion of variances, respectively, from among the SNPs associated with DM resistance (Supp. Table 2). Interestingly, in all 26 associations identified, the minor allele was found to contribute positively towards lowering SDM incidence (Supp. Table 2). Lines developed by crossing DM resistant lines with elite inbred lines (including CMLs) developed from the DM resistant populations were found to carry the maximum number of favourable alleles in the CAAM panel. A number of these SNPs were within the same chromosomal bins as the QTLs previously mapped for SDM resistance in multiple studies (Table 2). Annotated genes in the reference genome of B73 RefGen_V2 (available at http://ensembl.gramene.org/Zea_mays) carrying the 26 associated SNPs showed many genes reported to be associated with broad spectrum disease resistance or resistance specifically to mildews (Table 3).

(a) Inflation depicted by Q-Q plots of observed versus expected −log10 (P values) plots for SDM using the naïve association model (G-test), GLM (G + Q) and MLM (G + Q + K); G = genotype (fixed), Q = ten principal components (fixed), K = kinship matrix (random); (b) Highly significant SNPs identified from MLM model using Manhattan plot, plotted with the individual SNPs on the X-axis and −log10 P value of each SNP on the Y-axis. The blue vertical lines show the significant associations at previously reported QTLs.
Haplotype estimation and trend regression analysis
The 26 SNPs identified as being significantly associated with SDM incidence were spread across seven chromosomes. Some of these SNPs had physical coordinates (based on B73 RefGen_V2) that were in close proximity, and some of them were SNPs located inside the same annotated genes. Hence haplotype block estimation analysis was carried out to determine whether these SNPs form haplotype blocks using the haplotype defining algorithm to minimize historical recombinations. In this analysis, four haplotype blocks were detected on three chromosomes from among the 26 SNPs associated with SDM incidence. Based on this analysis, 16 independent variants (including both SNPs and haplotype blocks) were considered for further analysis. Trend regression analysis on these 16 variants against SDM incidence estimated the proportion of variance explained by each of these to range from 3.61 to 26.74% (Table 4). S2_6154311 contributed 26.74% of the variation, followed by Hap-1, which contributed 17.34% of the variation.
Discussion
The downy mildews of maize caused by Peronosclerospora species are a group of economically important and widespread diseases in many tropical and subtropical regions of the world5. The lack of adequate and genetically diverse sources of resistance to DMs has been a major constraint in tropical Asia, especially in the maize growing environments of South and Southeast Asia. Introgressing DM resistance in the breeding pipeline is one of the priorities of CIMMYT’s maize breeding programs as well as National Agricultural Research System (NARS) partners in Asia. Breeding for DM resistance, especially phenotyping under artificial epiphytotic conditions with high disease pressure is cost-intensive. Therefore, identifying, validating and deploying molecular markers with DM resistance can increase the efficiency of developing DM resistant tropical and subtropical maize germplasm especially in those countries where DM resistance is mandatory for the release of maize varieties.
The CAAM panel used in this study includes tropical/subtropical inbred lines from CIMMYT breeding programs in Asia, Mexico, Kenya, Zimbabwe and Colombia, that are also adapted to the Asian tropics. Derivatives of several lines developed by crossing CIMMYT populations with Suwan sources of DMR from Thailand, and the Asian mildew acid tolerant late (AMATL) populations are included in the CAAM panel. This panel is an excellent resource to study genomic regions for SDM resistance considering CIMMYT ARMP’s breeding efforts aimed at improving DM resistance of Asian maize germplasm. Heritability estimates of the trials evaluated at Mandya were high (0.74–0.92); for the two years (2012 and 2015) combined, the SDM scores ranged from 7.81 to 100%, indicating there is genetic variability for SDM in the CAAM panel. Genotypic variance was also highly significant for SDM resistance in the CAAM panel. Lines showing the lowest disease percentage were CML433, (CML474/S92145–2EV-7-3-B*5)-F2-25-1-B, CML472, CA14709-4-7-5-1, which were either developed for resistance to DM for the Asian region or derived from crosses with DMR lines.
A GBS protocol commonly used by the maize research community was applied in this study, which produced a partially imputed SNP dataset, calling about a million SNPs. For principal component and kinship analysis, high quality SNPs with filtering criteria of CR ≥0.9 and MAF ≥0.1, and pruned at an r2 threshold of 0.5 were used. It was necessary to reduce the number of SNPs for computational efficiency, but by selecting SNPs that have higher MAF and by LD-pruning, due care was taken to avoid confounding of the calculations due to large blocks of SNPs that have strong LD with each other33. The PCA showed only a moderate population structure within the CAAM panel, with no clear grouping of the lines into well-defined clusters. The first three PCs explained only 39.3% of the variation in the panel. The DMR lines did not fall into a particular cluster, but were scattered among the groups. This was also observed by George et al.14 who studied the diversity pattern of a group of Asian inbred lines along with CIMMYT’s tropical and subtropical lines, and suggested that the tropical and subtropical lines in the Asian region possess significant genetic diversity that did not allow a clear-cut distinction into well-defined clusters. Warburton et al.34 observed that the CIMMYT populations that served as germplasm sources for many Asian lines had a large amount of diversity within, rather than between, source populations. Due to this heterogeneous nature of the CIMMYT populations, they suggested that it would be difficult to find a well-defined population structure in the Asian maize lines. Genetic diversity is directly related to LD, which in turn affects LD based association mapping; the larger the LD blocks, the slower the LD decay, which will result in lower mapping resolution35,36. For estimating genome-wide LD, we used a subset of the SNP data filtered for CR ≥0.9 and MAF ≥0.3. This was also done for computational efficiency required for the analysis, with due care taken for SNP selection to represent all population structure groups by way of increasing MAF. The same set of SNPs used for PCA were not used in this analysis as the former set had already been pruned at an r2 threshold of 0.5. In the CAAM panel, the average LD decay of the panel was 15.53 kb at r2 = 0.1 and 5.39 kb at r2 = 0.2, with localized differences within and between chromosomes. The slowest LD decay was observed in chromosome 8, and a similar observation was made by Suwarno et al.35, while studying a tropical/subtropical carotenoid association mapping panel. LD decay in tropical maize germplasm is generally found to be high as compared to temperate maize germplasm. Lu et al.36 suggested the LD decay distance in temperate maize germplasm (10 to 100 kb) was 2 to 10 times higher than that of tropical maize germplasm (5 to 10 kb). Similarly, Romay et al.37 studied 2,815 inbred lines and found LD decay of 1 kb for tropical maize germplasm and 10 kb for temperate germplasm. The high LD decay of tropical maize germplasm suggested that it has a broader genetic base resulting from high recombination events and might have more rare alleles than temperate lines38; this provides an excellent opportunity for maize breeders to select germplasm that combines higher grain yield with disease resistance and relevant abiotic stress tolerance traits.
QTL mapping for DM resistance was reported by many groups on multiple DMs in diverse germplasm and environments5,15,16,17,18,19,31. A genome-wide study using high density marker information from widely adapted maize lines has not yet been reported. On Asia-adapted CIMMYT maize lines genotyped at high density, three methods of association testing were employed in this study; the single locus mixed linear model was eventually selected for reporting results, as it was successful in controlling genomic inflation by correcting for population structure and familial relatedness39. A set of 26 SNPs on seven chromosomes were found to be significantly associated with SDM resistance (Supp. Table 2). Since many of the SNPs identified were having strong LD in average pairwise LD analysis (Sup. Table 3), we carried out haplotype block detection among those SNPs. We estimated the haplotype frequencies and identified 16 genomic regions in linkage equilibrium, either as SNPs or haplotype blocks. The SNP alleles/haplotypes were regressed against the phenotype to ascertain the genomic regions that contributed most significantly to DM resistance. SNP/haplotype-trait associations identified were assessed by way of previously reported QTL in these genomic regions, and functional domains in the predicted gene models carrying the SNPs. This will further be followed up by validation in independent bi-parental populations, before deployment in breeding pipeline.
The most significant association was found to be on chromosome 2 (SNP S2_6154311; Bonferroni-corrected p value = 3.92E-23) contributing 26.70% to phenotypic variation. The SNP S2_6154311 was found to be located in bin 2.02 according to the Maize Genome Data Base (WWW.maizegdb.org, Maize B73 RefGen_V2). Lohithaswa et al.19 identified a QTL for SDM resistance in the chromosomal bin 2.02 between SSR markers umc2363 and umc1165 (Table 3). Nair et al.16 identified QTL on chromosome 3, bin 3.04/3.05 (between SSR markers umc1223 and bnlg420) in a backcross mapping population that was also phenotyped at Mandya for SDM resistance. Jampatong et al.18 also identified a QTL on chromosome 3.04 for SDM resistance in their study on an F2:3 mapping population. Our study also identified two highly significant (Bonferroni value ≤ 0.05) SNPs, S3_28841309 and S3_51326624, on chromosome 3 (bin 3.04) for SDM resistance, explaining 9.20 and 13.80% of the phenotypic variance, respectively. This particular chromosomal bin was found to be harbouring clusters of disease resistance genes40 for as many as six of the 11 diseases in a meta-analysis study41. Phumichai et al.30 identified significant SSR marker umc1033 linked with DM resistance on chromosome 9 (bin 9.02) in the association studies. In our study, SNP S9_16183526 (Bonferroni value ≤ 0.05) located on chromosome 9 in bin 9.02 was identified for DM resistance.
Another most important genomic region for DMR is on chromosome 6, which has been identified in many studies5,16,18. In our study, nine SNPs were found to be significantly associated with SDM resistance on chromosome 6, and seven of them formed two haplotype blocks. One SNP and two haplotype blocks were located on bin 6.05, where a significant QTL was identified by George et al.5 for multiple DM resistance against five different pathogens including the SDM pathogen. Nair et al.16 also identified a major QTL on chromosome 6.05 for SDM and RDM in their backcross population. The significant associations identified in this region in our GWAS add strength to the earlier findings, and could be taken up for further analysis and potential application in maize breeding programs. Another SNP identified on chromosome 6, SNP S6_28477908, is located in bin 6.01; two earlier studies identified SDM resistance QTL in this bin18,19. Chromosome 6 is also reported to harbour clusters of resistance genes for multiple biotic stresses, and bin 6.01 was reported to have an array of resistance genes, as in the case of chromosomal bin 3.0440. The other significant associations identified in this study seem to be novel, and were not found in the previous QTL mapping studies on SDM resistance (Table 4).
Candidate genes associated with SDM
Generally, plant disease resistance is achieved through various genes acting on mechanisms ranging from innate plant immunity to broad spectrum basal defence mechanisms to specialized mechanisms targeting specific pathogens. Many of the SNPs that were found to be associated with SDM resistance in this study were located in annotated genes (B73 RefGen_V2) with functional domains that were previously reported to influence disease resistance in various crops (Table 3). Mildews, including both downy mildews and powdery mildews, are caused by obligate biotrophic pathogens that require a live host for their survival. At least 10 out of the 26 SNPs identified as significant in the present study were localized within five different genes that had been previously implicated in mildew resistance in different crops. A novel haplotype identified on chromosome 1 was located within GRMZM2G040441, which has an active MLO-like domain. An MLO protein was cloned initially from barley as the resistance gene against powdery mildew42,43, and thereafter, many orthologs and paralogs for the MLO gene were identified and characterized in great detail in many plants44. MLO proteins are reported to have characteristic domain features which aid the defence signalling mechanism. Though we have not encountered reports on MLO-like proteins involved in downy mildew resistance, it would be worthwhile to investigate the role of MLO-like proteins in SDM resistance in maize.
Significantly associated SNPs on chromosome 6 were located in four genes, three of which are adjacent genes predicted on bin 6.05. Whether the association of many SNPs in close proximity is due to high LD in the region or whether there is a cluster of genes operating towards a resistance reaction needs further investigation. Three significant associations on chromosome 6 were discovered within the Cupin super family gene. Germins, which are part of this super family, have been reported to be involved in powdery mildew resistance in wheat and barley45,46. Similarly, three significantly associated SNPs were found be located within GRMZM2G108057, putatively coding for a cation transporting ATPase. Regulation of cation ATPase is involved in one of the earliest cellular responses during activation of plant immune responses by modulation of extracellular pH and acidification of the apoplast, which was observed in incompatible interactions of barley plants with the powdery mildew fungus Blumeria graminis 47. Another closely located gene, GRMZM2G479608, is predicted to code for a transmembrane Bax inhibitor motif containing protein 4. In barley, over-expression of the Bax Inhibitor 1 gene was found to breakdown mlo mediated penetration resistance against powdery mildew48. Watanabe et al.49 concluded that Bax Inhibitor 1 was a conserved plant cell death suppressor, which is a key molecular switch downstream from a range of stress signals. On chromosome bin 6.01, a significant association was detected with a SNP located in gene GRMZM2G124297 predicted to code for the HECT sub-class of E3 Ubiquitin ligases. This class of proteins have been reported to provide innate immunity in plants50. E3 Ubiquitin ligases were also found to be involved in SAR independent resistance to the downy mildew pathogen in Arabidopsis thaliana 51,52. As many as eight novel associations were identified in this study, and five of them were within predicted genes with functional domains implicated in disease resistance. Though the correlations identified in association studies cannot be dubbed as causations, associated SNPs on genes previously reported to be trait related add much value and strength to the results to carry forward with independent validation in biparental populations and functional studies of the candidate genes.
Conclusion
The present study is the first one to report significant SNPs and haplotypes associated with SDM resistance in maize, based on GWAS. Six of these SNPs/haplotypes are located within major QTL intervals reported in previous mapping studies on biparental populations, and hence, could be of value to maize breeding programs in tropical Asia that are intensively engaged in developing and deploying DM resistant elite maize varieties. Also, since ten marker-trait associations were identified within candidate genes implicated in disease resistance, especially mildew resistance, the findings from the present study could serve as a strong base for future functional studies to dissect resistance to downy mildews in maize.
Methods
Germplasm
The CAAM panel consisted of 419 inbred lines that were developed and adapted in Asia, based on a global collection of maize germplasm available from CIMMYT. It involved inbred lines with tolerance to abiotic stresses like drought, high temperature and excess moisture, besides Quality Protein Maize (QPM) lines, and lines developed from downy mildew resistant populations in Asia. The CAAM panel includes lines that are adapted to tropical, subtropical, lowland, mid-altitude and highland environments, and were classified as early maturing, intermediate maturing and late maturing based on growing degree days (GDD).
Phenotypic evaluation
The CAAM panel, with a set of 368 lines, was phenotyped for SDM (Peronosclerospora sorghi) at the hot-spot location of Mandya, Karnataka, India (12°N; 76°E; 695 masl; 705 mm/year average annual rainfall) during the monsoon season in 2012 and 2015. Mandya was also identified as the location with the highest DM susceptibility scores, along with Maros in Indonesia, in an earlier study that evaluated a common mapping population across five locations in Asia5. Artificial epiphytotic conditions were created following the infector row method by planting ‘infector rows’ of CM500, 30 days before planting the test entries (in the first week of July). The infector rows were planted on all sides of the experimental block, and also on one bed (2 m wide) after every two beds of test entries (each 3 m wide) to ensure uniform disease pressure on the test entries across the field. Fresh conidia were collected early in the morning (between 2 and 3 am) from 3- to 4-week-old systematically infected maize plants in the DM nursery by suspending the severely infected leaves in water. The conidia suspension was sprayed on seedlings (2–3 leaf stage) in the infector rows using hand-sprayers immediately after collection. The inoculation process was repeated continuously for 7–10 days to achieve good disease incidence on infector rows. Test entries were planted in one-row plots about 30 days after infector row planting (in the first week of August, when relative humidity was >90%), at the time when DM infection on infector row plants reached >70%. Each test entry was planted in standard 3-m-long rows with plant-to-plant spacing of 20 cm and row-to-row spacing of 75 cm in an alpha-lattice design with two replications. Similar to the infector rows, conidial suspension was collected from the DM nursery and sprayed on the test entries and disease indicator rows, starting from 7 to 8 days after seedling emergence, and during the following 7 days to avoid any chance of escape53. Severe SDM incidence (>95%) on all indicator rows of susceptible check entries planted along with the test entries was considered an indication of uniform and sufficient disease pressure throughout the trial.
Disease scoring
Percentage disease incidence was calculated as the number of SDM infected plants at 21 and 35 days after emergence compared to the total number of plants per plot. Disease percentage at 35 days was considered as the final score because the disease score remained constant after 35 days. A DM rating was recorded using a modified rating method suggested for maize54, i.e. 0% = highly resistant, 1–10% = resistant, 11–25% = moderately resistant, 26–50% = moderately susceptible, 51–75% = susceptible, and 76–100% = highly susceptible.
Phenotypic data analysis
The phenotypic data on percentage disease scores were skewed towards susceptibility in the CAAM panel. The data were subjected to Box-Cox power transformations55, which helped the data approach normality. Best Linear Unbiased Estimators (BLUEs) obtained using the software METAR-4.156 from multi-location data analysis were used for GWAS analysis. Analysis of variance components were estimated using Genstat (14th edition)57. Broad-sense heritability of the combined analysis across years was calculated as H2 = σ2 g/(σ2 g + σ2 ge/e + σ2 e/er), where σ2 g, σ2 ge and σ2 e are the genotypic, genotype-by-year interaction and error variance components, respectively, and e and r are the number of years and number of replicates within each year included in the corresponding analysis, respectively.
DNA isolation and genotyping
DNA of all inbred lines was isolated from the leaf samples of 3–4-week-old seedlings using the standard CIMMYT laboratory protocol58. Genotyping was carried out on the GBS platform59 at the Institute of Genomic Diversity, Cornell University, Ithaca, USA. Genomic DNA was digested with the restriction enzyme ApeK1. GBS libraries were constructed in 96-plex and sequenced on Illumina HiSeq. 2000. SNP calling was performed using the TASSEL-GBS pipeline with B73 as the reference genome60 to generate a comprehensive genotype collection, the AllZeaGBSv2.7 Production Build (www.panzea.org). The TASSEL-GBS pipeline is optimized for low sequencing depth (0.5 to 3×) over a large number of markers in a large sample of individuals. This collection included genotypes of more than 60,000 maize samples. In this study, we focused on the subset of 368 lines forming the CAAM panel, and partially imputed raw GBS data were used for further genetic analyses. The original partially imputed data set consisted of 955,690 SNPs across all the chromosomes, which included partially imputed data based on an algorithm that searched for the closest neighbour in small SNP windows across the entire maize database, allowing for a 5% mismatch37. If the requirements were not met, the SNP was not imputed, leaving only about 10% of the data unimputed. For GWAS, filtration criteria of call rate (CR) ≥0.7 and minor allele frequency (MAF) ≥0.03 were used, yielding 333,039 SNPs. For calculating PCA and kinship matrix, high quality SNPs with filtering criteria of CR ≥0.9 and MAF ≥0.1, and pruned at an r2 threshold of ≤0.5 were used for selecting 63,546 SNPs. Filtering criteria of CR ≥0.9 and MAF ≥0.3 generated 37,043 SNPs for the LD adjacent pair analysis used for calculating r2.
Principal component and kinship analysis
The PCA method described by Price et al.61 was implemented in the SNP & Variation Suite (SVS) V_8.6.0 (SVS, Golden Helix, Inc., Bozeman, MT, www. goldenhelix.com). A three-dimensional plot of the first three principal components was drawn to visualize the possible population stratification among the samples. A scree plot was plotted to determine the number of principal components to be included in the GWAS analysis. A kinship matrix was computed from identity-by-state distances matrix as executed in SVS V_8.6.0., where IBS distance = (no. of markers IBS2) + 0.5 × (no. of markers IBS1) no. non-missing markers, where IBS1 and IBS2 are the states in which the two inbred lines share one or two alleles, respectively at a marker62.
LD analysis and haplotype trend regression
The extent of genome-wide and chromosome-wise linkage disequilibrium (LD) was based on adjacent pairwise r2 values (the squared correlation coefficients among alleles at two adjacent SNP markers) between adjacent SNPs among 37,043 high quality SNPs from the GBS data and physical distances between those SNPs63. Nonlinear models with r2 as responses (y) and pairwise distances (x) as predictors were fitted into the genome-wide and chromosome-wise LD data using the ‘nlin’ function in R64. Average pairwise distances in which LD decayed at r2 = 0.2 and r2 = 0.1 were then calculated based on the model given by Hill & Weir65.
LD analysis and haplotype frequency estimation were also done among all the SNPs found to be associated with SDM resistance using the Expectation Maximisation (EM) algorithm66 as implemented in SVS V_8.6.0. Haplotype frequencies were estimated from among the SNPs associated with the trait using 50 EM iterations, an EM convergence tolerance of 0.0001 and a frequency threshold of 0.01. Haplotype blocks were detected based on the block-defining algorithm to minimise historical recombinations67. Trend regression analysis of the haplotypes and SNPs was carried out based on a stepwise regression of the SDM phenotype with the pre-estimated haplotypes with forward elimination and a P-value cut-off of 0.001.
GWAS
GWAS was carried out on the SDM phenotype based on three methods: using only genotype data (G; Naïve model), using genotype data corrected for population structure (G + Q; General linear model (GLM)) and using genotype data corrected for both population structure and kinship (G + Q + K; single locus mixed linear model (MLM)). These analyses used genotype association tests with an additive model for the first two analyses and a mixed model using single locus (EMMAX)68 for the third analysis as implemented in SVS V_8.6.0. The mixed association mapping model used was Y = SNP*β + PC*α + K *µ + ε, where Y = response of the dependent variable (SDM percent), SNP = SNP marker (fixed effects), PC = principal component coordinate from the PCA (fixed effects), K = kinship matrix (random effects), α is the vector of PC, β and µ are the vectors of SNP and K, respectively, and ε is the error. In the linear models, the first 10 principal components were used as covariates. Manhattan plots were plotted using the −log 10 P values of all SNPs used in analysis; Q-Q plots were plotted of the observed −log 10 P values and the expected −log 10 P values to study the genomic inflation.
References
FAO. Food and Agricultural Organisation of the United Nations (FAO), FAO statistical database. http://faostat.faao.org (2013).
Shiferaw, B., Prasanna, B. M., Hellin, J. & Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 3, 307–327 (2011).
Prasanna, B. Maize research-for-development Scenario: Challenges and Opportunities for Asia. (eds Prasanna, B.M., Vivek, B. S., Sadananda, A. R., Zaidi, P. H., Boeber, C., Erenstein, O., Babu, R., Nair, S. K., Gerarad, B., Jat, M. L., Palacios, N., Pixley, K.) 2–11 (12th Asian Maize conference and Expert consultation on Maize for Food, Feed, Nutrition and Environmental Security. Bangkok, Thailand, 2014).
Jeffers, D. et al. Status in breeding for resistance to maize diseases atCIMMYT. (eds Vasal, S. K., Gonzalez, C., XingMing, F.) 257–266 (7th Asian Regional Maize Workshop. PCARRD, Los Baños, Philippines 2000).
George, M. L. C. et al. Identification of QTLs conferring resistance to downy mildews of maize in Asia. 544–551 https://doi.org/10.1007/s00122-003-1280-6 (2003).
Broyles,W. J. Observations on time and location of penetration in relation to amount of damage and chemical control of Physoderma maydis. Phytopathology 8 (1956).
Wongkaew, A. & Phumichai, C. Detection of candidate R genes and single nucleotide polymorphisms for downy mildew resistance in maize inbred lines by association analysis. 109–118 https://doi.org/10.1007/s10681-013-1056-2 (2014).
Lukman, R., Afifuddin, A. & Lubberstedt, T. Plant Pathology & Microbiology Unraveling the Genetic Diversity of Maize Downy Mildew in Indonesia. 4, 2–9 (2013).
Krishnappa, M., Naidu, B. S. & Seetharam, A. Inheritance of host resistance to downy mildew in maize. Crop Improv. 22, 33–37 (1995).
Dalmacio, S. Importance of and growing concern for maize diseases in theAsian region (eds Vasal, S. K., Gonzalez, C., XingMing, F.) 267–276 (7th Asian Regional Maize Workshop. PCARRD, Los Baños, Philippines 2000).
Raymundo, A. D. Downy mildew of maize in Asia: new perspectives in resistance breeding (eds Vasal, S. K., Gonzalez, C., XingMing, F.). 277–284 (7th Asian Regional Maize Workshop. PCARRD, Los Baños, Philippines 2000).
Nair, S. K., Prasanna, B. M., Rathore, R. S. & Setty, T. A. S. Genetic analysis of resistance to sorghum downy mildew and Rajasthan downy mildew in maize (Zea mays L.). Field crop Res 89, 379–387 (2004).
Yen, T. T. O., Prasanna, B. M., Setty, T. A. S., Setty & Rathore, R. S. Genetic variability for resistance to sorghum downy mildew (Peronosclerospora sorghi) and Rajasthan downy mildew (P. heteropogoni) in the tropical/sub-tropical Asian maize germplasm. Euphytica 138, 23–31 (2004).
George, M. L. C., Regalado, E., Warburton, M., Vasal, S. & Hoisington, D. Genetic diversity of maize inbred lines in relation to downy mildew. Euphytica 135, 145–155 (2004).
Agrama, H. A., Moussa, M. E., Naser, M. E., Tarek, M. A. & Ibrahim, A. H. Mapping of QTL for downy mildew resistance in maize. Theor Appl Genet 99, 519–523 (1999).
Nair, S. K. et al. Identification and validation of QTLs conferring resistance to sorghum downy mildew (Peronosclerospora sorghi) and Rajasthan downy mildew (P. heteropogoni) in maize. 1384–1392 https://doi.org/10.1007/s00122-005-1936-5 (2005).
Sabry, A., Jeffers, D., Vasal, S., Frederiksen, R. & Magill, C. A region of maize chromosome 2 affects response to downy mildew pathogens. Theor. Appl. Genet. 113, 321–330 (2006).
Jampatong, C. et al. Mapping of QTL affecting resistance against sorghum downy mildew (Peronosclerospora sorghi) in maize (Zea mays L.). Maydica 58, 119–126 (2013).
Lohithaswa, H. C., Jyothi, K., Kumar, K. R. S., Puttaramanaik & Hittalmani, S. Identification and introgression of QTLs implicated in resistance to sorghum downy mildew (Peronosclerospora sorghi (Weston and Uppal) C. G. Shaw) in maize through marker-assisted selection. J. Genet 94, 741–748 (2015).
Holland, J. B. Genetic architecture of complex traits in plants. Curr. Opin. Plant Biol. 10, 156–161 (2007).
Zhu, C., Gore, M., Buckler, E. S. & Yu, J. Status and Prospects of Association Mapping in Plants. Plant Genome 1, 5 (2008).
Zila, C. T. et al. Genome-wide association study of Fusarium ear rot disease in the USA maize inbred line collection. BMC Plant Biol. 14, 372–376 (2014).
Shi, L. et al. Genetic characterization and linkage disequilibrium mapping of resistance to gray leaf spot in maize (Zea mays L.). Crop J. 2, 132–143 (2014).
Weng, J. et al. Molecular Mapping of the Major Resistance Quantitative Trait Locus qHS2.09 with Simple Sequence Repeat and Single Nucleotide Polymorphism Markers in Maize. Phytopathology 692–699 (2012).
Poland, J. A., Bradbury, P. J., Buckler, E. S. & Nelson, R. J. Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc. Natl. Acad. Sci. USA 108, 6893–8 (2011).
Kump, K. L. et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 43, 163–168 (2011).
Tao, Y. et al. Combined linkage and association mapping reveals candidates for Scmv1, a major locus involved in resistance to sugarcane mosaic virus (SCMV) in maize. BMC Plant Biol. 13, 162 (2013).
Nair, S. K. et al. Fine mapping of Msv1, a major QTL for resistance to Maize Streak Virus leads to development of production markers for breeding pipelines. Theor. Appl. Genet. 128, 1839–1854 (2015).
Gowda, M. et al. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 128, 1957–1968 (2015).
Phumichai, C. et al. Detection and integration of gene mapping of downy mildew resistance in maize inbred lines though linkage and association. Euphytica 187, 369–379 (2012).
Yu, J. & Buckler, E. S. Genetic association mapping and genome organization of maize. Curr. Opin. Plant Biol. 17, 155–160, https://doi.org/10.1016/j.copbio.2006.02.003 (2006).
De Leon, C. & Lothrop, J. Lowland Tropical Germplasm Development in Asia with Emphasis on Selection for Downy Mildew. Vasal, S. K. & S. McLean (eds) The Lowland Tropical Maize Subprogram. Maize Program Special Report. Mexico, D.F.: CIMMYT., 30–46 (1994).
Patterson, N. Price, A. L. Reich, D. Population Structure and Eigenanalysis. PLoS Genet(12) e190. https://doi.org/10.1371/journal.pgen.0020190 (2006).
Warburton, M. L. et al. Genetic characterization of CIMMYT maize inbred lines and open-pollinated populations using large-scale fingerprinting methods. Crop Sci. 42, 1832–1840 (2002).
Suwarno, W. B., Pixley, K. V., Palacios-Rojas, N., Kaeppler, S. M. & Babu, R. Genome-wide association analysis reveals new targets for carotenoid biofortification in maize. Theor. Appl. Genet. 128, 851–864 (2015).
Lu, Y. et al. Comparative SNP and haplotype analysis reveals a higher genetic diversity and rapider LD decay in tropical than temperate germplasm in maize. PLoS One 6 (2011).
Romay, M. C. et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol 14, R55 (2013).
Lu, Y. et al. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor Appl Genet 120, 93–115 (2009).
Yu, J., Holland, J. B., McMullen, M. D. & Buckler, E. S. Genetic design and statistical power of nested association mapping in maize. Genetics 178, 539–551 (2008).
McMullen, M. D. & Simcox, K. D. Genomic organization of disease and insect resistance genes in maize. Mol. Plant Microbe Interact 811–815 (1995).
Wisser, R. J., Balint-Kurti, P. J. & Nelson, R. J. The Genetic Architecture of Disease Resistance in Maize: A Synthesis of Published Studies. Phytopathology 96, 120–129 (2006).
Ablazov, A. & Tombuloglu, H. Genome-wide identification of the mildew resistance locus O (MLO) gene family in novel cereal model species Brachypodium distachyon. Eur. J. Plant Pathol. 239–253 https://doi.org/10.1007/s10658-015-0833-2 (2016).
Feechan, A., Jermakow, A. M., Torregrosa, L., Panstruga, R. & Dry, I. B. Identification of grapevine MLO gene candidates involved in susceptibility to powdery mildew. Funct. Plant Biol. 1255–1266 (2008).
Acevedo-Garcia, J., Kusch, S. & Panstruga, R. Magical mystery tour: MLO proteins in plant immunity and beyond. New Phytologist 204, 273–281 (2014).
Himmelbach, A. et al. Promoters of the Barley Germin-Like GER4 Gene Cluster Enable Strong Transgene Expression in Response to Pathogen. The Plant Cell 22, 937–952 (2010).
Hurkman, W. J. & Tanaka, C. K. Germin Gene Expression is Induced in Wheat Leaves by Powdery Mildew Infection. Plant Physiol. 111, 735–739 (1996).
Elmore, J. M. & Coaker, G. The Role of the Plasma Membrane H 1 -ATPase in Plant – Microbe Interactions. Mol. Plant 4, 416–427 (2011).
Hückelhoven, R., Dechert, C. & Kogel, K. Overexpression of barley BAX inhibitor 1 induces breakdown of mlo-mediated penetration resistance to Blumeria graminis. Proc. Natl. Acad. Sci. USA 100, 5555–60 (2003).
Watanabe, N. & Lam, E. Bax Inhibitor-1, a Conserved Cell Death Suppressor, Is a Key Molecular Switch Downstream from a Variety of Biotic and Abiotic Stress Signals in Plants. Inter. J. Molecular. Sci. 10, 3149–3167, https://doi.org/10.3390/ijms10073149 (2009).
Craig, A., Ewan, R., Mesmar, J., Gudipati, V. & Sadanandom, A. E3 ubiquitin ligases and plant innate immunity. J. Ext. Bot. 4, 1123–32, https://doi.org/10.1093/jxb/erp059 (2009).
Kim, H. S. & Delaney, T. P. Over-expression of TGA5, which encodes a bZIP transcription factor that interacts with NIM1/NPR1, confers SAR-independent resistance in Arabidopsis thaliana to Peronospora parasitica. Plant j. 2, 151–163 (2002).
Dreher, K. & Callis, J. Ubiquitin. Hormones and biotic stress in plants. Annals of Botany 99, 787–822 (2007).
Rashid, Z., Zaidi, P. H., Vinayan, M. T., Sharma, S. S. & Srirama Setty, T. A. Downy mildew resistance in maize (Zea mays L.) across Peronosclerospora species in lowland tropical Asia. Crop Prot. 43, 183–191 (2013).
Craig, J., Bockholt, A. J., Frederiksen, R. A. & Zuber, M. S. Reaction of important corn inbred lines to Sclerospora sorghi. Plant Dis. Rep. 61, 563–564 (1977).
Box, G. E. P. & Cox, D. R. An Analysis of Transformations. J. R. Stat. Soc. Ser. B 26, 211–252 (1964).
Alvarado, G. et al. “META-R (Multi Environment Trial Analysis with R for Windows) Version 4.1”, 6–8 (2015) http://hdl.handle.net/11529/10201.
Payne, R. W., Murray, D. A., Harding, S. A., Baird, D. B. & Soutar, D. An Introduction to GenStat for Windows (14th Edition). (2011).
CIMMYT. Laboratory protocols: CIMMYT applied molecular genetics laboratory protocols. CIMMYT, Mex (2001).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, 1–10 (2011).
Glaubitz, J. C. et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9, e90346, https://doi.org/10.1371/journal.pone.0090346 (2014).
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E. & Shadick, N. A. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38, 904–909 (2006).
Bishop, D. T. & Williamson, J. A. The power of identity-by-state methods for linkage analysis. Am. J. Hum. Genet. 46, 254–265 (1990).
Remington, D. L. et al. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 98, 11479–11484 (2001).
R Core Team. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. at http://www.r-project.org/ (2014).
Hill, W. G. & Weir, B. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 31, 54–78 (1988).
Excoffier, L. & Slatkin, M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 12, 921–927 (1995).
Gabriel, S. B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
Kang, H. M. H. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Wisser, R. J. et al. Multivariate analysis of maize disease resistances suggests a pleiotropic genetic basis and implicates a GSTgene. Proc Natl. Acad. Sci. https://doi.org/10.1073/pnas.1011739108 (2011).
Poppenberger, B. et al. Detoxification of the Fusarium mycotoxin deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana. J. Biol. Chem. 278, 47905–14 (2003).
Zhong, R. & Ye, Z. Unraveling the functions of glycosyltransferase family 47 in plants. Trends Plant Sci. 8, 565–568 (2003).
Bisgrove, S. R., Hable, W. E. & Kropf, D. L. +TIPs and Microtubule Regulation. The Beginning of the Plus End in Plants. Plant Physiol. 136, 3855–3863 (2004).
Chan, J., Calder, G., Fox, S. & Lloyd, C. Localization of the Microtubule End Binding Protein EB1 Reveals Alternative Pathways of Spindle Development in Arabidopsis Suspension Cells. The Plant Cell 17, 1737–1748 (2005).
Guo, L. et al. Evaluating the microtubule cytoskeleton and its interacting proteins in monocots by mining the rice genome. Anna. Bot. 3, 387–402, https://doi.org/10.1093/aob/mcn248 (2009).
Sekhwal, M. K., Li, P., Lam, I., Wang, X. & Cloutier, S. Disease Resistance Gene Analogs (RGAs) in Plants. Int. J. Mol. Sci. 8, 19248–19290, https://doi.org/10.3390/ijms160819248 (2015).
Regina, I. et al. Peroxidase activity in maize inbred lines resistant or susceptible to maize dwarf mosaic virus. Revista brasileira de milho e sorgo 2, 1–8 (2003).
Hiraga, S., Sasaki, K., Ito, H., Ohashi, Y. & Matsui, H. A Large Family of Class III Plant Peroxidases. Plant Cell Physiol. 42, 462–468 (2001).
Johal, G. & Briggs, S. Reductase activity encoded by the HM1 disease resistance gene in maize. Science 258, 985–987 (1992).
Ayliffe, M. A & Lagudah, E. S. Molecular Genetics of Disease Resistance in Cereals. Ann Bot. 6, 765–773, https://doi.org/10.1093/aob/mch207 (2004).
Nwugo, C. C., Duan, Y. & Lin, H. Study on Citrus Response to Huanglongbing Highlights a Down-Regulation of Defense-Related Proteins in Lemon Plants Upon ‘Ca. Liberibacter asiaticus’ Infection. PLoS One 8, 1–13 (2013).
Acknowledgements
The authors gratefully acknowledge the financial support received from the CGIAR Research Program (CRP) on MAIZE for sponsoring this research work. Authors would also like to acknowledge the technical assistance of Mr. Boregowda and Sanjaya, CIMMYT, in carrying out this research.
Author information
Authors and Affiliations
Contributions
S.N., P.H.Z. designed the experiment; Z.R., P.K.S. generated phenotyping data; Z.R., H.V., S.N. analysed data; B.M.P. S.N., Z.R. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rashid, Z., Singh, P.K., Vemuri, H. et al. Genome-wide association study in Asia-adapted tropical maize reveals novel and explored genomic regions for sorghum downy mildew resistance. Sci Rep 8, 366 (2018). https://doi.org/10.1038/s41598-017-18690-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18690-3
- Springer Nature Limited
This article is cited by
-
Identification and validation of a key genomic region on chromosome 6 for resistance to Fusarium stalk rot in tropical maize
Theoretical and Applied Genetics (2022)
-
Genome-wide association study and its applications in the non-model crop Sesamum indicum
BMC Plant Biology (2021)
-
Genome wide association mapping for heat tolerance in sub-tropical maize
BMC Genomics (2021)
-
Genome wide association study and genomic prediction for stover quality traits in tropical maize (Zea mays L.)
Scientific Reports (2021)
-
Comparative Breeding potential of two crosses for response to late wilt disease (LWD) in maize (Zea mays L.)
Genetic Resources and Crop Evolution (2021)