
Abstract
Cattleyak are interspecific hybrids between cattle and yak, exhibiting the same prominent adaptability as yak and much higher performances than yak. However, male infertility of cattleyak resulted from spermatogenic arrest has greatly restricted their effective utilization in yak breeding. In past decades, much work has been done to investigate the mechanisms of spermatogenic arrest, but little is known about the differences of the post-transcriptional regulators between cattleyak and yak, which may contribute to the impaired spermatogenesis. MiRNAs, a class of endogenous non-coding small RNA, were revealed to play crucial roles in regulating gene expression at post-transcriptional level. In the present study, we identified 50 differentially expressed (DE) known miRNAs and 11 novel miRNAs by using Illumina HISeq and bioinformatic analysis. A total of 50 putative target sites for the 13 DE known miRNAs and 30 for the 6 DE novel miRNAs were identified, respectively. GO and KEGG analyses were performed to reveal the functions of target genes for DE miRNAs. In addition, RT-qPCR was performed to validate the expression of the DE miRNAs and its targets. The identification of these miRNAs may provide valuable information for a better understanding of spermatogenic arrest in cattleyak.
Similar content being viewed by others


Introduction
Cattleyak are interspecific hybrids between cattle (♂) and yak (♀), which exhibit the same prominent adaptability to harsh environment on Qinghai-Tibetan Plateau as yak. Moreover, cattleyak display much higher performances than yak in such economic traits as meat and milk production, and the hybrids have played important roles in promoting the social and economic development in the plateau regions. However, the F1 males of cattleyak are infertile due to spermatogenic arrest and numerous valuable genes cannot be fixed and passed down to next generations1. Therefore, spermatogenic arrest of cattleyak has greatly restricted the effective utilization of the hybrids for decades of years. To investigate the mechanisms of spermatogenic arrest of cattleyak, we have compared the testis transcriptomes and proteomes between cattleyak and yak, and identified 2960 DE genes and 645 DE proteins between cattleyak and yak, respectively2,3,4. The significant divergences between DE genes and proteins demonstrated that the expression of a large number of genes may have been inhibited or silenced during post-transcriptional process. MiRNAs are a class of endogenous non-coding small RNA molecules with ~22 nucleotides in length, regulating gene expression at a post-transcriptional level by binding to the 3′-untranslated regions (3′-UTR) of target mRNAs5,6,7. Since the first identified miRNA lin 4 in Caenorhabditis elegans8,9, hundreds of miRNAs have been discovered in mammalian species and accumulating evidences show that miRNAs have been involved in diverse biological processes, such as cell proliferation10, differentiation11 and apoptosis12. In addition, many miRNAs are evolutionarily conserved across species13, but several miRNAs are expressed in tissue-specific patterns14.
Spermatogenesis is a highly-regulated process comprising three main continuous stages of germ cell differentiation: mitotic proliferation of spermatogonia, meiosis of spermatocytes and spermiogenesis of haploid spermatids. In mammals, the differentiation of germ cells to sperms is implicated in the sequential gene expression regulated by extrinsic and intrinsic factors15. Dysregulation at any stage of spermatogenesis may cause infertility16,17,18. Several studies have revealed that miRNAs play important roles in regulating the distinct process in mammalian spermatogenesis. MiR-20 and miR-106a were identified to promote the renewal of mouse spermatogonial stem cells (SSCs) at the post-transcriptional level via targeting STAT3 and Ccnd119, and miR-221/222 were found to play a crucial role in maintaining the undifferentiated state of spermatogonia through repression of KIT expression20. In addition, miR-469 were reported to repress the expression of TP2 and Prm2 at the translational level with minor effect on mRNA degradation, which is essential in pachytene spermatocytes for their timely transition to spermiogenesis at later times of spermiogenesis21. MiR-122 was shown to regulate chromatin remodeling during sperm development by suppressing the expression of Tnp222,23. MiR-355 and miR-181b/c were all up-regulated in the adult testis by targeting Rsbn1, a novel homeobox-like protein gene involved in the transcriptional regulation in haploid germ cells24,25. As the emerging roles of miRNAs discovered in regulating gene expression at the post-transcriptional level, investigation of the regulatory functions of miRNAs involved in spermatogenesis will contribute to a better understanding of the mechanisms of spermatogenic arrest of cattleyak. In this study, we therefore performed small RNA sequencing and compared the miRNA expression profiles between cattleyak and yak testis. For the first time, we identified the DE miRNAs between the testis samples of cattleyak and yak, some of which were involved in gene regulation during mitotic proliferation, meiosis and spermiogenesis processes. The further characterization of these miRNAs is helpful to reveal the mechanisms of spermatogenic arrest of cattleyak and the identified miRNAs together with their target genes may serve as effective molecular markers in resolving the problems of male infertility of cattleyak in the future.
Results
Deep sequencing data of testis small RNAs for cattleyak and yak
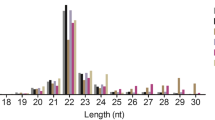
In order to identify DE miRNAs involved in spermatogenic arrest of cattleyak, small RNA libraries were constructed from testis total RNAs of cattleyak and yak individuals for Illumina HISeq and miRNA screening. After removing the low quality reads and adaptors, the overall length distribution of clean reads was shown schematically in Fig. 1. All the reads identified from each library ranged from 10 to 50 nt, with two distinctly distributional peaks approximating to 20 and 30 nt. Illumina HISeq provided a total of 20671097, 23154701 and 35289072 reads from the library of CY1 (CY, cattleyak), CY2, and CY3, respectively; while a total of 55509326, 29394401 and 34144367 reads from the library of YK1 (YK, yak), YK2 and YK3, respectively (Fig. 1). After matched to the latest Sanger miRBase and other non-coding RNA database like ncRNA, piRNA and Rfam database using software CLC genomics_workbench 5.5, the clean reads derived from the six samples ranged from 20560465 to 55291115, with the rate of annotated reads ranging from 3.3% to 12.8% and the ambiguously annotated reads ranging from 0.5% to 2.6% (Table 1). The numbers of small RNAs obtained from the six samples ranged from 4920921 to 11294549, with the annotated percentages ranging from 0.2% to 0.8% and ambiguously annotated percentages ranging from 0.1% to 0.2% (Table 1). Subsequently, the annotated small RNAs varying from 18 to 35 nt were classified into several RNA categories such as the known miRNAs, precursors, tRNAs, rRNAs, misc-RNAs, mRNAs, snoRNAs/snRNAs, lincRNAs and unannotated RNAs, in which unannotated RNAs accounted for most of the fractions with the amount ranging from 14157462 to 37310411 and the amount of miRNAs (ranging 20 to 24 nt) varied from 1076993 to 3975469 (Fig. 2, Supplementary Table S1).

The number of reads from cattleyak and yak testis libraries with different length.

The classification of small RNAs for cattleyak and yak testis miRNA-seq.
Novel miRNAs identified from testis of cattleyak and yak
The special hairpin-like structure of miRNA precursor can be used to predict novel miRNAs. Identification of novel miRNAs by software sRNA Toolkit was based on its secondary structure, the Dicer enzyme cleavage site and the minimum free energy index (MFEI) of the unannotated reads (Table 1) that could be mapped to bovine genome. A total of 168 novel miRNAs matching to 615 hits in the bovine genome were identified from the unannotated reads with the MFEI varying from −161.81 to −13.2 and the novel miRNAs were named according to the species and categories they belonged to. The length of novel miRNAs ranged from 18 to 23 nt, with a distribution peak at 22 nt. Of the 168 novel miRNAs, 88 were identified from cattleyak and 120 from yak libraries, and 40 miRNAs were identified from both cattleyak and yak libraries (Supplementary Table S2). Furthermore, 11 novel miRNAs were identified to be differentially expressed between cattleyak and yak groups, in which 8 miRNAs were significantly upregulated and 3 were downregulated (Fig. 3A, Table 2).

Volcano plot: Log2 values of relative expression of known (A) and novel (B) testis miRNAs (cattleyak related to yak) versus −Log10 of false discovery rate (FDR). Horizontal line is at P = 0.05 and vertical lines are at fold change (FC) = ±2.
DE miRNAs between testis of cattleyak and yak
To gain insight into the possible roles of miRNAs involved in spermatogenic arrest of cattleyak, it is crucial to investigate the DE miRNA profiles of cattleyak and yak testis. The software edgeR was used to analyze the expression level of miRNAs obtained from both the cattleyak and yak, and DE miRNAs were screened between cattleyak and yak after the number of reads were normalized to transcripts per million (TPM). Volcano plot indicated the pattern of 50 DE known miRNAs (Fig. 3B) between cattleyak and yak, of which 11 were up-regulated and 39 were down-regulated (Table 3). However, of these DE miRNAs, the top three up-regulated miRNAs were bta-miR-592, bta-miR-1247-5p, bta-miR-2484, with an increased fold-change of 11.94, 4.114338, 3.319338, respectively; the top three down-regulated miRNAs were bta-miR-449a, bta-miR-34c, bta-miR-449b, with a decreased fold-change of 0.001319, 0.006306, 0.007821, respectively.
Prediction of target genes for DE miRNAs
To identify the potential function roles of these miRNAs, target gene prediction was performed using software miRanda. The potential targets of the DE miRNAs were screened out with total score ≧172 and total energy ≦−20.00. Target genes for the 13 DE known miRNAs between cattlyak and yak were shown in Supplementary Table S3 and for the 6 DE novel miRNAs were shown in Supplementary Table S4. A total of 50 putative target sites for the 13 DE known miRNAs and 30 for the 6 DE novel miRNAs were identified, respectively. Figure 4 indicated the target sites for 6 DE miRNAs. The results showed that most miRNAs had multiple distinct target genes possessing diverse functions. For instance, a total of 18 putative target genes for bta-miR-135a were identified and 9 putative target genes for bta-novel-miR-160 were identified. The putative target sites for bta-miR-135a were zinc finger family genes, CSPP1, CUBN and so on.

Predicted targets and binding sites of the DE known miRNAs between cattleyak and yak.
GO and KEGG enrichment of the target genes for DE miRNAs
To understand the roles of DE miRNAs between cattleyak and yak, a total of 46887 target genes for DE known miRNAs and 181138 for DE novel miRNAs were subjected to GO and KEGG enrichment. As a result, a total of 114 significantly enriched GO terms were obtained for target genes of DE known miRNAs, in which G-protein coupled receptor signaling pathway (p = 6.88E-09), structural constituent of ribosome (p = 1.77E-10) and ribosome (p = 4.32E-10) were the top listed GO terms involved in biological process, molecular function and cellular component, respectively (Fig. 5A, Supplementary Table S5). Meanwhile, 575 GO terms were significantly enriched for target genes of DE novel miRNAs, in which G-protein coupled receptor signaling pathway (p = 2.99E-25), G-protein coupled receptor activity (p = 1.24E-33) and cytoskeleton (p = 3.69E-10) were the top listed GO terms involved in biological process, molecular function and cellular component, respectively (Fig. 5B, Supplementary Table S6). KEGG enrichment results showed that only 8 significantly enriched pathways were obtained for target genes of DE known miRNAs, in which MicroRNAs in cancer (p = 2.20E-06), Ribosome (p = 4.32E-05) and Oxidative phosphorylation (p = 0.000827) were the top listed three pathways (Fig. 5C, Supplementary Table S7). Meanwhile, 53 pathways were significantly enriched for target genes of DE novel miRNAs, in which ECM-receptor interaction (p = 2.00E-05), Oxidative phosphorylation (p = 4.67E-05) and Focal adhesion (p = 8.21E-05) were the top listed three pathways (Fig. 5D, Supplementary Table S8).

Enrichments of target genes for DE miRNAs. The top 10 items of GO enrichments of target genes for DE known miRNAs (A) and for DE novel miRNA (B) were based on biological process, molecular function and cellular component, respectively. GO terms in each ontological category were ranked according to decreased −log10 of p values listed on the y-axis. The top 10 pathways of KEGG enrichments of target genes for DE known miRNAs (C) and for DE novel miRNAs (D) were ranked according to decreased −log10 of p values listed on the y-axis.
Further analysis revealed that hundreds or thousands of target genes were enriched in each GO term or KEGG pathway (Supplementary Tables S5–S8), and these genes were mainly involved in such biological processes as cellular signal transduction, cellular development, cellular differentiation and protein modification. For example, 3024 target genes were involved in developmental process, 3178 in single-organism developmental process, 1551 in intracellular signal transduction, 2059 in cell differentiation and 855 in cell cycle (Supplementary Table S5); 112 genes were involved in MicroRNAs in cancer, 163 in Focal adhesion and 73 in ECM-receptor interaction (Supplementary Table S7).
RT-qPCR validation of DE miRNAs and their target genes
To validate the expression level of DE miRNAs, stem-loop RT-qPCR was performed on 6 known miRNAs (bta-miRNA-449a/b, bta-miRNA-135, bta-miRNA-19b, bta-miRNA-378 and bta-miRNA-184) and 2 novel miRNAs (bta-novel 10 and bta-novel 11). RPS18 was used as the internal reference and the sequences of primers were listed in the Supplementary Table S9. As shown in Fig. 6, comparison of miRNA expression revealed that the expression of all the miRNAs selected were downregulated in cattleyak with respect to yak except the upregulation of bta-miRNA-19b, which was fully consistent with their expression patterns obtained from sequencing data. In addition, 7 target genes (E2F5, ZNF226, CCNT2, MAPK1IP1L, NCOR2, MAP3K8 and JAMIP1) of these miRNAs were selected to validate their expression levels. β-actin was used as the internal reference and the sequences of primers were listed in the Supplementary Table S10. As shown in Fig. 7, the expression levels of all the target genes were upregulated in cattleyak except E2F5, which indicated that expression of the target genes were almost inhibited by the corresponding miRNAs. Meanwhile, the expression of E2F5 and CCNT2 were not found to be significantly inhibited by bta-miRNA-449a/b and bta-miRNA-19b, respectively.

The expression of miRNAs in yak (Y) and cattleyak(C) testis tissues were validated by RT-qPCR.

The expression of target genes for miRNAs in yak (Y) and cattleyak (C) testis tissues were detected by RT-qPCR.
Discussion
Since the discovery of miRNAs and their roles in post-transcriptional gene regulation, many studies have shown that they were implicated in the regulation of diverse biological processes10,11,12 and several miRNAs have been found to be involved in multiple processes of spermatogenesis such as mitotic proliferation of spermatogonia, meiosis of spermatocytes and spermiogenesis of haploid spermatids26,27,28,29. However, no study was conducted to characterize miRNAs involved in spermatogenic arrest of cattleyak. In our study, 50 DE known miRNAs and 11 novel miRNAs were identified between the testis samples of cattleyak and yak by using Illumina HISeq and bioinformatic screening. The majority of miRNAs were 21–23 nt in length, which was consistent with previous reports on the length distribution of miRNA identified in the adipose tissue and mammary gland of bovine30,31. Stem-loop RT-qPCR was performed to validate the expression level of known and novel miRNAs, and the results were highly consisted with the sequencing data. The identification of these miRNAs may provide valuable information to a better understanding of the mechanisms of spermatogenic arrest of cattleyak.
In this work, the target genes for DE miRNAs were identified to be involved in such biological processes as cellular signal transduction, cellular development, cellular differentiation and protein modification (Supplementary Tables S5, S6), which was bound to be associated with specific spermatogenic processes in cattleyak. During spermatogenesis, the regulation of balance between maintenance of undifferentiated SSCs and transition to a differentiating state could ensure adequate numbers of spermatogonia undergoing spermatogenesis and lifelong supply of spermatozoa. The downregulation of miR-135a was revealed to activate FoxO1 gene involved in cellular proliferation, which resulted in the decreased number of SSCs and failure of spermatogonial stem cell maintenance in cryptorchid testes32. The expression of miR-378 was also significantly downregulated in prostate cancer tissues, which was identified to suppress cell growth through downregulation of MAPK1 gene involved in cell proliferation process33. Meanwhile, miR-184 was reported to downregulate Ncor2 gene and result in significantly lower number of cells in the G1 phase in mouse spermatogenesis34. In the present study, the downregulation of bta-miR-135a, bta-miR-378 and bta-miR-184 could contribute to the decreased number of SSCs and spermatogenic alterations in cattleyak, which was consistent with the cellular distribution characteristics in cattleyak testis revealed by histological analysis1,2,3. On the other hand, the over expression of miR-19b-3p was reported to repress the expression of USP32, RAB18 and Dusp6 involved in cell differentiation process and contribute to the inhibited proliferation and slow-downed cycle of SH-SY5Y cells35. Over expression of miR-592 could promote the cell proliferation by downregulating the expression of FOXO3 involved in cell cycle process and promote the proliferation of prostate cancer cells36. Therefore, inhibited expression of USP32, RAB18 and Dusp6 caused by bta-miR-19b-3p may be conducive to the maintaining of undifferentiated SSCs in cattleyak, while downregulation of FOXO3 by bta-miR-592 could contribute to the transition of SSCs to a differentiating state, which was consistent with the conclusions from our previous study that spermatogenic arrest of cattleyak started at the stage of spermatogonial differentiation and get aggravated during meiosis3.
Meiosis and spermiogenesis, the two successive phases of spermatogenesis, must be tightly regulated and any mistake in these processes will lead to spermatogenic arrest. As known, the progression of these processes implicates complex gene expression regulation at post-transcriptional levels15,16,17,18. To date, few miRNAs have been shown to play critical roles in these processes. Upon the initiation of meiosis, the miR-449 cluster and miR-34b/c functioned redundantly in down-regulating the activities of the E2F-pRb pathway, which allowed male germ cells to exit from the mitotic cycle and to enter the meiotic program in murine testes37. MiRNA-34c was mainly expressed in the late stages of meiosis and was likely to influence germ cell fate during this period via downregulation of TGIF2 and NOTCH238. Over expression of miR-34c was revealed to promote the expression of Nanos3, Scp3, and Stra8 genes involved in meiosis in mouse spermatogenesis39. As the essential roles played by these miRNAs in meiosis, the downregulation of bta-miR-34b/c and bta-miR-449 cluster in cattleyak could repress the expression of Nanos3, Scp3, and Stra8 and contribute to the failure of the transition from mitosis to meiosis and the subsequent spermiogensis, which was consistent with our previous finding that spermatogenic arrest of cattleyak got aggravated during meiosis3. Although the spermatogenic arrest occurred during spermatogonial differentiation, the balance between undifferentiated SSCs and differentiated spermatogonic cells should be partially maintained in cattleyak.
In conclusion, for the first time we provided a useful resource for further elucidation of the regulatory role of potential miRNAs in spermatogenic arrest of cattleyak. Using Illumina HISeq and bioinformatic analysis, 50 DE miRNAs and 11 novel miRNAs were identified between yak and cattleyak. The GO and KEGG enrichment for the predicted target genes indicated the functional complexity of miRNAs. The downregulation of bta-miR-135a, bta-miR-378 and bta-miR-184 could contribute to the decreased number of SSCs and spermatogenic alteration in cattleyak. Furthermore, the downregulation of bta-miR-34b/c and bta-miR-449 cluster may have caused the transition failure from mitosis to meiosis and the subsequent spermiogensis in cattleyak. In the future, much effort should be exerted to investigate the role of individual miRNA involved in spermatogenic arrest of cattleyak and explore the pathways involved to reveal the mechanisms of cattleyak infertility.
Materials and Methods
Animals and testis processing
Three cattleyak (named C1, C2 and C3) and three yak (named Y1, Y2 and Y3) aged 12 months were sampled from a Maiwa yak population fed on a pasture in Hongyuan county, Sichuan province of China, in which cattleyak were F1 generation of Simmental and Maiwa yak. Testis of each animal was obtained by veterinary surgical operation and the caudal epididymis was firstly resected. Then fat and fascia surrounding the testis were resected and two crosscut slices of the testicular tissues in the middle of testis were obtained by fine scale dissection. All crosscut slices were snap frozen in liquid nitrogen (−196 °C), transported to laboratory and stored at −80 °C until RNA isolation.
Ethics statement
Sample collection was carried out under the license in accordance with the Guideline for Care and Use of Laboratory Animals of China and all protocols were approved by the institution Review Board of Southwest University of Science and Technology.
RNA preparation, miRNA cloning and sequencing
Small RNAs were extracted from the six frozen testis samples using mirVana™ miRNA Isolation Kit (Cat #. AM1561, Austin TX, USA) according to the manufacturer’s instructions. The integrity and concentration of total RNA were assessed using Agilent 2100 Bioanalyzer (Agilent technologies Santa Clara, USA) to meet the requirement of the Illumina HiSeqTM 2000 sequencing platform. The RNA 3′-adapter was specifically modified to target miRNAs and other small RNAs that have 3′-hydroxyl groups resulted from enzymatic cleavage by Dicer or other RNA processing enzymes. The adapters were ligated to each end of the RNA molecule and a RT reaction was used to create single stranded cDNA. The cDNA is amplified though PCR and followed by gel purification to generate library products. The libraries were then sequenced separately using Illumina HISeq and generated a total of 20732767, 23210984 and 35385105 reads from three testis libraries of cattleyak, and a total of 55707150, 29469263 and 34228475 reads from three testis libraries of yak.
Statistical analysis
Fastx (Fastx_toolkit-0.0.13.2) was used to remove the low quality reads and adaptors obtained from the six libraries. The clean reads of these libraries were matched to the latest Sanger miRBase (http://www.mirbase.org/) and other noncoding databases like ncRNA (http://asia.ensembl.org/index.html), piRNA (http://www.ncbi.nlm.nih.gov/and http://pirnabank.ibab.ac.in/) and Rfam database (http://rfam.sanger.ac.uk/) with a tolerance of two mismatches using the software of CLC genomics_workbench 5.5. Sequences that did not overlap with any annotated sequence were classified as unannotated reads. Next, the annotated small RNAs varying between 18 to 35 nt were classified into several RNA categories such as the known miRNAs, precursors, tRNAs, rRNAs, misc-RNAs, mRNAs, snoRNAs/snRNAs, lincRNAs and unannotated RNAs. The software sRNA Toolkit was used to identify the novel miRNAs based on its secondary structure, Dicer enzyme cleavage site and minimum free energy indexes (MFEI) of the unannotated reads which could be mapped to bovine genome.
Differentially expression analysis
To compare the miRNA expression levels between the two samples to determine the DE miRNAs, the software edgeR was utilized to analyze the expression level of miRNAs obtained from the two samples and DE miRNAs were screened between catlleyak and yak after the number of reads were normalized to transcripts per million (Actual miRNA count/Total count of clean reads*1000000, TPM). Fold-change formula: Fold_change = log2 (CYTPM/YKTPM). P-value formula:
N1 and X represent the total number of clean reads and normalized expression level of a given miRNA in the small RNA library obtained from cattleyak, respectively, and N2 and Y represent the total number of clean reads and normalized expression level of a given miRNA in the small RNA library generated from yak, respectively. The significant differential expressed miRNAs were selected according to Fold change and P-value with the criteria: Fold change >2 or <−2 and P-value < 0.05.
In silico analysis of identified miRNAs and target prediction
Target prediction was performed using software miRanda and the rules were used for target prediction in term of the following criteria: targets located in the 3′-UTR region, seed length of at least 7 base pairs (bp), and the minimum free energy (MFE). The potential targets of the DE miRNAs were screened out with total score ≧172 and MFE ≦−20.09.
GO and KEGG enrichment of the target genes for DE miRNAs
GO enrichment is an effective and efficient tool to classify the target genes for DE miRNAs based on the specific biological functions. KEGG pathways are applied to elucidate in vivo comprehensive inferences of reactions such as metabolic pathways or signal transduction pathways. To depict the interaction of the target genes for DE miRNAs involved in the same certain biological functions of spermatogenic arrest of cattleyak, we employed enrichment of GO and KEGG pathway. These two methods calculated the target gene numbers for every term or pathway after comparing to a genome background and then used the hypergeometric test to filter the significantly enriched term or pathway. The same calculating formula was developed to do the analyses described as follows:
N is the number of all genes with GO or KEGG annotation; n is the number of target genes in N; M is the number of all genes that are annotated to certain GO terms or specific pathways; m is the number of target genes in M. The significantly enriched GO terms or pathways were defined after the calculated p-value went though Bonferroni Correction, taking corrected p-value ≦0.05 as a threshold.
RT-qPCR validation of DE miRNAs and their target genes
Total RNA was extracted from the six frozen testis samples using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s instructions. The integrity and concentration of total RNA were assessed using theAgilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Stem-loop reverse transcription (RT) and real-time polymerase chain reaction (qPCR) with SYBR Green were used for the quantification of miRNA expression. Total RNA (1 µg) was reversely transcribed with PrimeScriptTM RT reagent Kitwith gDNA Eraser (TaKaRa, Dalian, China) using a stem-loop RT primer for each tested miRNA. The cDNA was then used for real-time PCR quantification of miRNA using the miRNA specific forward primer and the universal reverse primer. The bovine ribosomal protein S18 (RPS18) (GenBank NO. NM_001033614.1) gene was used as an endogenous control. The primers for stem-loop RT and miRNA quantification were listed in Supplementary Table S4. Real-time quantitative PCR was performed in triplicate using a Bio-Rad CFX 96™ Real Time Detection System in a 20 µL reaction comprising 100 ng cDNA for each miRNA, 0.4 µM forward and reverse primers, and 10 µL of 2 × SYBR®Premix Ex TaqTM II (TaKaRa, Dalian, China). Reactions were performed at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s, 60 °C for 10 s, and 68 °C for 20 s. Individual samples were run in triplicate. In the end of the PCR cycles, melting curve analysis was performed to validate the specific generation of the expected PCR products. The quantification of each miRNA relative to RPS18 gene was calculated using the2−ΔΔCt method.
Real time PCR was also carried out to validate the expression of the target genes E2F5, ZNF226, CCNT2, MAPK1IP1L, NCOR2, MAP3K and JAKMIP1 in the testis of cattleyak and yak according to the methods described previously3. The primers for the quantification of these genes were listed in Supplementary Table S5. The relative expression level of mRNA for each gene was calculated using b-actin as an endogenous reference gene using the 2−ΔΔCt method.
Statistical analysis
All experiments were performed in triplicate and all data were presented as mean ± SEM. The significantly differentially expressed miRNAs between cattleyak and yak were selected according to Fold change and P-value with the criteria: Fold change >2 or <−2 and P-value < 0.05, using the analysis of variance (ANOVA) and a 2-tailed t-test.
Availability of data and material
All analyzed data in this study are included in this published article and all RNA-Seq data in this study are available in NCBI under accession number: SRP117203.
References
Li, X. et al. Sequence analysis and study on the expression level of Dmc1 mRNA in yak and cattleyaks testis. Scientia Agricultura Sinica. 43, 3221–3229 (In Chinese with English abstract) (2010).
Yu, S. et al. Comparative iTRAQ proteomics revealed proteins associated with spermatogenic arrest of cattleyak. J Proteomics. 142, 102–113 (2016).
Cai, X. et al. Comparative analysis of testis transcriptomes associated with male infertility in cattleyak. Theriogenology. 88, 28–42 (2017).
Sun, L. et al. Comparative testis proteome of cattleyak from different developmental stages. Animal. 11, 101–111 (2017).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 116, 281–297 (2004).
Olde Loohuis, N. F. et al. MicroRNA networks direct neuronal development and plasticity. Cell Mol Life Sci. 69, 89–102 (2012).
Lund, E., Guttinger, S., Calado, A., Dahlberg, J. E. & Kutay, U. Nuclear export of microRNA precursors. Science. 303, 95–98 (2004).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C.elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell. 75, 843–854 (1993).
Wightman, B., Ha, I. & Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern-formation in C. Elegans. Cell. 75, 855–862 (1993).
Pogue, A. I. et al. MicroRNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci Lett. 476, 18–22 (2010).
Song, L. & Tuan, R. S. MicroRNAs and cell differentiation in mammalian development. Birth Defects Res C Embryo Today. 78, 140–149 (2006).
Xiao, C. & Rajewsky, K. MicroRNA control in the immune system: basic principles. Cell. 136, 26–36 (2009).
Pasquinelli, A. E. et al. Conservation of the sequence and temporal expression of let-7 heterochronicregulatory RNA. Nature. 408, 86–89 (2000).
Lagos-Quintana, M. et al. Identification of tissue-specific microRNAs from mouse. Current Bio. 12, 735–739 (2002).
Mei, X. X., Wang, J. & Wu, J. Extrinsic and intrinsic factors controlling spermatogonial stem cell self-renewal and differentiation. Asian J Androl. 17, 347–354 (2015).
Jaiswal, D., Trivedi, S., Agrawal, N. K. & Singh, K. Dysregulation of apoptotic pathway candidate genes and proteins in infertile azoospermia patients. Fertil Steril. 104, 736–43 (2015).
Zhang, X. & Lui, W. Y. Dysregulation of nectin-2 in the testicular cells: an explanation of cadmium-induced male infertility. Biochim Biophys Acta. 1839, 873–884 (2014).
Larder, R., Clark, D. D., Miller, N. L. & Mellon, P. L. Hypothalamic dysregulation and infertility in mice lacking the homeodomain protein Six6. J Neurosci. 31, 426–438 (2011).
He, Z. et al. MiRNA-20 and mirna-106a regulate spermatogonial stem cell renewal at the post-transcriptional level via targeting STAT3 and Ccnd1. Stem Cells. 31, 2205–2217 (2013).
Yang, Q. E. et al. MicroRNAs 221 and 222 regulate the undifferentiated state in mammalian male germ cells. Development. 140, 280–290 (2013).
Dai, L. Testis-specific miRNA-469 up-regulated in gonadotropin-regulated testicular RNA helicase(GRTH/DDX25)-null mice silences transition protein 2 and protamine 2 messages at sites within coding region: implications of its role in germ cell development. J Biol Chem. 286, 44306–44318 (2011).
Yu, Z., Raabe, T. & Hecht, N. B. MicroRNA Mirn122a reduces expression of the posttranscriptionally regulated germ cell transition protein 2 (Tnp2) messenger RNA (mRNA) by mRNA cleavage. Biol Reprod. 73, 427–33 (2005).
Liu, T. et al. MicroRNA-122 influences the development of sperm abnormalities from human induced pluripotent stem cells by regulating TNP2 expression. Stem Cells and Development. 22, 1839–1850 (2013).
Yan, N. et al. A microarray for microRNA profiling in mouse testis tissues. Reproduction. 134, 73–79 (2007).
McIver, S. C., Roman, S. D., Nixon, B. & McLaughlin, E. A. miRNA and mammalian male germ cells. Hum Reprod Update. 18, 44–59 (2012).
Ambros, V. et al. Auniform system for microRNA annotation. RNA. 9, 277–279 (2003).
Brennecke, J., Hipfner, D. R., Stark, A., Russell, R. B. & Cohen, S. M. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 113, 25–36 (2003).
He, L. et al. A microRNA polycistron as a potential human oncogene. Nature. 435, 828–833 (2005).
Yao, C. et al. MicroRNAs and DNA methylation as epigenetic regulators of mitosis, meiosis and spermiogenesis. Reproduction. 150, R25–R34 (2015).
Gu, Z., Eleswarapu, S. & Jiang, H. Identification and characterization of microRNAs from the bovine adipose tissue and mammary gland. FEBS Lett. 581, 981–988 (2007).
Chen, X. et al. Identification and characterization of microRNAs in raw milk during different periods of lactation,commercial fluid, and powdered milk products. Cell Res. 20, 1128–1137 (2010).
Moritoki, Y. et al. Expression profiling of microRNA in cryptorchid testes: miR-135a contributes to the maintenance of spermatogonial stem cells by regulating FoxO1. J Urol. 191, 1174–1180 (2014).
Chen, Q. G. et al. MiR-378 suppresses prostate cancer cell growth through downregulation of MAPK1 in vitro and in vivo. Tumour Biol. 37, 2095–2103 (2016).
Wu, J., Bao, J., Wang, L., Hu, Y. & Xu, C. MicroRNA-184 downregulates nuclear receptor corepressor 2 in mouse spermatogenesis. BMC Dev Biol. 11, 64 (2011).
Ma, Y. et al. The mechanism of action of FXR1P-related miR-19b-3p in SH-SY5Y. Gene. 588, 62–68 (2016).
Lv, Z., Rao, P. & Li, W. MiR-592 represses FOXO3 expression and promotes the proliferation of prostate cancer cells. Int J Clin Exp Med. 8, 15246–15253 (2015).
Bao, J. et al. MicroRNA-449 and microRNA-34b/c function redundantly in murine testes by targeting E2F transcription factor-retinoblastoma protein (E2F-pRb) pathway. J Biol Chem. 287, 21686–21698 (2012).
Yu, M. et al. miR-34c enhances mouse spermatogonial stem cells differentiation by targeting Nanos2. J Cell Biochem. 115, 232–242 (2014).
Yu, M. et al. miR-34c enhances mouse spermatogonial stem cells differentiation by targeting Nanos2. J Cell Biochem. 115, 232–242 (2014).
Acknowledgements
The authors thank Shurong Li from Shanghai Biotechnology Corporation (Shanghai, China) for coordinating RNASeq and data analysis. The authors also gratefully acknowledge the financial support of the grant from the National Natural Science Foundation of China (31572396).
Author information
Authors and Affiliations
Contributions
C.X. and S.W. conducted the experiment, participated in data analysis and prepared the manuscript. W.Z. and T.M. participated in sampling and experiment. J.L., W.L., C.Y., M.A.S. and S.Y. participated in the design of the study, data analysis, and drafting of the manuscript. X.C. conceived of the study, designed and supervised the all aspects of the experiments, data analysis and manuscript drafting, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, C., Wu, S., Zhao, W. et al. Differentially expressed microRNAs between cattleyak and yak testis. Sci Rep 8, 592 (2018). https://doi.org/10.1038/s41598-017-18607-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18607-0
- Springer Nature Limited
This article is cited by
-
Identification and profiling of microRNAs during yak’s testicular development
BMC Veterinary Research (2023)
-
Comparative iTRAQ proteomics identified proteins associated with sperm maturation between yak and cattleyak epididymis
BMC Veterinary Research (2021)
-
Testis transcriptome profiling identified lncRNAs involved in spermatogenic arrest of cattleyak
Functional & Integrative Genomics (2021)