
Abstract
Members of the broadly distributed Rid/YER057c/UK114 protein family have imine/enamine deaminase activity, notably on 2-aminoacrylate (2AA). Strains of Salmonella enterica, and other organisms lacking RidA, have diverse growth phenotypes, attributed to the accumulation of 2AA. In S. enterica, 2AA inactivates a number of pyridoxal 5’-phosephate(PLP)-dependent enzymes, some of which have been linked to the growth phenotypes of a ridA mutant. This study used transcriptional differences between S. enterica wild-type and ridA strains to explore the breadth of the cellular consequences that resulted from accumulation of 2AA. Accumulation of endogenously generated 2AA in a ridA mutant resulted in lower expression of genes encoding many flagellar assembly components, which led to a motility defect. qRT-PCR results were consistent with the motility phenotype of a ridA mutant resulting from a defect in FlhD4C2 activity. In total, the results of comparative transcriptomics correctly predicted a 2AA-dependent motility defect and identified additional areas of metabolism impacted by the metabolic stress of 2AA in Salmonella enterica. Further, the data emphasized the value of integrating global approaches with biochemical genetic approaches to understand the complex system of microbial metabolism.
Similar content being viewed by others

Introduction
Microbes inhabit environments that are often dynamic with respect to nutrient availability and the presence of chemical and/or physical stresses. A hallmark of microbes is their ability to respond to changing conditions by altering their metabolic network and/or physical characteristics to survive and thrive under new conditions1,2. A common response to stresses and/or changing environmental conditions is the coordinated regulation of genes that ameliorate the stress and exploit changing nutrient circumstances3,4,5. Numerous examples of global responses to environmental stress and nutrient availability that are mediated by transcriptional regulation have been described1,2. Characterization of these responses has contributed to our understanding of not only metabolic strategies but mechanisms of regulation, effector binding, and gene function. Metabolism can be regulated at many levels, but perturbations in the metabolic network are ultimately expected to, directly or indirectly, impact the transcriptome. Thus, the global expression pattern of a microbe can be used to decipher the environment perceived by the cell and understand conditions both internal and external to the organism6,7,8.
Metabolic imbalance caused by inhibition of a specific enzyme, for instance by exposure to antimicrobial agents or toxic metabolites, often result in transcriptional changes that are triggered as a consequence of the resultant metabolic imbalance9,10,11,12. In this scenario, if multiple insults generated similar or overlapping transcriptional profiles, it would indicate the perturbations ultimately affected the same part of metabolism. For instance, in a Bacillus subtilis strain harboring a purA mutation that diminishes flux toward adenine biosynthesis, the cell senses a purine limitation and attempts to overcome it via de-repression of the PurR regulon13. Treatment of Bacillus subtilis with the dihydrofolate reductase (FolA; EC 1.5.1.3) inhibitor, Trimethoprim, causes a disruption of C1 unit transfer in the cell, resulting in a bottleneck in purine biosynthesis at the AICAR transformylase (PurH; EC 2.1.2.3) enzyme. In this case, the cell perceives a purine limitation and attempts to overcome it via de-repression of the PurR regulon14. Thus, the shared features of the transcriptional response to both perturbations indicated that the perceived stress in each case was purine limitation.
In Salmonella enterica, accumulation of the reactive enamine species 2-aminoacrylate (2AA) perturbs the metabolic network by partially inactivating a number of PLP-dependent enzymes, which results in growth phenotypes15,16,17,18,19. 2AA is generated as a mechanistic intermediate of multiple PLP-dependent enzymes in central metabolic pathways (e.g., serine/threonine dehydratases EC 4.3.1.19)20,21. Thus far, the validated targets of 2AA in vivo are PLP enzymes where the enamine attacks the internal aldimine and results in the covalent modification of the PLP cofactor. Depending on the mechanism of attack, an inactive pyruvate-PLP adduct is formed in the active site, or the enzyme itself forms an irreversible bond with the modified PLP17,19,22. In either scenario, the affected enzyme is irreversibly damaged, and flux through the pathway to which it belongs is constrained. A summary of the described RidA paradigm of 2AA stress in S. enterica is provided in Fig. 1. Most bacteria, including S. enterica and E. coli, have multiple RidA paralogs, but to date no physiological role has been identified for these proteins.

RidA paradigm of 2AA stress in S. enterica. Multiple pyridoxal 5′-phosphate (PLP)-dependent enzymes can catalyze β-elimination reactions of 3-carbon alpha amino acids containing favorable leaving groups on the beta-carbon (1). This generates the reactive enamine intermediate 2-aminoacrylate (2AA), which goes into solution. 2AA has three possible fates following its release from the active site: First, 2AA can tautomerize to its imine form, producing 2-iminiopropanoate, which is non-enzymatically hydrolyzed, forming the stable keto-acid pyruvate (2). Second, RidA catalyzes the deamination of 2AA to pyruvate (3). Finally, 2AA can attack the internal aldimine of various PLP enzymes, irreversibly damaging the target enzyme. 2AA attack results in the formation of an inactive pyruvate-PLP adduct (4) or covalent modification of the enzyme by the 2AA-PLP aldimine (5).
Members of the RidA subfamily of the Rid/YER057c/UK114 protein superfamily, are enamine deaminases that quench 2AA and thus prevent the accumulation of this metabolite in vivo 16,23. The combination of in vitro and in vivo studies in S. enterica showed that at least the PLP-dependent enzymes serine hydroxymethyltransferase (GlyA; EC 2.1.2.1), branched-chain amino acid aminotransferase (IlvE; EC 2.6.1.42) and alanine racemase (Alr/DadX; EC 5.1.1.1) were targets of free 2AA in vivo and their activity is between 40–80% decreased in strains lacking RidA17,19,22,24,25. The presence of RidA proteins across all domains of life supports a conserved role for this family in quenching reactive metabolites, including 2AA, to prevent damage to metabolic components.
The current understanding of 2AA targets in vivo is the result of biochemical genetic studies, primarily in S. enterica. Although powerful, this approach demanded that the damage caused by 2AA generated a detectable growth phenotype. It was therefore likely that the impact of free 2AA on the metabolic network was broader than what has been defined by this approach. Additionally, the impact that 2AA damage of known targets has on the global network configuration remains unexplored. For example, while it is known that the growth defect of an S. enterica ridA mutant grown in minimal medium is caused by 2AA damage to serine hydroxymethyl transferase (GlyA; EC 2.1.2.1), it is unclear how the cell perceives the resulting reduction in C1 unit production24. The current study was initiated to generate a global view of metabolic changes that result from the accumulation of 2AA in S. enterica. The premise of this work was that a transcriptional profile reflects expression changes that result, directly or indirectly, from perceived changes in the internal or external environment6,8,26.
Results
Deletion of ridA alters the global transcription profile
The transcriptomes of a S. enterica ridA mutant and the isogenic wild-type strain were defined after growth on minimal glucose medium to provide a snapshot of the global metabolic state of the cells. The transcriptome data were viewed with two questions in mind; i) are previously characterized metabolic defects caused by elevated 2AA discoverable, and ii) do the data generate new insights about the global impact of 2AA accumulation?
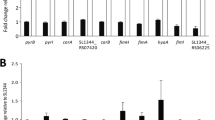
Pair-wise comparison of gene expression identified 413 genes with changes (186 higher expression, and 227 lower expression) in transcript abundance between ridA mutant and wild-type strains, using a false discovery rate (FDR) less than 0.05 (Table S1)27. Of these loci, genes with no known function (STMxxxx and yxxX locus tags) and those that showed less than a two-fold difference in expression were excluded to generate the dataset of 84 genes shown in Fig. 2. There was no evidence that the biosynthetic genes for isoleucine (ilv) were de-repressed in a ridA mutant, despite the fact that the specific activity of isoleucine transaminase (IlvE, the last step in isoleucine synthesis) is more than 50% reduced in the absence of RidA17,18. These data corroborated the conclusion reached with a single reporter, that lack of RidA did not result in a starvation for isoleucine18. Similarly, no differential expression was simply attributable to the decreased activity of serine hydroxymethyl transferase (GlyA) found in ridA mutants. The transcription of dadAX was increased in the ridA mutant and potentially reflected a consequence of the decreased alanine racemase activity of DadX, caused by 2AA accumulation22, since increased accumulation of L-alanine may contribute to the complex transcriptional regulation of dadAX 28.

Gene expression differs between a ridA mutant and wild type strain. Summary of relative expression levels of genes in S. Typhimurium LT2 found to differ significantly (FDR < 0.05, Fold-change >2) between ridA and wild-type strains. A value of 1 indicates no detectable difference in expression, values > 1 indicate higher expression in the ridA mutant, and values < 1 indicate higher expression in wild-type. Arrows to the left of the gene name indicate whether that gene is contained within an operon, with the order of the operon arranged following the arrow base to the arrowhead.
Results from the global RNA-seq experiment were independently validated with real-time qRT-PCR. The relative expression level of six select genes was measured using the comparative threshold cycle (ΔΔCT) method29 (Fig. 3). Four of the six genes chosen met the two-fold expression threshold in the RNA-seq experiment (napF, metH, dadX, and fliI), one (thiF) had a statistically significant difference in expression that was less than two-fold (1.88-fold, Supplementary Table 1), and the last (sdaC) showed no statistically significant change in gene expression between mutant and wild-type. For the six genes, a strong positive correlation between results from the qRT-PCR and global RNA-seq approaches was supported by a Pearson’s correlation coefficient of r2 = 0.96. Of the four genes with a greater than two-fold expression difference, only napF had a statistically significant (p-value < 0.05) discrepancy between the RNA-seq and qRT-PCR data. However, both approaches detected a greater than ten-fold decrease in the napF transcript levels in a ridA mutant compared to wild-type. Finally, although RNA-seq transcriptome analysis detected increased sdaC expression in a ridA strain, the expression difference did not meet the statistical cut off (FDR < 0.05). The qRT-PCR data similarly found this transcript increased in a ridA mutant, but the difference was statistically significant using this technique, suggesting that the DEseq algorithm, used to call differentially expressed genes from the RNAseq dataset, performed conservatively30.

qTR-PCR supports RNAseq differential expression results for a subset of genes. Differential expression of a subset of genes for S. enterica LT2 ridA strain (DM3480) relative to its isogenic wild-type (DM9404) as determined by qRT-PCR (dark grey bars). The RNAseq differential expression values (light grey bars) were included as a reference for comparison. Error bars on qRT-PCR data represent the 95% confidence interval. All genes presented, excluding sdaC, displayed differential expression (ridA/wild-type) with FDR < 0.05, following RNAseq analysis; the corresponding FDR value for sdaC is provided above the bar representative of the RNAseq data.
Features of the transcriptome data beyond metabolic gene expression were noted. For instance, multiple genes in the Salmonella pathogenicity islands (SPIs) were differentially expressed in the two strains. Genes in SPI1 and SPI5 generally had lower expression and those in SPI2 had higher expression in the ridA mutant compared to the wild-type strain, which was reminiscent of the expression pattern during S. enterica serovar Typhimurium replication within murine macrophages7. This similarity was extended with the decrease in expression of the flagellar biosynthetic operons and several genes involved in chemotaxis in the ridA mutant.
Transcription profiling uncovers a role for RidA in motility
Without exception, expression of the genes involved in assembly of flagella was lower, with the majority being more than two-fold lower, in a ridA mutant than wild-type strain. Expression of genes involved in chemotaxis, including cheM, cheA and cheY, was also decreased in a ridA mutant (2.34, 1.68, 1.78, -fold, respectively). This level of differential expression can generate a detectable motility defect31, and the swimming motility of the wild-type and ridA mutant strains was tested. As predicted by the transcript levels, the ridA mutant was less motile than the wild-type strain on minimal medium (Fig. 4). Quantification of the zone of swimming showed motility in ridA mutants was ~40% less than wildtype (Table 1), and was restored to wild-type by the expression of ridA in trans (Table 1, ln 5–8). It was formally possible that the slight growth defect of a ridA mutant on minimal medium was responsible for the ridA decrease in motility. The addition of glycine, which restored wild-type growth to a ridA strain24, failed to restore full motility to the mutant (Table 1, ln 3–4). This result supported the conclusion that reduced motility reflected a new consequence of a ridA mutation.

A ridA mutation leads to a defect in swimming motility. Representative image of swimming motility halos for ridA and wild-type strains grown on minimal glucose (11 mM) motility plates (0.25% agar). Following inoculation using 1 uL overnight cultures grown in LB medium, plates were allowed to incubate 20 h at 30 °C. Diameter using the outermost edge for each halo was measured to determine zone of swimming motility. Two biological replicates are shown for each strain.
Accumulation of 2-aminoacrylate (2AA) generates a motility defect
Metabolic phenotypes of a ridA mutant have been attributed to the accumulation of 2-aminoacrylate, which is generated by the serine/threonine dehydratase IlvA, using serine as a substrate17. Three lines of evidence supported the role of IlvA generated 2AA in compromising motility of a ridA mutant (Table 2). First, exogenous threonine restored motility (ln 1–4), presumably by outcompeting serine for the active site of IlvA, as described for other ridA mutant phenotypes16,32. Second, addition of isoleucine restored motility (ln 5,6), presumably by allosteric inhibition of IlvA33, that decreased the formation of 2AA25. The latter conclusion was further supported by strains carrying the ilvA219 allele, which encodes an IlvAL447F variant, that is resistant to allosteric inhibition by isoleucine9. When the ilvA219 allele replaced wild-type ilvA, data in Table 2 (ln 7–10) showed that with or without isoleucine, a ridA mutation decreased motility by ~35%. Finally, by increasing the production of 2AA, the motility was further decreased (Table 2, ln 11–16). In strains carrying a 2AA generating system (ABcc1), there are two copies of ilvA, the wild-type allele in the chromosome and the ilvA219 allele under the control of an inducible P BAD in the chromosome34. Isogenic ridA and ridA+ strains carrying the ABcc1 construct were generated. In this experiment, without induction of ilvA219, the ridA mutant had 20% less motility than the wild-type strain. When expression of ilvA219 was induced, the ridA mutant had a greater than 40% defect in relative motility. Addition of isoleucine partially restored motility, consistent with the presence of the wild-type IlvA, which is sensitive to allosteric inhibition. Taken together the data support the conclusion that like previously characterized phenotypes, the effect of the ridA mutation on motility is due to the accumulation of 2AA generated by IlvA.
Lowered FlhD4C2 activity can explain reduced motility but not SPI expression
Biosynthesis of the S. enterica flagellar apparatus is controlled by a well-characterized regulatory cascade, and expression/activity of the master regulator FlhD4C2 is tightly regulated. Figure 5 schematically represents the relevant genes and their general position in this cascade. The ridA mutant had decreased expression of several operons that are directly, or indirectly, activated by the FlhD4C2 master regulator. These genes included napFDAG, flg and fli genes and those in SPI I, as indicated in Figure 2 35,36. A simple scenario would suggest the function of the FlhD4C2 activator complex was reduced in a ridA mutant, resulting in the decreased expression of its regulon. However, the expression of flhD and flhC were significantly (FDR < 0.05) higher in the ridA strain (1.64, 1.58 –fold, respectively). These two results could be reconciled if the ridA mutation generated a post-transcriptional effect that resulted in differential function of FlhD4C2 in the two strains.

General regulatory cascade involving FlhD4C2. Schematic of the model for regulation involving FlhD4C2 in E. coli is depicted. Arrows and blocked arrows indicate positive and negative regulation, respectively. The four small Hfq-dependent non-coding regulatory RNAs arcZ, omrA, omrB, and oxyS, negatively regulate motility by binding with the 5′ untranslated region of flhDC and preventing translation43. YdiV acts as an anti-FlhD4C2 factor to disrupt promoter binding37. FliZ can counterbalance this affect by repressing ydiV expression as well as increasing FlhD4C2 protein abundance in an YdiV-independent manner39. FliZ also promotes increased Salmonella pathogenicity island 1 (SPI1) transcription58.
If accumulation of 2AA impacted one of the characterized regulators of flhDC translation or FlhD4C2-dependent promoter binding, elimination of the respective regulatory component would nullify the effect of the ridA mutation. Lesions in the small regulatory RNAs omrBA, micA, arcZ, oxyS, as well as rpoS, each of which post-transcriptionally regulate FlhD4C2, were introduced to isogenic ridA strains. The motility of the resulting strains was determined, and the data are in Table 3. Mutations in arcZ and rpoS caused no detectable defect in motility compared to wild-type and did not impact the effect of the ridA mutation (Table 3, ln 5–6, 13–14). Mutations in micA, omrBA, or oxyS decreased motility slightly (~20–25%) with a ridA mutation further decreasing the motility by ~30% (Table 3, ln 3–4, 7–10). The proteins YdiV and FliZ also regulate FlhD4C2 activity post-transcriptionally. YdiV acts on FlhD4C2 to disrupt promoter binding37 (Fig. 5). FliZ can repress ydiV expression, indirectly activating FlhD4C2 function38; as well as increase FlhD4C2 protein levels through an unknown YdiV-independent mechanism39 (Fig. 5). In the absence of FliZ, expression of genes activated by FlhD4C2 is reduced ~2-fold31, similar to the magnitude resulting in the ridA mutant (Fig. 2). Deletion of fliZ decreased motility of S. enterica by ~43%. Inclusion of a ridA mutation resulted in a strain that had 56% the motility of the parental fliZ strain (Table 3, ln 15–16) and suggested these two mutations had independent (additive) effects on motility. Deletion of ydiV slightly (~7%) increased motility of wild-type S. enterica and the impact of a ridA mutation introduced into this background was consistent with the ~30% decrease repeatedly observed (Table 3, ln 11–12). Taken together, these data did not support a role for 2AA acting via a known regulator of FlhD4C2 translation or promoter binding of the complex. Representative images for the motility measurements given in Tables 1–3 were compiled and incorporated into Supplementary Figure S1.
To investigate the scope of flhDC involvement of the differential expression of motility, chemotaxis, and SPI-encoded genes in a ridA mutant, qRT-PCR compared the expression of representative loci (napF, fliI, fliC, cheM, invA, ssaV, and sopB) between a ridA flhDC::cat mutant (DM15818) and flhDC::cat wild-type (DM15817) (Fig. 6). Welch’s two-tailed t-test revealed that elimination of flhDC significantly (p-value < 0.05) changed the relative expression (ridA:wild-type) of the napF, fliI, and cheM genes. Strikingly, expression of fliC was not significantly (p < 0.05) different between the flhDC and flhDC ridA strains. These data supported a model that a single perturbation affecting FlhD4C2 activity, directly or indirectly, could be responsible for the expression pattern of genes involved in flagellar biosynthesis, nitrate reduction, and chemotaxis. It is formally possible that 2AA interacts directly with FlhD4C2 to reduce its activity, but since free 2AA is not known to damage enzymes lacking a PLP cofactor, a more likely model suggests a disruption of the metabolic network to which a post-transcriptional regulator of flhDC responds.

Differential expression is impacted by status of FlhD4C2 and growth medium. Expression of a subset of genes was monitored in a ridA strain (DM3480) relative to the isogenic wild-type strain (DM9404) by qRT-PCR. The strains were grown in minimal glucose medium (white bars) and minimal medium with 1 mM L-glycine (dark grey bars). Gene expression was similarly measured in an flhDC ridA strain (DM15817) relative to the isogenic flhDC strain (DM15818) when they were grown in minimal glucose medium (light grey bars). Error bars represent the 95% confidence interval.
Exogenous glycine overrides ridA mutant effect on SPI expression
The flhDC mutation did not alter the influence a ridA mutation had on transcription of genes located within SPI1 (invA), SPI2 (ssaV), and SPI5 (sopB). These data suggested that the accumulation of 2AA, caused by the ridA mutation, affected expression of genes in SPI1, SPI2, and SPI5 by an FlhD4C2-independent mechanism. Exogenous glycine eliminates many growth phenotypes of a ridA mutant because it bypasses the need for serine hydroxymethyl transferase (GlyA), which is a primary target of 2AA in S. enterica 24. qRT-PCR compared the expression of relevant loci (napF, fliI, fliC, cheM, invA, ssaV, and sopB) between ridA (DM3480) and wild-type (DM9404) strains grown on minimal medium supplemented with 1 mM glycine (Fig. 6, dark grey bars). Importantly, based on Welch’s two-tailed t-test, the relative expression (ridA:wild-type) of the napF, fliI, cheM, invA, and sopB loci was significantly different (p-value < 0.05) when the strains were grown in the presence vs. absence of glycine in the medium. Further, when considered as an isolated experiment, expression of cheM, invA, ssaV, and sopB was not significantly (p-value < 0.05) different between the ridA and wild-type backgrounds, when the cells were grown in medium with glycine. Taken together, these data suggested that exogenous glycine eliminated the effect of a ridA mutation on the expression of cheM, invA, ssaV, and sopB, but napF, fliI, and fliC expression was still significantly (p-value < 0.05) reduced in the ridA mutant. The latter result was consistent with the finding that glycine did not completely restore motility to the ridA mutant (Table 1, ln 3–4). The effect a ridA mutation had on the transcription of napF, fliI, and cheM was affected by both an flhDC deletion and glycine addition, suggesting both FlhD4C2 activity and glycine/5,10-methylene-tetrahydrofolate influence the expression of these genes19,24.
Discussion
Differential gene expression was used to probe the global consequences of eliminating the RidA protein in S. enterica. The transcription dataset described herein made a few points. First, the biochemical defects previously identified in ridA mutants, with the possible exception of DadX, did not alter the transcription of genes in the respective metabolic pathways affected. This result was consistent with genetic data that ridA mutants have compromised enzyme activity but, in the absence of further perturbations, maintain a robust and functional metabolism. This observation is consistent with the capacity of microbes to adapt to environment changes by maintaining a robust metabolic network.
Second, the expression pattern of ridA vs. wild-type paralleled the expressional changes found with cells replicating in a macrophage, compared to the same strain grown in vitro 7. Similar expressional changes were seen for the genes associated with nitrate reduction, methionine biosynthesis, chemotaxis, flagellar biosynthesis, and genes encoded on SPI1, SPI2, and SPI5 (Fig. 2). These results hint at similarities between the metabolic state of S. enterica cells that accumulate 2AA and what the cells face during infection of a host. This similarity was emphasized by the coordinated response of genes encoded by the three different SPIs, which are all required for Salmonella enteropathogenicity40,41. Serine hydroxymethyl transferase (GlyA) is compromised, (i.e., glycine and C1 units are reduced) in strains lacking ridA 24. By adding glycine and restoring one-carbon unit abundance in a ridA mutant, the differential expression of these SPI loci was minimized. Further work will be needed to extend this correlation and determine whether the ridA paradigm can provide a model to extend the understanding of the intracellular environment and the factors influencing the response of S. enterica to the intracellular environment of the macrophage.
A third feature of the data was the reduced expression of flagellar biosynthesis genes, chemotaxis genes, and periplasmic nitrate reductase (nap) genes in a ridA mutant. Consistent with the expression pattern, ridA mutants had decreased motility, when compared to the parental strain. As with previously characterized phenotypes of a ridA mutant, the lowered motility was a consequence of 2AA accumulation, although the specific target and resulting mechanism were not clear. The only targets of 2AA characterized thus far are PLP-dependent enzymes, where the reactive 2AA enamine attacks the PLP in an active site to lock the respective enzyme in an inactive state17,19,22. There are no PLP-dependent enzymes directly involved in motility, suggesting either a new class of targets for 2AA, or an indirect response to a targeted PLP-dependent enzyme(s). The master activator, FlhD4C2, controls a significant number of the genes that are differentially regulated in a ridA mutant, including the periplasmic nitrate reductase (nap) associated genes. The qRT-PCR data showed that the transcriptional remodeling in a ridA mutant is multifaceted, even when only considering pathogenesis-associated genes (Fig. 5). Significantly, the differential expression of the nap operon, chemotaxis genes, and flagellar biosynthesis genes in a ridA mutant required an intact flhDC locus, while the differences in SPI gene expression were flhDC-independent. The flhDC-dependent link between the effect of 2AA on the nap operon and motility-associated gene expression suggested a need to coordinate nitrate reduction and motility44.
In total, the comparative transcription analyses herein identified a new motility phenotype that results from the accumulation of 2AA in S. enterica strains lacking ridA. The enamine deaminase activity of RidA is conserved across all domains of life, suggesting 2AA accumulation is a metabolic concern in other Enterobacteriaceae and an even wider set of organisms16,23,34,45 E. coli RidA has been reported to act as a chaperon to protect against reactive oxygen species (ROS) toxicity, specifically hypochlorite42. Significantly, ROS stress acts to repress the flagellar biosynthesis machinery43, making it a formal possibility that the chaperon role of RidA is involved in the motility defect described here. However, the genetic data linking the motility defect to 2AA production appeared to eliminate this possibility.
This study emphasized the ability of transcriptome analyses to provide global metabolic snapshots that may not result in a detectable growth phenotype. It is likely that some of the differentially expressed genes that failed to meet the arbitrary two-fold threshold will prove to be physiologically relevant. These data provided insights into the endogenous environment resulting from the accumulation of 2AA in Salmonella and uncovered areas of metabolism that may be targeted by metabolic stress caused by 2AA. Identifying additional enzymes that are affected by 2AA will require targeted genetic and biochemical approaches that are driven by global datasets, like the one described here.
Methods
Bacterial Strains, Plasmids, and Primers
Strains and plasmids used in this work were derivatives of Salmonella enterica subsp. enterica serovar Typhimurium LT2 and are listed in Table S2. Transductional crosses were carried out using the high-frequency general transducing mutant of bacteriophage P22 (HT105/1, int−201)46. New gene disruptions constructed during this study were made using the λ-Red recombinase system described by Datsenko and Wanner47 using the primers listed in Table S3, transduced into relevant genetic backgrounds by selecting chloramphenicol resistance, and verified by PCR. Plasmid pDM1439 was constructed using the high-efficiency cloning method48 to clone the LT2 ridA gene into plasmid pCV1. The resulting construct was confirmed by sequence. pCV1 is a modified pBAD24 plasmid containing BspQ1 sites, conferring ampicillin resistance, and the cloned gene’s expression is inducible by L-(+)-arabinose49. Gene-specific primers for cloning and qRT-PCR analysis were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA) and are listed in Table S3. Primers were designed using Primer 3 software, and evaluated for specificity by melting curve analysis using S. enterica LT2 gDNA prior to running the qPCR experiment.
Culture Media and Chemicals
Minimal medium was no-carbon E medium (NCE) supplemented with 1 mM MgSO4 50, trace minerals51, and 11 mM D-glucose as the sole carbon source. L-(+)-arabinose was added to a final concentration of 0.2 g/L when needed. Difco nutrient browth (NB) (8 g/L) containing NaCL (5 g/L) was used as rich medium. Difco BiTek agar (15 g/L) was added for solid medium. Ampicillin was added when required for plasmid maintenance to rich and minimal medium at 150 and 7.5 μg/mL, respectively. Chloramphenicol (20 μg/mL) and Ampicillin (150 μg/mL) were used in rich medium, during strain construction. All chemicals were obtained from Sigma-Aldrich Chemical Company (St. Louis, MO).
RNA isolation
Relevant strains were grown overnight with shaking at 37°C in quadruplicate (RNA-seq) or triplicate (qRT-PCR) in 2 mL NB. Following incubation, strains we diluted 1:100 into 5 mL fresh minimal medium, containing any additional supplements indicated in the text, and allowed to grow with shaking at 37°C to an optical density at 650 nm (OD650) of 0.6. Each 5 mL sample was quickly centrifuged at 16,000 × g in 1.5 mL Eppendorf tubes, supernatant removed and pellets flash-frozen in liquid nitrogen and kept on dry ice. Total RNA was extracted using the RNAsnap TM method52. Pellets were resuspended in 110 μl boil solution (95% [vol/vol] RNA-grade formamide, 18 mM EDTA, 0.025% [wt/vol] SDS, 1% 2-mercaptoethanol in UltraPureTM (ThermoFisher) distilled water). Samples were incubated at 95°C for 7 min before centrifuging at 16,000 × g for 5 min while still hot and 100 μl supernatant was transferred to a new 1.5 mL Eppendorf tube. 300 μl UltraPureTM water and 40 μl sodium acetate (3 M [pH5.2]) was added and mixed into each sample before adding 400 μl ice-cold ethanol (100%) and incubating on ice 15 min. Samples were centrifuged at 16,000 × g for 15 min at 4°C and ethanol was decanted. 400 μl ethanol (70% [vol/vol]) was added and pellets were immediately centrifuged at 12,000 × g for 10 min at 4°C before decanting ethanol and pellets were allowed to dry 20 min at 25°C. DNA/RNA pellets were resuspended in 90 μl UltraPureTM water at 4°C overnight prior to treatment with RNase-free Turbo DNase (Ambion), and being precipitated once more by sodium acetate-ethanol treatment. Resulting RNA was resuspended at 4°C for 3 h and stored at −80°C until use.
RNA-Seq expression profiling and data analysis
The total RNA was extracted from four independent replicates of S. enterica wild-type and ridA mutant strains. In each of the resulting eight samples, rRNA was removed from 5 μg total RNA using the Ribo-Zero rRNA removal kit (Bacteria; Illumina) following manufacturer’s recommendations. A cDNA library was created from samples containing < 10% rRNA contamination, as measured by the RNA 6000 pico kit for the Agilent 2100 bioanalyzer, and sequenced by the University of Georgia Genomics Facility (GGF) from these RNA samples. Specifically, one hundred nanograms of the rRNA-free RNA was used to create each sequencing library using the KAPA stranded RNA-seq kit (KAPA Biosystems). The RNA libraries were sequenced on an Illumina NextSeq. 500 (150 cycles) Mid Output Flowcell in the paired-end mode with a read length of 75 bp. Processing and differential expression determination of the sequencing data were performed by the Georgia Advanced Computing Resource Center (GACRC) at the University of Georgia. Adaptor removal and quality trimming (Phred score, ~20) of the raw reads were done using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) and Trimmomatic (Usadel). The trimmed reads were mapped to Salmonella enterica subsp. Enterica serovar Typhimurium str. LT2 chromosome, complete genome (RefSeq Accession Number: NC_003197.1) and read counts were generated using the prokaryotic-tailored gene alignment tool EDGE-Pro53. Genes differentially expressed (false discovery rate, FDR < 0.05) between the respective wild-type and ridA strains of S. enterica were identified using DEseq27,30,54. Briefly, an FDR < 0.05 cutoff controls the expected proportion of falsely discovered differentially expressed genes (false positives) to 5% (21 genes) of the identified positive hits meeting this threshold (413 genes). The procedures used by DEseq software to determine the FDR value for each gene is described by Benjamini and Yekutieli27.
qRT-PCR
The total RNA from three independent replicates of each S. enterica strain tested, as described in the text, was extracted. An aliquot of each sample was sent to GGF for quality control analysis and quantification using the RNA 6000 nano kit for the Agilent 2100 bioanalyzer and only samples with an RNA integrity number (RIN) over 5.0 were used55. DNase-treated RNA (600ng) was subjected to first strand cDNA synthesis, using the iScript cDNA synthesis kit (Bio-Rad Laboratories) according to the manufacturer’s protocol. PCRs were performed in an Applied Biosystems 7500 Fast real-time (RT) PCR system. Each reaction was carried out in a total volume of 20 μl on a 96-well optical reaction plate (Applied Biosystems) containing 10 μl FastStart Universal SYBR green Master (ROX) mix (Roche Applied Science), 8 ng cDNA, and two gene-specific primers at a final concentration of 0.5 μM each. The real-time cycling conditions were as follows: 95°C for 20 s, and 40 cycles of 95°C for 3 s and 60°C for 30 s. Melting-curve analysis verified that each reaction mixture contained a single PCR product. The threshold cycle (CT) values of gyrB and rpoB were used as internal controls56,57. Fold changes were calculated using the comparative threshold cycle (ΔΔCT) method29. Briefly, the fold-change in gene expression was calculated using the formula: ridA/wild-type = 2ΔΔCT, where ΔΔCT = ΔCT ridA - ΔCTwild-type and ΔCT = CTtarget gene - CTnormalization gene (gyrB). Gaussian error propagation was used to determine the standard error of the mean (SEM) for ΔΔCT from the ΔCT SEM values, and this value used to calculate the 95% confidence interval for each gene. Welch’s two-tailed t-test was performed on log2 transformed 2ΔΔCT values, using GraphPad Prism 6.0b software. To ensure gyrB was constant under the conditions tested, the relative fold change for the internal control rpoB, whose expression was expected to remain constant across treatments, was calculated as described above. Only those samples with an rpoB fold change between 0.8 and 1.25 were used.
Motility Assays
Motility was monitored on minimal medium motility plates (0.25% agar) using either D-glucose (11 mM) or glycerol (20 mM) as the sole carbon source. The plates were prepared in the morning and allowed to solidify at room temperature un-stacked on the benchtop ~6 h before use in the evening. S. enterica cells were grown to full-density in LB and 1 μl of the culture was inoculated onto the plate and incubated at 30 °C inside of a sealed 6 quart plastic Sterilite container to maintain constant moisture content. Motility halos were measured after 20 h using a ruler with markings every mm; if the halo diameter fell between mm markings, the recorded halo diameter was rounded to the nearest marking. Technical replicates (i.e., plates) were prepared from each of four biological replicates and the data were averaged and expressed as mean (mm) ± SD.
Data Availability
Sequencing data were deposited at the NCBI Gene Expression Omnibus (GSE103146), and can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103146. All other datasets generated in the current study are available from the corresponding author on reasonable request.
References
Bailey, A. M. et al. Exposure of Escherichia coli and Salmonella enterica serovar Typhimurium to triclosan induces a species-specific response, including drug detoxification. J. Antimicrob. Chemoth. 64, 973–985, https://doi.org/10.1093/jac/dkp320 (2009).
Shimizu, K. In Bioreactor Engineering Research and Industrial Applications I Vol. 155 (eds Qin Ye, Jie Bao, & Jian-Jiang Zhong) 1–54 (Springer Berlin Heidelberg, 2015).
Dandekar, T. et al. Salmonella-how a metabolic generalist adopts an intracellular lifestyle during infection. Front. Cell. Infect. Microbiol. 4, 1–11, https://doi.org/10.3389/fcimb.2014.00191 (2015).
Blair, J. M. A., Richmond, G. E., Bailey, A. M., Ivens, A. & Piddock, L. J. V. Choice of bacterial growth medium alters the transcriptome and phenotype of Salmonella enterica Serovar Typhimurium. PLoS ONE 8, e63912, https://doi.org/10.1371/journal.pone.0063912 (2013).
Ishii, N. et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 316, 593–597, https://doi.org/10.1126/science.1136031 (2007).
Alves, P. C. et al. Transcriptomic and metabolomic profiling of ionic liquid stimuli unveils enhanced secondary metabolism in Aspergillus nidulans. BMC Genomics 17, https://doi.org/10.1186/s12864-016-2577-6 (2016).
Srikumar, S. et al. RNA-seq brings new insights to the intra-macrophage transcriptome of Salmonella Typhimurium. PLoS Pathog. 11, e1005262, https://doi.org/10.1371/journal.ppat.1005262 (2015).
Tognetti, V. B. et al. Perturbation of indole-3-butyric acid homeostasis by the UDP-glucosyltransferase UGT74E2 modulates Arabidopsis architecture and water stress tolerance. Plant Cell 22, 2660–2679, https://doi.org/10.1105/tpc.109.071316 (2010).
LaRossa, R. A., Van Dyk, T. K. & Smulski, D. R. Toxic accumulation of alpha-ketobutyrate caused by inhibition of the branched-chain amino acid biosynthetic enzyme acetolactate synthase in Salmonella typhimurium. J. Bacteriol. 169, 1372–1378, https://doi.org/10.1128/jb.169.4.1372-1378.1987 (1987).
Hama, H., Kayahara, T., Tsuda, M. & Tsuchiya, T. Inhibition of homserine dehydrogenase I by L-serine in Escherichia coli. J.Biochem. 109, 604–608, https://doi.org/10.1093/oxfordjournals.jbchem.a123427 (1991).
Zhang, X., El-Hajj, Z. W. & Newman, E. Deficiency in L-serine deaminase interferes with one-carbon metabolism and cell wall synthesis in Escherichia coli K-12. J. Bacteriol. 192, 5515–5525, https://doi.org/10.1128/JB.00748-10 (2010).
Kim, J., Kershner, J. P., Novikov, Y., Shoemaker, R. K. & Copley, S. D. Three serendipitous pathways in E. coli can bypass a block in pyridoxal-5′-phosphate synthesis. Mol. Syst. Biol. 6, 436, https://doi.org/10.1038/msb.2010.88 (2010).
Saxild, H. H. & Nygaard, P. Regulation of levels of purine biosynthetic enzymes in Bacillus subtilis: effects of changing purine nucleotide pools. Microbiology 137, 2387–2394, https://doi.org/10.1186/s12934-014-0101-8 (1991).
Stepanek, J. J., Schäkermann, S., Wenzel, M., Prochnow, P. & Bandow, J. E. Purine biosynthesis is the bottleneck in trimethoprim-treated Bacillus subtilis. Proteom. Clin. Appl. 10, 1036–1048, https://doi.org/10.1002/prca.201600039 (2016).
Ernst, D. C., Lambrecht, J. A., Schomer, R. A. & Downs, D. M. Endogenous synthesis of 2-aminoacrylate contributes to cysteine sensitivity in Salmonella enterica. J. Bacteriol. 196, 3335–3342, https://doi.org/10.1128/jb.01960-14 (2014).
Lambrecht, J. A., Flynn, J. M. & Downs, D. M. Conserved YjgF protein family deaminates reactive enamine/imine intermediates of pyridoxal 5′-phosphate (PLP)-dependent enzyme reactions. J. Biol. Chem. 287, 3454–3461, https://doi.org/10.1074/jbc.M111.304477 (2012).
Lambrecht, J. A., Schmitz, G. E. & Downs, D. M. RidA proteins prevent metabolic damage inflicted by PLP-dependent dehydratases in all domains of life. MBio 4, e00033–00013, https://doi.org/10.1128/mBio.00033-13 (2013).
Schmitz, G. & Downs, D. M. Reduced transaminase B (IlvE) activity caused by the lack of yjgF Is dependent on the status of threonine deaminase (IlvA) in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 186, 803–810, https://doi.org/10.1128/jb.186.3.803-810.2004 (2004).
Flynn, J. M., Christopherson, M. R. & Downs, D. M. Decreased coenzyme A levels in ridA mutant strains of Salmonella enterica result from inactivated serine hydroxymethyltransferase. Mol. Microbiol. 89, 751–759, https://doi.org/10.1111/mmi.12313 (2013).
Chargaff, E. & Sprinson, D. B. Studies on the mechanism of deamination of serine and threonine in biological systems. J. Biol. Chem. 151, 273–280 (1943).
Phillips, A. T. & Wood, W. A. The mechanism of action of 5′-adenylic acid-activated threonine dehydratase. J. Biol. Chem. 240, 4703–4709, PMID: 5321308 (1965).
Flynn, J. M. & Downs, D. M. In the absence of RidA, endogenous 2-aminoacrylate inactivates alanine racemases by modifying the pyridoxal 5’-phosphate cofactor. J. Bacteriol. 195, 3603–3609, https://doi.org/10.1128/JB.00463-13 (2013).
Niehaus, T. D. et al. Genomic and experimental evidence for multiple metabolic functions in the RidA/YjgF/YER057c/UK114 (Rid) protein family. BMC Genomics 16, 382, https://doi.org/10.1186/s12864-015-1584-3 (2015).
Ernst, D. C. & Downs, D. M. 2-Aminoacrylate stress induces a context-dependent glycine requirement in ridA strains of Salmonella enterica. J. Bacteriol. 198, 536–543, https://doi.org/10.1128/JB.00804-15 (2016).
Christopherson, M. R., Schmitz, G. E. & Downs, D. M. YjgF is required for isoleucine biosynthesis when Salmonella enterica is grown on pyruvate medium. J. Bacteriol. 190, 3057–3062, https://doi.org/10.1128/JB.01700-07 (2008).
Davletova, S. The zinc-finger protein Zat12 Plays a central role in reactive oxygen and abiotic stress signaling in Arabidopsis. Plant Physiol 139, 847–856, https://doi.org/10.1104/pp.105.068254 (2005).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat Methodol 57, 289–300, https://doi.org/10.2307/2346101 (1995).
Zhi, J., Mathew, E. & Freundlich, M. Lrp binds to two regions in the dadAX promoter region of Escherichia coli to repress and activate transcription directly. Mol. Microbiol. 32, 29–40, https://doi.org/10.1128/JB.185.6.1886-1894.2003 (1999).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Robles, J. A. et al. Efficient experimental design and analysis strategies for the detection of differential expression using RNA-sequencing. BMC Genomics 13, 484, https://doi.org/10.1186/1471-2164-13-484 (2012).
Saini, S., Brown, J. D., Aldridge, P. D. & Rao, C. V. FliZ Is a posttranslational activator of FlhD4C2-dependent flagellar gene expression. J. Bacteriol. 190, 4979–4988, https://doi.org/10.1128/JB.01996-07 (2008).
Christopherson, M. R., Lambrecht, J. A., Downs, D. & Downs, D. M. Suppressor analyses identify threonine as a modulator of ridA mutant phenotypes in Salmonella enterica. PLoS One 7, e43082, https://doi.org/10.1371/journal.pone.0043082 (2012).
Umbarger, H. E. & Brown, B. Threonine deamination in Escherichia coli. II. Evidence for two L-threonine deaminases. J Bacteriol. 73, 105–112, PMID: 13405870 (1957).
Borchert, A. J. & Downs, D. M. The response to 2-aminoacrylate differs in E. coli and S. enterica, despite shared metabolic components. J. Bacteriol. JB.00140–00117 https://doi.org/10.1128/JB.00140-17 (2017).
Prüss, B. M. et al. FlhD/FlhC is a regulator of anaerobic respiration and the Entner-Doudoroff pathway through induction of the methyl-accepting chemotaxis protein Aer. J. Bacteriol. 185, 534–543, https://doi.org/10.1128/JB.185.2.534-543.2003 (2003).
Iyoda, S., Kamidoi, T., Hirose, K., Kutsukake, K. & Watanabe, H. A flagellar gene fliZ regulates the expression of invasion genes and virulence phenotype in Salmonella enterica serovar Typhimurium. Microb. Pathogenesis 30, 81–90, https://doi.org/10.1006/mpat.2000.0409 (2001).
Wada, T. et al. EAL domain protein YdiV acts as an anti-FlhD4C2 factor responsible for nutritional control of the flagellar regulon in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 193, 1600–1611, https://doi.org/10.1128/JB.01494-10 (2011).
Wada, T., Tanabe, Y. & Kutsukake, K. FliZ acts as a repressor of the ydiV gene, which encodes an anti-FlhD4C2 factor of the flagellar regulon in Salmonella enterica serovar Typhimurium. J. Bacteriol. 193, 5191–5198, https://doi.org/10.1128/JB.05441-11 (2011).
Stewart, M. K. & Cookson, B. T. Mutually repressing repressor functions and multi-layered cellular heterogeneity regulate the bistable Salmonella fliC census. Molecular Microbiology 94, 1272–1284, https://doi.org/10.1111/mmi.12828 (2014).
Rice, C. J., Ramachandran, V. K., Shearer, N. & Thompson, A. Transcriptional and post-transcriptional modulation of SPI1 and SPI2 expression by ppGpp, RpoS and DksA in Salmonella enterica sv Typhimurium. PLoS ONE 10, e0127523, https://doi.org/10.1371/journal.pone.0127523 (2015).
Wood, M. W. et al. Identification of a pathogenicity island required for Salmonella enteropathogenicity. Mol. Microbiol. 29, 883–891, https://doi.org/10.1046/j.1365-2958.1998.00984.x (1998).
Müller, A. et al. Activation of RidA chaperone function by N-chlorination. Nat. Commun. 5, 5804, https://doi.org/10.1038/ncomms6804 (2014).
De Lay, N. & Gottesman, S. A complex network of small non-coding RNAs regulate motility in Escherichia coli. Mol. Microbiol. 86, 524–538, https://doi.org/10.1111/j.1365-2958.2012.08209.x (2012).
Prüss, B. M., Liu, X., Hendrickson, W. & Matsumura, P. FlhD/FlhC-regulated promoters analyzed by gene array and lacZ gene fusions. FEMS Microbiol. Lett. 197, 91–97, https://doi.org/10.1111/j.1574-6968.2001.tb10588.x (2001).
Niehaus, T. D. et al. Arabidopsis and Maize RidA proteins preempt reactive enamine/imine damage to branched-chain amino acid biosynthesis in plastids. Plant Cell 26, 3010–3022, https://doi.org/10.1105/tpc.114.126854 (2014).
Schmieger, H. Phage P22-mutants with increased or decreased transduction abilities. Mol. Gen. Genet. 119, 75–88, https://doi.org/10.1007/BF00270447 (1972).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645, https://doi.org/10.1073/pnas.120163297 (2000).
Galloway, N. R., Toutkoushian, H., Nune, M., Bose, N. & Momany, C. Rapid cloning for protein crystallography using type IIS restriction enzymes. Cryst. Growth Des. 13, 2833–2839, https://doi.org/10.1021/cg400171z (2013).
VanDrisse, C. M. & Escalante-Semerena, J. C. New high-cloning-efficiency vectors for complementation studies and recombinant protein overproduction in Escherichia coli and Salmonella enterica. Plasmid 86, 1–6, https://doi.org/10.1016/j.plasmid.2016.05.001 (2016).
Vogel, H. J. & Bonner, D. M. Acetylornithase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218, 97–106, PMID: 13278318 (1956).
Balch, W. E. & Wolfe, R. S. New approach to the cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminantium in a pressurized atmosphere. Appl. Environ. Microbiol 32, 781–791, PMID: 827241 (1976).
Stead, M. B. et al. RNAsnap: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 40, e156, https://doi.org/10.1093/nar/gks680 (2012).
Magoc, T., Wood, D. & Salzberg, S. L. EDGE-pro: estimated degree of gene expression in prokaryotic genomes. Evol. Bioinform. Online 9, 127–136, https://doi.org/10.4137/EBO.S11250 (2013).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106, https://doi.org/10.1186/gb-2010-11-10-r106 (2010).
Fleige, S. & Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Aspects Med. 27, 126–139, https://doi.org/10.1016/j.mam.2005.12.003 (2006).
Heng, S. S. J., Chan, O. Y. W., Keng, B. M. H. & Ling, M. H. T. Glucan biosynthesis protein G is a suitable reference gene in Escherichia coli K-12. ISRN Microbiol. 2011, 1–6, https://doi.org/10.5402/2011/469053 (2011).
Czapski, T. R. & Trun, N. Expression of csp genes in E. coli K-12 in defined rich and defined minimal media during normal growth, and after cold-shock. Gene 547, 91–97, https://doi.org/10.1016/j.gene.2014.06.033 (2014).
Chubiz, J. E. C., Golubeva, Y. A., Lin, D., Miller, L. D. & Slauch, J. M. FliZ regulates expression of the Salmonella pathogenicity island 1 invasion locus by controlling HilD protein activity in Salmonella enterica serovar Typhimurium. J. Bacteriol. 192, 6261–6270, https://doi.org/10.1128/JB.00635-10 (2010).
Acknowledgements
This work was supported by competitive grant GM095837 from the National Institutes of Health (DMD). The authors acknowledge valuable discussions with Magdy Alabady of the Georgia Genome Facility and Walt Lorenz of the Georgia Advanced Computing Resource Center. We thank Jorge Escalante-Semerena for use of the Applied Biosystems 7500 Fast RT-PCR system.
Author information
Authors and Affiliations
Contributions
A.J.B. and D.M.D. designed the research; A.J.B. performed the experiments, analyzed the data, and prepared the manuscript; D.M.D. revised the manuscript; A.J.B. and D.M.D. reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borchert, A.J., Downs, D.M. Endogenously generated 2-aminoacrylate inhibits motility in Salmonella enterica . Sci Rep 7, 12971 (2017). https://doi.org/10.1038/s41598-017-13030-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13030-x
- Springer Nature Limited