
Abstract
The NTR system is the major regulator of nitrogen metabolism in Bacteria. Despite its broad and well-known role in the assimilation, biosynthesis and recycling of nitrogenous molecules, little is known about its role in carbon metabolism. In this work, we present a new facet of the NTR system in the control of NADPH concentration and the biosynthesis of molecules dependent on reduced coenzyme in Herbaspirillum seropedicae SmR1. We demonstrated that a ntrC mutant strain accumulated high levels of polyhydroxybutyrate (PHB), reaching levels up to 2-fold higher than the parental strain. In the absence of NtrC, the activity of glucose-6-phosphate dehydrogenase (encoded by zwf) increased by 2.8-fold, consequently leading to a 2.1-fold increase in the NADPH/NADP+ ratio. A GFP fusion showed that expression of zwf is likewise controlled by NtrC. The increase in NADPH availability stimulated the production of polyhydroxybutyrate regardless the C/N ratio in the medium. The mutant ntrC was more resistant to H2O2 exposure and controlled the propagation of ROS when facing the oxidative condition, a phenotype associated with the increase in PHB content.
Similar content being viewed by others

Introduction
Poly-3-hydroxybutyrate (PHB) is an aliphatic polyester member of the polyhydroxyalkanoates (PHA) family synthesised by some bacteria as carbon and reducing equivalents storage1,2. Usually, bacteria produce PHB under conditions of carbon excess and low levels nitrogen, phosphate and oxygen3. At least three enzymes are involved in its synthesis: 3-ketothiolase, acetoacetyl-CoA reductase and PHA synthase encoded by phaA, phaB and phaC respectively4. These enzymes catalyse the condensation of acetyl-CoA forming acetoacetyl-CoA, then reduction of acetoacetyl-CoA to 3-hydroxybutyryl-CoA (3HB-CoA) and finally polymerisation of 3HB-CoA to yield PHB5.
PHB is a thermoplastic biodegradable polymer with physicochemical properties comparable to recalcitrant oil-based plastics such as polypropylene and polystyrene6. Although PHB is a sustainable alternative to such plastics, its production cost is still considerably higher, creating a necessity for engineering of PHB overproducer strains and process optimisation. So far, several studies of metabolic engineering for improving microbial PHB production have been reported7,8,9,10,11. The majority of these studies focused on carbon metabolism pathways engineering and the improvement of NAD(P)H availability. Although the carbon to nitrogen (C/N) ratio is a major factor controlling PHB accumulation in several bacteria12, little attention has been given to re-engineering the nitrogen metabolism. In bacterial cultivation for the production of PHB, the addition of high ammonium concentration to the medium improves the cell growth but reduces the production of PHB13. The negative effect is due to that with a high concentration of ammonium the bacterium diverts much of the carbon skeleton to produce amino acids and other nitrogenous molecules. Therefore, a better understanding of the carbon and nitrogen metabolism interrelationships can contribute to engineering better PHB producers.
Herbaspirillum seropedicae SmR1 is a diazotrophic β-Proteobacterium that associates beneficially with economically relevant species of Poaceae14 and produces PHB as means of carbon and energy storage15,16,17. Thirteen genes probably involved in PHB metabolism were identified in H. seropedicae SmR1 by genome analysis18, including four phaC, two phaZ and two phaP genes, encoding PHA synthases, PHA depolymerases and phasins, respectively. The PHB synthesis in H. seropedicae SmR1 seems to be entirely supported by the PHA synthase encoded by phaC1 since its deletion abolishes PHB accumulation19. The PHA depolymerases are the enzymes that mobilise the PHB granules releasing 3-hydroxybutyrate (3HB) or oligos of 3HB. The phasins are the proteins more abundant on the surface of PHB. They are important to avoid the coalescence of the granules and also to control their number and size. Recently, the role of the two phasins (PhaP) was described in H. seropedicae SmR1, showing that PhaP1 is the main phasin and highly expressed in conditions of PHB production, on the other hand, PhaP2 acts as backup phasin when phaP1 is not expressed17. The nitrogen metabolism and its regulation have been extensively studied in H. seropedicae SmR120, turning H. seropedicae a model to investigate the impact of C/N ratio on PHB production. Specifically, several components of the NTR system, the master nitrogen regulatory system, were studied in H. seropedicae SmR1. The NTR system is composed of a cascade of regulatory proteins controlling the nitrogen assimilation in Bacteria21 (Fig. 1). Five proteins are involved: GlnD, a uridylyl transferase/uridylyl-removing enzyme; the signal-transducing proteins GlnB (PII) and GlnK; NtrB, a histidine protein kinase; and the response regulator, NtrC21,22. Under limiting nitrogen conditions, GlnD uridylylates PII which as PII-UMP enables NtrB to phosphorylate NtrC. Phosphorylated NtrC positively activates several nitrogen assimilation operons dependent of the sigma 54 (RpoN) factor, including glnAntrBntrC23. Under high nitrogen conditions, GlnD deuridylylates PII-UMP, activating in turn, the dephosphorylation of NtrC by NtrB and hence rendering it inactive. The influence of the NTR system on carbon metabolism, specifically on PHB metabolism, was already studied in Azospirillum brasilense Sp724,25. The authors found that the ntrC mutant could produce PHB in both low- and high-C/N ratio media, while the wild-type had no significant PHB production in low-C/N ratio24. Also, mutants in both PII proteins genes (glnB and glnZ) or glnD had higher contents of PHB than the wild type under low-C/N ratio25. These findings indicate that PHB synthesis is coupled with the nitrogen levels via the components of the NTR system. To better understand how the NTR system affects PHB production in bacteria, we investigated the PHB synthesis in NTR mutants of H. seropedicae SmR1. The results obtained in this work reveal a new facet of how the NTR system can influence the carbon metabolism, especially in the synthesis and accumulation of PHB.

The NTR system and its regulation. In nitrogen excess, high glutamine concentration activates the deuridylylation of GlnB by GlnD. The unmodified form of GlnB stimulates the adenylyl transfer activity of GlnE, resulting in glutamine synthetase adenylation (GS-AMP) and therefore its inactivation. GlnB also interacts with the NtrB stimulating its phosphatase activity, resulting the dephosphorylation of the transcriptionally active form of NtrC. On the other hand, when the bacterium faces a nitrogen-limiting condition, the low glutamine concentration leads to the uridylylation of GlnB (GlnB-UMP) by GlnD. GlnB-UMP stimulates the deadenylylation of GS-AMP, turning GS active. The 2-oxoglutarate (2-OG) is an effector of GlnB, and its concentration is high under nitrogen-limiting conditions.
Results
The ntrC mutant of H. seropedicae SmR1 produces more PHB than the parental strain
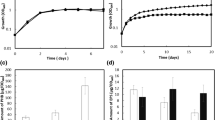
The C/N ratio is one of the key factors controlling PHB accumulation in bacteria26,27. Accordingly, we anticipated that mutant strains of the NTR system would be a useful tool to investigate its involvement in PHB synthesis. We measured the content of PHB in glnB, glnK, glnD, amtB and ntrC knock-out mutants as well as in the parental strain H. seropedicae SmR1. The strains were grown in low C/N ratio (37 mM DL-malate and 20 mM NH4Cl) and high C/N ratio (37 mM DL-malate and 5 mM NH4Cl). The cultures were sampled in four stages of growth: early-exponential phase (OD600 0.4), mid-exponential phase (OD600 0.8), early stationary phase (OD600 1.2) and late stationary phase (OD600 1.6). The PHB contents of the parental, glnK and glnD strains were very similar since their maximum PHB content was around 25%/cdw (cell dry weight) in high C/N (Fig. 2). The PHB was reduced to 16%/cdw when the strains grew in low C/N. Interestingly, the glnB and amtB mutants presented a significant reduction of PHB accumulation compared to the parental strain (Fig. 2). This effect was remarkable in low C/N where glnB and amtB mutants produced only half of the PHB level of the parental strain. In contrast, PHB production in the ntrC mutant was unregulated and presented the highest content of PHB amongst all strains analysed, reaching 32% of PHB regardless the initial C/N ratio (Fig. 2). This higher PHB production regardless the nitrogen level in the medium suggests that ntrC knock-out decouples the synthesis of PHB of the C/N ratio.

PHB accumulation profiles of H. seropedicae SmR1 (parental strain) and the NTR system mutants. Strains grew in NFbHP medium amended with 37 mM DL-malate and 20 mM (A) or 5 mM (B) of ammonium chloride at 30 °C (orbital agitation at 120 rpm). The PHB contents were determined in three independent samples. nd – non-detected.
The ntrC mutant also produces more PHB with monosaccharides as carbon source
As malate is not a conventional substrate for biotechnological applications due to its high price compared to other sources, we measured PHB accumulation in the ntrC mutant on D-glucose and D-xylose, which are monosaccharides highly abundant in cheap feedstocks and agro-industrial residues. The C/N ratios were the same. When grown in the presence of D-glucose, the maximum PHB production of the ntrC mutant was 1.8- and 1.7-fold higher than the parental production, in low and high C/N ratio, respectively (Fig. 3A and D). On D-fructose, the maximum PHB production of the ntrC mutant was 2.0- and 2.15-fold higher than the parental production, in low and high C/N ratio respectively (Fig. 3B and E). Interestingly, when D-xylose was used as the carbon source, in low C/N ratio there was no difference of PHB between both strains, but in high C/N ratio, the ntrC mutant produced 1.5-fold more PHB (Fig. 3C and F). Table 1 shows the maximum content of PHB and the productivities obtained for both strains in the different conditions assayed. The profile of sugar consumption and the data of PHB concentration (g/L) and yield of g PHB/g of substrate for the SmR1 and ntrC strains are shown in the supplemental information (Figure S1 and Tables S1 and S2).

PHB accumulation profiles of H. seropedicae SmR1 and ntrC mutants in D-glucose, D-fructose and D-xylose as sole carbon sources. Strains grew in NFbHP medium amended with 25 mM D-glucose (A and D), 25 mM D-fructose (B and E) or 30 mM D-xylose (C and F). Low C/N ratio media had 20 mM NH4Cl (blue circles), while high C/N ratio media had 5 mM NH4Cl (red circles). PHB contents were determined in three independent samples. Strains were cultivated at 30 °C at 120 rpm (orbital shaking).
Complementation of the ntrC mutant restores PHB production to the parental level
The ntrC gene is clustered in an operon downstream from glnA and ntrB18,28. The ntrC mutant was complemented through pKRT1 conjugation. The pKRT1 is a pLAFR3-derivative containing a glnAntrBC operon copy from H. seropedicae SmR123,29. PHB accumulated in strains harbouring pKRT1 was reduced when grown in malate or glucose, regardless the C/N ratio applied (Fig. 4). Therefore, the complementation of the ntrC mutant demonstrates that the NtrC is directly involved in the higher PHB production observed.

PHB content of ntrC mutant strain carrying an additional copy of the operon glnAntrBntrC of H. seropedicae SmR1. Strains harbouring the plasmids pLAFR3 (empty vector) or pKRT1 (pLAFR3 with the glnAntrBntrC as an insert) grew in NFbHP medium amended with 25 mM D-glucose. Graphs A and C represent the data for SmR1, while B and D for the ntrC mutant at low and high C/N ratio, respectively. Red symbols are non-complemented strains, while blue symbols correspond to the complemented ones. Data represent the average ± standard deviation of three independent samples.
The derepression of the zwf (glucose-6-phosphate dehydrogenase) gene results in high NADPH in the ntrC mutant
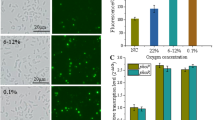
Previous works have shown that NtrC is involved in the regulation of the expression of glucose-6-phosphate dehydrogenase (G6PDH) and glutamate dehydrogenase (GDH)30,31. Specifically, the G6PDH activity is widely implicated in the generation and maintenance of the NADPH/NADP+ ratio32,33. The activity of the NADP+-dependent malic enzyme (ME) is also correlated with NADPH/NADP+ balance34. This observation led us to determine the activity of these enzymes in the parental strain SmR1 and ntrC mutant. The G6PDH and NADP+-dependent ME activities were 2.3- and 1.6-fold higher in the ntrC mutant when grown in NFb-glucose with 20 mM NH4Cl (Fig. 5A and C). GDH activity did not differ statistically between both strains (Fig. 5B). The specific activities of G6PDH, ME and GDH enzymes for the SmR1 and ntrC strains are shown in the Supplemental Figure S2. The repression of zwf transcription by NtrC has been previously reported for the bacterium Pseudomonas putida3,30. The zwf of H. seropedicae is located downstream from pgi (Hsero1099, encoding a phosphoglucoisomerase) and upstream from a gene encoding a transcriptional regulator of the HexR family (Hsero1097) and talB (Hsero1096, encoding a transaldolase). Since the 76 bp region between pgi and zwf seems not to contain a promoter region, the upstream region of pgi was cloned with GFP in the pEKGFP01 to determine the expression profile of the operon carrying zwf in H. seropedicae SmR1 and ntrC mutant. The activity of the Ppgi-gfp fusion in the ntrC mutant was higher than that of the parental strain during all growth phases, achieving a maximum difference of 2.8-fold (Fig. 5D). The transcription of zwf and other genes involved in the Entner-Doudoroff pathway and in the PHB metabolism were compared between the SmR1 and ntrC strains through RNA-seq analysis. The data corroborate the higher expression of zwf in the ntrC mutant (Table S3). Also, the transcription of genes involved in the PHB metabolism was lower in the ntrC mutant (Table S4), indicating that the higher PHB production measured is a consequence of a metabolic factor. This prompted us to measure the NAD(P)H/NAD(P)+ ratio in both strains. The NADH/NAD+ ratio had no statistically significant difference between the parental strain SmR1 complemented or not with an additional copy of the operon glnAntrBntrC cloned into pLAFR3 (pKRT1) (Fig. 5E). The same was observed for the NADH/NAD+ ratio in the ntrC mutant (Fig. 5F). However, the NADPH/NADP+ ratio was statistically higher for both strains when they were not complemented with pKRT1. For the ntrC mutant, the NADPH/NADP+ ratio was 2.1-fold greater than in the parental strain (Fig. 5E and F, comparing the red bars). Taken together these results indicate that the knock-out in ntrC increases the expression of G6PDH and the regeneration of NADP+ in NADPH.

The ntrC mutant presents increased G6PDH activity and NADPH/NADP+ ratio. The G6PDH (A), GDH (B) and ME (C) activities were measured in crude extracts of the SmR1 (black bars) and ntrC (red bars) strains cultivated at 30 °C in NFbHP with 25 mM of glucose and 20 mM of ammonium chloride. (C) The transcriptional activity of the Ppgi-gfp fusion was measured during SmR1 (red circles) and ntrC mutant (blue circles) cultivation at 30 °C in NFbHP with 25 mM of glucose and 20 mM of ammonium chloride. The NADH/NAD+ (black bars) and NADPH/NADP+ (red bars) ratios were measured for strains SmR1 (E), ntrC (F) and both strains complemented with pKRT1. Where appropriate, statistical significance is shown (*−p-value ≤ 0.05, independent two-sample t-test).
The ntrC mutant of H. seropedicae SmR1 is more resistant to oxidative stress caused by hydrogen peroxide
A high NADPH/NADP+ ratio stimulates PHB production35,36. High [NADPH] increases the flux towards PhaB (NADPH-dependent acetoacetyl-CoA reductase), yielding PHB not only as a carbon stock but also a redox sink2,36. Furthermore, NADPH is crucial to anti-oxidative defences in most organisms, ensuring a reductive cellular environment to mitigate the deleterious effects of oxidative species as hydrogen peroxide, hydroxyl radical and superoxide37. Therefore we determine the resistance to oxidative stress of both strains. Cellular growth was assessed in media amended with increasing concentrations of hydrogen peroxide (H2O2). The addition of the oxidant at 6 h of cultivation impaired the growth of the parental strain in concentrations above 0.2 mM, while the ntrC mutant resisted the oxidative shock, recovering growth after one hour even at 5 mM of H2O2, as observed in Fig. 6B. The serial dilution onto agar plates containing H2O2 showed that the ntrC mutant was able to grow in dilution of 104-fold while the parental SmR1 growth only until 102-fold dilution at 0.1 mM H2O2 (Fig. 6C, lower panel). The ROS measurement applying the fluorescent probes H2-DCDFDA showed that the ntrC mutant was able to control ROS propagation, while the parental strain could not maintain the ROS at low levels (Fig. 6D). The fluorescence value of the oxidised H2-DCDFDA increased 2.5-fold from 0 to 2 mM of H2O2 in the parental SmR1, while in the ntrC mutant the oxidation of the probe increased 1.3-fold applying the same treatment with H2O2. Taken these results together, we conclude that the ntrC mutant has a more efficient defence against the oxidative stress, probably due to the NADPH accumulation which can be applied to mitigate the deleterious effects of the oxidative insult. The high NADPH generation also stimulated the PHB synthesis observed in the mutant.

The effect of H2O2 on growth and ROS propagation in the parental strain and the ntrC mutant of H. seropedicae SmR1. Growth kinetics of parental strain SmR1 (A) and ntrC mutant (B) in NFbHP-malate medium in the presence of increasing hydrogen peroxide concentrations. The growth kinetics were determined in three independent samples for each strain. At the time of 6 h hydrogen peroxide was added to the cultures. (C) SmR1 and ntrC mutant grew to stationary phase and serial dilutions were plated onto NFbHP-malate medium containing H2O2 at the indicated concentrations. After two days of incubation at 30 °C, the SmR1 strain presented higher hypersensitivity to H2O2 than the ntrC mutant. (D) Median fluorescence of H2-DCFDA treated cultures during H2O2 stress in SmR1 and ntrC strains. The median fluorescence was determined in three independent samples for each strain. Where appropriate, statistical significance is shown (*−p-value ≤ 0.05, independent two-sample t-test).
Discussion
The nitrogen level in the growth medium is a key factor interfering with PHB production in bacteria38,39,40. Therefore, the interruption of nitrogen regulatory systems can be a useful strategy to reduce the C/N ratio effect on PHB production and ultimately to improve PHB production. Since the NTR system is the master regulator of the nitrogen metabolism in several bacterial species41, wherein we investigate the PHB accumulation using as a model the bacterium H. seropedicae SmR1 and a set of mutants defective in the expression of regulatory proteins of the NTR system. Among all strains evaluated only the ntrC mutant produced higher PHB contents than the parental strain. The ntrC mutant produced around the double of PHB than the parental strain (in % PHB/mg of cdw), in all tested conditions. Similar results were also reported to ntrB and ntrC mutants of A. brasilense Sp724, suggesting that possibly the effect of NtrC on PHB synthesis is widespread among prokaryotes. Based on these findings, we anticipated some possible explanations for our results.
The synthesis of PHB is largely dependent on high levels of acetyl-CoA and NADPH. Normally, both metabolites are in high intracellular concentrations when the bacterium faces a condition of carbon excess and limitation in another nutrient, such as nitrogen1. This condition is permissive for PHB synthesis due to elevating acetyl-CoA level generated by carbon overflow and high NADPH/NADP+ ratio as consequence of nitrogen limitation7,9,35.
Considering that H. seropedicae ntrC mutant produced more PHB than the parental strain even under unfavourable conditions (low C/N ratio) suggests that NtrC somehow is modulating acetyl-CoA and/or NADPH concentrations. A role of NtrC on directly activating transcription of genes involved in PHB synthesis is unlikely since PHB production is higher in the ntrC mutant even under an excess of ammonium, a condition where NtrC is mainly dephosphorylated and therefore inactive22 (Fig. 2A and B). A transcriptomic analysis of the ∆ntrC mutant of the PHA-producing bacterium Pseudomonas putida KT2442 brought important elements to understand the link between NtrC and PHA metabolism3. In the ∆ntrC strain, the transcription of zwf-1 (encoding glucose-6-phosphate dehydrogenase – locus tag PP1022) and gap-1 (glyceraldehyde-3-phosphate dehydrogenase - PP1009) was upregulated 5.7- and 2.6-fold, indicating that NtrC represses their expression. Hervás et al. suggested that repression of zwf shows that NtrC controls hexose catabolism in bacteria, likely to prevent a carbon overflow under nitrogen-limiting conditions for growth3. In another work, the ntrC deletion in P. putida KT2440 also rendered cells more resistant to oxidative stress30. Furthermore, zwf-1 (PP1022) was also up-regulated in the ∆ntrC mutant of P. putida KT2440, as determined by transcriptomic analysis. H. seropedicae SmR1 and P. putida have the same incomplete glycolytic pathway since it lacks 6-phosphofructokinase (PFK-1). As a consequence, the Entner-Doudoroff (ED) pathway converts glucose and fructose into pyruvate and glyceraldehyde-3-phosphate. Particularly, in mutants defective in ntrC expression, it is expected a higher metabolic flux through glucose-6-phosphate dehydrogenase, generating more acetyl-CoA and NADPH, which in turn increases PHB accumulation.
These findings suggest that the up-expression of zwf could be a major factor leading ntrC defective mutants to accumulate more PHB under permissive conditions. The observation that the H. seropedicae ntrC mutant produced more PHB even in unfavourable conditions (low C/N ratio) suggests a higher pool of reducing power as compared to the parental strain. Therefore is likely that the bacterium switches the synthesis of PHB as an electron sink to avoid the deleterious effect of a high reductive environment.
In fact, we have already reported that mutant strains defective in PHB synthesis or accumulation presented a severe growth penalty on glucose17. Probably, as a consequence of the redox imbalance caused by the inability of those mutants to divert electrons towards PHB synthesis.
NtrC is widely recognised as a transcriptional regulator restricted to regulate genes involved in nitrogen metabolism. Indeed, a significant portion of genes corresponding to 2% of the E. coli genome was determined to be under control of the NtrC transcriptional activity, most of them involved in amino acids and ammonium transport, nitrogen assimilation and nitrogen recycling42. Our results demonstrate that NtrC interferes with PHB synthesis, bring to the discussion if NtrC could be one of the connection points between nitrogen and carbon metabolism. Alternatively, the NtrC control of the NADPH recycling may help cells dealing with a stressful condition such as nitrogen limitation. In fact, the glutamate synthase (GOGAT) and the glutamate dehydrogenase (GDH), which are important reactions for ammonium assimilation require NADPH as an electron donor to catalyse the amination of 2-oxoglutarate43. In E. coli, NADPH concentration was homeostatic after ammonium upshift44. Since NADPH consumption increases when ammonium assimilation is higher, it is likely that other pathways are generating the NADPH demanding44. Whether or not the phosphorylation state of NtrC interferes in NADPH production through zwf expression is uncertain. The repression of gdhA encoding the glutamate dehydrogenase in P. putida KT2442 by NtrC was shown to be independent of the phosphorylation state, since both wild-type and the mutant NtrCD55E,S161F (mimicking the phosphorylated protein) can bind to the gdhA promoter and repress transcription31. The effect of phosphorylation could be further studied employing a ntrB mutant of H. seropedicae and complementation of the ntrC mutant with NtrC variants unable to be phosphorylated. Such experiments will determine if the reduction in PHB production observed in the glnB and amtB mutants derived from the phosphorylation state of NtrC.
It would be interesting to investigate further the role of NtrC on PHB synthesis in other PHB-producing models, such as Ralstonia eutropha, Azotobacter vinelandii and P. putida. It would point if the involvement of NtrC on PHB synthesis is conserved among other classes of bacteria, serving as a strategy for metabolic engineering aiming to improve PHB production.
Methods
Bacterial Strains, Plasmids, and Growth Conditions
Strains and plasmids used are listed in Table 2. Escherichia coli strain Top10 (Thermo Fisher Scientific Inc., Waltham, MA, USA) and E. coli S17-145 were used for cloning and conjugational procedures, respectively. E. coli strains grew in LB medium at 37 °C and shaken at 160 rpm. H. seropedicae parental strain SmR146 and mutant strains were cultured in NFbHP media with 37 mM DL-malate and the indicated concentration of NH4Cl at 30 °C and shaken at 120 rpm49. D-glucose at 25 mM, D-fructose at 25 mM and D-xylose at 30 mM were applied as alternative carbon sources for PHB production measurement as a replacement for malate. Two regimes of carbon-to-nitrogen (C/N) ratio were used at the start of cultivation: high C/N ratio with 5 mM of NH4Cl and low C/N with 20 mM of NH4Cl.
Quantification of PHB
PHB was quantified by methanolysis, followed by GC-FID (gas chromatography coupled to a flame ionisation detector) analyses as described previously50 with 5 to 10 mg of lyophilised bacteria. Amounts of PHB in each sample were normalised to cell dry weight (cdw; the weight of the lyophilised bacterial pellet) and expressed as % of PHB cell dry weight−1.
Construction of Ppgi-gfp transcriptional fusion
The intergenic region of the pgi (locus-tag Hsero1099) was amplified using the primers Fw_Ppgi_Hs 5′TATCTCGAGTGTCGGGTTCCTGTTAGCGT 3′, containing a XhoI site (underlined) and Rev_Ppgi_Hs 5′TATACTAGTCATATGGGTCTGGTGTCGGTTGGCGG 3′, containing a SpeI site (underlined) as previously described51. The amplified product was cloned into the sites XhoI and SpeI of the pBBR1MCS-352, generating the pEK07. The reporter gene gfp containing upstream the rbs site B0034 and downstream the double terminator B0015 was extracted from the plasmid BBa_I13504 (Registry of Standard Biological Parts, partsregistry.org) digested with the EcoRI and SpeI enzymes and cloned into pBlueScript II KS(+) digested with EcoRI and XbaI. Then, the gfp cassette was removed by digestion with the XbaI and SacI enzymes and cloned into the pEK07 digested with the same enzymes, generating the pEKGFP01. The pEKGFP01 was transformed in E. coli S17-1 and conjugated to H. seropedicae by bi-parental mating.
Measurement of Ppgi-gfp transcriptional activity
To measure the transcriptional activity of the pgi promoter, 2 μL of saturated cultures containing either the transcriptional fusion pEKGFP01 or an empty pBBR1MCS-3 vector were inoculated in a 96-well microplate containing 200 μL of NFbHP-glucose with 20 mM of NH4Cl as a nitrogen source. The microplate was incubated in an orbital shaker (Incubator Shaker Series I26, New Brunswick™) at 30 °C and 120 rpm. The fluorescence was measured using a Berthold™ TriStar LB 941 using 355 nm filter for excitation and a 535 nm for the emission wavelength. Arbitrary fluorescence units were normalised by OD readings at 600 nm using a Bio-Rad iMark™ Microplate Reader.
Complementation of ntrC mutant
The pKRT1 cosmid was conjugated by bi-parental mating between E. coli S17-1 and H. seropedicae strains. The transconjugant colonies were selected in NFbHP-malate with 20 mM NH4Cl agar with 10 µg/mL of tetracycline. The complemented strains were cultivated in NFbHP-malate and NFbHP-glucose at high and low-C/N ratios.
Determination of enzymatic activities
Glucose-6-phosphate dehydrogenase (G6PDH), glutamate dehydrogenase (GDH) and malic enzyme (ME) activity assays were performed by measuring the formazan production at 585 nm, as previously described52. Formazan is the insoluble product formed by MTT (3-(4,5-dimethyl-thiazolyl-2)-2,5-diphenyl-tetrazolium bromide) reduction. Cells were lysed by sonication on an ice bath. The lysates were centrifuged at 4,000 × g for 10 min at 4 °C. The supernatants were maintained on ice until assay setup. The reaction contained 300 μM NADP+, 300 μM of the substrate (D-glucose-6-phosphate, DL-malate or L-glutamate), 300 μM MTT and 30 μM PES (phenazine ethosulphate). The components were dilute up to 900 µL in 50 mM Tris-HCl buffer at pH 8 with 0.13% (m/v) gelatin. The gelatin was employed to prevent the formazan precipitation. The reactions were carried out in 1 mL-cuvettes, starting by addition of 100 μL of the supernatant. The reactions were monitored for 5 min at 585 nm in a Shimadzu™ spectrophotometer. The activity was expressed as µmol of formazan/min of reaction per OD600 of the culture.
Determination of NAD(P)H/NAD(P)+ ratio
Intracellular levels of NAD+, NADP+, NADH and NADPH were determined by the improved cyclic assay using either ADH (Sigma #A3263) or G6PDH (Sigma #G6378), respectively53. The dinucleotides were extracted using cell pellets from 1 mL of culture, cultivated either until the mid-log (OD600 of 0.4-0.5) or late-log (OD600 of 1.0-1.2) phases. Reduced and oxidised nicotinamide adenine dinucleotides were differentially extracted by treatment with alkali or acid, respectively, followed by extract neutralisation. The assays were performed in 200 μL in a water bath for 30 minutes at 37 °C containing the following components: 0.1 M Tricine–NaOH buffer (pH 8.0); 4.2 mM MTT; 40 mM EDTA (disodium salt); 16.6 mM PES; 5 M ethanol as substrate for alcohol dehydrogenase to determine NADH/NAD+ or 25 mM glucose 6-phosphate (dipotassium salt) as substrate for G6PDH to determine NADPH/NADP+. To determine NADPH/NADP+ and NADH/NAD+, 10 µL of a baker’s yeast G6PDH solution (14 units/mL) or 10 µL of a baker’s yeast alcohol dehydrogenase solution (100 units/mL) were added per reaction, respectively. Reactions were stopped by adding 100 μL of 5 M NaCl followed by 5 minutes of ice incubation. The precipitated formazan was centrifuged for 5 min at 14,000 x g and solubilised in 500 μL of 96% ethanol. The formazan was quantified as a function of the absorbance at 550 nm of 200 µL of sample in 96-well plates in a Biotek ELX-800 microplate reader. The standard calibration curve was run in triplicate using up to 30 pmol/assay of either NAD(P)H or NAD(P)+ standards.
Analysis of intracellular ROS levels using flow cytometry
Cells from 1 mL of culture were collected by centrifugation at 14,000 × g for 1 min and then resuspended in 500 μL of PBS buffer supplemented with 1 mM EDTA, 0.01% Tween 20 and 0.1% Triton X-100. Cells were subsequently incubated with 50 μM 2′-7′-dichlorofluorescein diacetate (H2-DCFDA) for 30 min at 30 °C in the dark. Control experiments without H2-DCFDA addition were also set up under the same conditions. Treatment with H2O2 was performed by pre-incubation of cells with increasing concentration of H2O2 for 30 min at 120 rpm and 30 °C, before addition of H2-DCFDA. The samples were analysed by flow cytometry using a BD AccuriTM C5 flow cytometer equipped with a 488 nm argon laser and a 533/30 nm bandpass filter (FL1-H). The median fluorescence intensity was used to determine the intracellular ROS levels.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Change history
22 June 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Anderson, A. J. & Dawes, E. A. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev 54, 450–472 (1990).
Madison, L. L. & Huisman, G. W. Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiol Mol Biol Rev 63, 21–53 (1999).
Hervas, A. B., Canosa, I., Little, R., Dixon, R. & Santero, E. NtrC-dependent regulatory network for nitrogen assimilation in Pseudomonas putida. J Bacteriol 191, 6123–6135, https://doi.org/10.1128/jb.00744-09 (2009).
Babel, W., Ackermann, J. U. & Breuer, U. Physiology, regulation, and limits of the synthesis of poly(3HB). Adv Biochem Eng Biotechnol 71, 125–157 (2001).
Steinbuchel, A. & Hein, S. Biochemical and molecular basis of microbial synthesis of polyhydroxyalkanoates in microorganisms. Adv Biochem Eng Biotechnol 71, 81–123 (2001).
Reddy, C. S., Ghai, R., Rashmi & Kalia, V. C. Polyhydroxyalkanoates: an overview. Bioresour Technol 87, 137–146 (2003).
Kocharin, K., Chen, Y., Siewers, V. & Nielsen, J. Engineering of acetyl-CoA metabolism for the improved production of polyhydroxybutyrate in Saccharomyces cerevisiae. AMB Express 2, 52, https://doi.org/10.1186/2191-0855-2-52 (2012).
Kocharin, K., Siewers, V. & Nielsen, J. Improved polyhydroxybutyrate production by Saccharomyces cerevisiae through the use of the phosphoketolase pathway. Biotechnol Bioeng 110, 2216–2224, https://doi.org/10.1002/bit.24888 (2013).
Lee, I. Y., Kim, M. K., Chang, H. N. & Park, Y. H. Regulation of Poly-Beta-Hydroxybutyrate Biosynthesis by Nicotinamide Nucleotide in Alcaligenes-Eutrophus. Fems Microbiology Letters 131, 35–39, https://doi.org/10.1111/j.1574-6968.1995.tb07750.x (1995).
Lee, J. N., Shin, H. D. & Lee, Y. H. Metabolic engineering of pentose phosphate pathway in Ralstoniaeutropha for enhanced biosynthesis of poly-beta-hydroxybutyrate. Biotechnol Prog 19, 1444–1449, https://doi.org/10.1021/bp034060v (2003).
Lim, S. J., Jung, Y. M., Shin, H. D. & Lee, Y. H. Amplification of the NADPH-related genes zwf and gnd for the oddball biosynthesis of PHB in an E. coli transformant harboring a cloned phbCAB operon. J Biosci Bioeng 93, 543–549, https://doi.org/10.1016/S1389-1723(02)80235-3 (2002).
Johnson, K., Kleerebezem, R. & van Loosdrecht, M. C. Influence of the C/N ratio on the performance of polyhydroxybutyrate (PHB) producing sequencing batch reactors at short SRTs. Water Res 44, 2141–2152, https://doi.org/10.1016/j.watres.2009.12.031 (2010).
Kalaiyezhini, D. & Ramachandran, K. B. Biosynthesis of poly-3-hydroxybutyrate (PHB) from glycerol by Paracoccus denitrificans in a batch bioreactor: effect of process variables. Prep Biochem Biotechnol 45, 69–83, https://doi.org/10.1080/10826068.2014.887582 (2015).
Baldani, J. I., Baldani, V. L. D. & Seldin, L. & Dobereiner. J. Characterization of Herbaspirillum-Seropedicae Gen-Nov, Sp-Nov, a Root-Associated Nitrogen-Fixing Bacterium. International Journal of Systematic Bacteriology 36, 86–93 (1986).
Catalan, A. I., Ferreira, F., Gill, P. R. & Batista, S. Production of polyhydroxyalkanoates by Herbaspirillum seropedicae grown with different sole carbon sources and on lactose when engineered to express the lacZlacY genes. Enzyme Microb Tech 40, 1352–1357, https://doi.org/10.1016/j.enzmictec.2006.10.008 (2007).
Kadowaki, M. A. et al. Identification and characterization of PhbF: A DNA binding protein with regulatory role in the PHB metabolism of Herbaspirillum seropedicae SmR1. BMC Microbiol 11, 230, https://doi.org/10.1186/1471-2180-11-230 (2011).
Alves, L. P. et al. Backup Expression of the PhaP2 Phasin Compensates for phaP1 Deletion in Herbaspirillum seropedicae, Maintaining Fitness and PHB Accumulation. Front Microbiol 7, 739, https://doi.org/10.3389/fmicb.2016.00739 (2016).
Pedrosa, F. O. et al. Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. PLoS Genet 7, e1002064, https://doi.org/10.1371/journal.pgen.1002064 (2011).
Tirapelle, E. F. et al. Identification of Proteins Associated with Polyhydroxybutyrate Granules from Herbaspirillum seropedicae SmR1 - Old Partners, New Players. PLoS One 8, e75066, https://doi.org/10.1371/journal.pone.0075066 (2013).
Chubatsu, L. S. et al. Nitrogen fixation control in Herbaspirillum seropedicae. Plant Soil 356, 197–207, https://doi.org/10.1007/s11104-011-0819-6 (2011).
Merrick, M. J. & Edwards, R. A. Nitrogen control in bacteria. Microbiol Rev 59, 604–622 (1995).
Ninfa, A. J. & Atkinson, M. R. PII signal transduction proteins. Trends Microbiol 8, 172–179, doi:S0966-842X(00)01709-1 (2000).
Persuhn, D. C. et al. The transcriptional activator NtrC controls the expression and activity of glutamine synthetase in Herbaspirillum seropedicae. FEMS Microbiol Lett 192, 217–221 (2000).
Sun, J., Peng, X., Van Impe, J. & Vanderleyden, J. The ntrB and ntrC genes are involved in the regulation of poly-3-hydroxybutyrate biosynthesis by ammonia in Azospirillum brasilense Sp7. Appl Environ Microbiol 66, 113–117 (2000).
Sun, J., Van Dommelen, A., Van Impe, J. & Vanderleyden, J. Involvement of glnB, glnZ, and glnD genes in the regulation of poly-3-hydroxybutyrate biosynthesis by ammonia in Azospirillum brasilense Sp7. Appl Environ Microbiol 68, 985–988 (2002).
Khanna, S. & Srivastava, A. K. Statistical media optimization studies for growth and PHB production by Ralstonia eutropha. Process Biochemistry 40, 2173–2182, https://doi.org/10.1016/j.procbio.2004.08.011 (2005).
Verlinden, R. A. J. et al. Production of polyhydroxyalkanoates from waste frying oil by Cupriavidus necator. AMB Express 1, 11–11, https://doi.org/10.1186/2191-0855-1-11 (2011).
Schwab, S. et al. The glnAntrBC operon of Herbaspirillum seropedicae is transcribed by two oppositely regulated promoters upstream of glnA. Can J Microbiol 53, 100–105, https://doi.org/10.1139/w06-113 (2007).
Staskawicz, B., Dahlbeck, D., Keen, N. & Napoli, C. Molecular characterization of cloned avirulence genes from race 0 and race 1 of Pseudomonas syringae pv. glycinea. J Bacteriol 169, 5789–5794 (1987).
Yeom, S., Yeom, J. & Park, W. NtrC-sensed nitrogen availability is important for oxidative stress defense in Pseudomonas putida KT2440. J Microbiol 48, 153–159, https://doi.org/10.1007/s12275-010-0075-0 (2010).
Hervas, A. B., Canosa, I. & Santero, E. Regulation of glutamate dehydrogenase expression in Pseudomonas putida results from its direct repression by NtrC under nitrogen-limiting conditions. Mol Microbiol 78, 305–319, https://doi.org/10.1111/j.1365-2958.2010.07329.x (2010).
Spaans, S. K., Weusthuis, R. A., van der Oost, J. & Kengen, S. W. NADPH-generating systems in bacteria and archaea. Front Microbiol 6, 742, https://doi.org/10.3389/fmicb.2015.00742 (2015).
Cai, D. et al. A novel approach to improve poly-gamma-glutamic acid production by NADPH Regeneration in Bacillus licheniformis WX-02. Sci Rep 7, 43404, https://doi.org/10.1038/srep43404 (2017).
Zhang, Y., Smallbone, L. A., diCenzo, G. C., Morton, R. & Finan, T. M. Loss of malic enzymes leads to metabolic imbalance and altered levels of trehalose and putrescine in the bacterium Sinorhizobium meliloti. BMC Microbiol 16, 163, https://doi.org/10.1186/s12866-016-0780-x (2016).
Dawes, E. A. & Senior, P. J. The role and regulation of energy reserve polymers in micro-organisms. Adv Microb Physiol 10, 135–266 (1973).
Carlson, R., Wlaschin, A. & Srienc, F. Kinetic Studies and Biochemical Pathway Analysis of Anaerobic Poly-(R)-3-Hydroxybutyric Acid Synthesis in Escherichia coli. Appl Environ Microb 71, 713–720, https://doi.org/10.1128/aem.71.2.713-720.2005 (2005).
Singh, R., Lemire, J., Mailloux, R. J. & Appanna, V. D. A Novel Strategy Involved Anti-Oxidative Defense: The Conversion of NADH into NADPH by a Metabolic Network. PLoS One 3, e2682, https://doi.org/10.1371/journal.pone.0002682 (2008).
Schlegel, H. G., Gottschalk, G. & Von Bartha, R. Formation and utilization of poly-beta-hydroxybutyric acid by Knallgas bacteria (Hydrogenomonas). Nature 191, 463–465 (1961).
Haywood, G. W., Anderson, A. J., Chu, L. & Dawes, E. A. Characterization of two 3-ketothiolases possessing differing substrate specificities in the polyhydroxyalkanoate synthesizing organism Alcaligenes eutrophus. FEMS Microbiology Letters 52, 91–96 (1988).
Haywood, G. W., Anderson, A. J., Chu, L. & Dawes, E. A. The role of NADH- and NADPH-linked acetoacetyl-CoA reductases in the poly-3-hydroxybutyrate synthesizing organism Alcaligenes eutrophus. FEMS Microbiology Letters 52, 259–264 (1988).
Reitzer, L. Nitrogen assimilation and global regulation in Escherichia coli. Annu Rev Microbiol 57, 155–176, https://doi.org/10.1146/annurev.micro.57.030502.090820 (2003).
Zimmer, D. P. et al. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc Natl Acad Sci USA 97, 14674–14679, https://doi.org/10.1073/pnas.97.26.14674 (2000).
Helling, R. B. Why does Escherichia coli have two primary pathways for synthesis of glutamate? J Bacteriol 176, 4664–4668 (1994).
Yuan, J. et al. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol Syst Biol 5, 302, https://doi.org/10.1038/msb.2009.60 (2009).
Simon, R., Priefer, U. & Puhler, A. A Broad Host Range Mobilization System for Invivo Genetic-Engineering - Transposon Mutagenesis in Gram-Negative Bacteria. Bio-Technol 1, 784–791, https://doi.org/10.1038/Nbt1183-784 (1983).
Souza, E. M., Pedrosa, F. O., Rigo, L. U., Machado, H. B. & Yates, M. G. Expression of the nifA gene of Herbaspirillum seropedicae: role of the NtrC and NifA binding sites and of the -24/-12 promoter element. Microbiology 146(Pt 6), 1407–1418, https://doi.org/10.1099/00221287-146-6-1407 (2000).
Noindorf, L. et al. Role of PII proteins in nitrogen fixation control of Herbaspirillum seropedicae strain SmR1. BMC Microbiol 11, 8, https://doi.org/10.1186/1471-2180-11-8 (2011).
Kovach, M. E. et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176 (1995).
Pedrosa, F. O. & Yates, M. G. Regulation of Nitrogen-Fixation (Nif) Genes of Azospirillum-Brasilense by Nifa and Ntr (Gln) Type Gene-Products. FEMS Microbiology Letters 23, 95–101, https://doi.org/10.1111/j.1574-6968.1984.tb01042.x (1984).
Braunegg, G., Sonnleimer, B. & Lafferty, R. A rapid gas chromatographic method for the determination of poly-β-hydroxybutyric acid in microbial biomass. European J Appl Microbiol Biotechnol 6, https://doi.org/10.1007/bf00500854 (1978).
Norholm, M. H. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol 10, 21, https://doi.org/10.1186/1472-6750-10-21 (2010).
Mayer, K. M. & Arnold, F. H. A colorimetric assay to quantify dehydrogenase activity in crude cell lysates. J Biomol Screen 7, 135–140, https://doi.org/10.1177/108705710200700206 (2002).
Gibon, Y. & Larher, F. Cycling assay for nicotinamide adenine dinucleotides: NaCl precipitation and ethanol solubilization of the reduced tetrazolium. Anal Biochem 251, 153–157, https://doi.org/10.1006/abio.1997.2283 (1997).
Acknowledgements
We are very grateful to CNPq (National Council for Scientific and Technological Development) and Fundação Araucária for financial support (Projects CNPq #458417/2014-9, Fundação Araucária #312/2014). ENMS, EYSK and HLRC are very grateful for CNPq and CAPES for scholarships received during the performing of the experiments.
Author information
Authors and Affiliations
Contributions
E.N.M.S., E.Y.S.K. and H.L.R.C. designed, performed the experiments and analysed the data. M.M.S., L.S.C., E.M.S., and F.O.P. conceived, supervised the study and analysed the data. M.M.S. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sacomboio, E.N.M., Kim, E.Y.S., Ruchaud Correa, H.L. et al. The transcriptional regulator NtrC controls glucose-6-phosphate dehydrogenase expression and polyhydroxybutyrate synthesis through NADPH availability in Herbaspirillum seropedicae. Sci Rep 7, 13546 (2017). https://doi.org/10.1038/s41598-017-12649-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12649-0
- Springer Nature Limited
This article is cited by
-
Phosphate-inducible poly-hydroxy butyrate production dynamics in CO2 supplemented upscaled cultivation of engineered Phaeodactylum tricornutum
Journal of Applied Phycology (2022)
-
Redox cofactor engineering in industrial microorganisms: strategies, recent applications and future directions
Journal of Industrial Microbiology and Biotechnology (2018)