
Abstract
Vibrio fischeri uses the AinS/AinR pheromone-signaling system to control bioluminescence and other symbiotic colonization factors. The Ain system is thought to initiate cell-cell signaling at moderate cell densities and to prime the LuxI/LuxR signaling system. Here we compared and analyzed the ain locus from two V. fischeri strains and a Vibrio salmonicida strain to explore ain regulation. The ainS and ainR genes were predicted to constitute an operon, which we corroborated using RT-PCR. Comparisons between strains revealed a stark area of conservation across the ainS-ainR junction, including a large inverted repeat in ainR. We found that this inverted repeat in cis can affect accumulation of the AinS-generated pheromone N-octanoyl homoserine lactone, which may account for the previously unexplained low-signal phenotype of a ∆ainR mutant, although the mechanism behind this regulation remains elusive. We also extended the previous observation of a possible “lux box” LuxR binding site upstream of ainS by showing the conservation of this site as well as a second putative lux box. Using a plasmid-based reporter we found that LuxR can mediate repression of ainS, providing a negative feedback mechanism in the Ain/Lux signaling cascade. Our results provide new insights into the regulation, expression, and evolution of ainSR.
Similar content being viewed by others


Introduction
The light-organ symbiont Vibrio fischeri ES114 uses pheromone signaling (PS) to regulate behaviors essential for colonizing its host squid, Euprymna scolopes 1,2,3,4,5,6,7. One of these behaviors, bioluminescence, was among the first phenotypes discovered to be regulated by PS, and its examination over the last fifty years has been fundamental to understanding bacterial cell-cell communication8. Of V. fischeri’s three integrated PS systems, two acyl-homoserine lactone (AHL)-based systems are primarily responsible for regulating bioluminescence and other colonization factors, while the autoinducer-2 (AI-2) signal, which is conserved across many bacteria, only modestly influences these phenotypes under the conditions tested2.
The two AHL-based PS systems in V. fischeri that regulate bioluminescence and other symbiotic factors are comprised of the signal synthase/receptor pairs LuxI/LuxR and AinS/AinR1,4,5. LuxI and AinS produce AHL molecules that can diffuse through membranes and mediate cell-cell signaling once they reach stimulatory concentrations9. LuxI synthesizes the pheromone N-3-oxohexanoyl homoserine lactone (3OC6-HSL)10,11, which binds LuxR, promoting LuxR dimerization and association with a regulatory element upstream of luxI called the “lux box”9,12. AHL-LuxR complexes activate transcription of the luxICDABEG operon, which results in more LuxI, more 3OC6-HSL, and bioluminescence. LuxI and LuxR are well studied and are the archetype for similar PS systems throughout the Proteobacteria.
Although less well known, the structurally distinct AinS/AinR PS system also uses an AHL signal and plays key roles in luminescence induction and symbiotic competence3. AinS synthesizes the pheromone N-octanoyl homoserine lactone (C8-HSL), which is sensed by AinR13,14 but can also activate LuxR15,16. Information about local C8-HSL concentration is funneled by AinR into a core PS system conserved among the Vibrionaceae, converging with the AI-2 system, to affect bioluminescence through a regulatory cascade comprised of LuxU, LuxO, the sRNA Qrr, and the terminal regulator of the system, which is called LitR in V. fischeri 17,18,19,20. LitR activates LuxR expression and, as noted above, C8-HSL can activate LuxR directly, albeit more weakly than 3OC6-HSL. Thus, in more than one way the Ain system serves to activate lux expression and “prime” 3OC6-HSL-based signaling3,15,16,17. However, at certain AHL ratios, C8-HSL can actually inhibit 3OC6-HSL-based activation15,21,22,23.
In V. fischeri strain ES114, which was isolated from the light organ of E. scolopes, the Ain system is critical for induction of luminescence in broth culture and underlies regulation of symbiotic colonization factors. Given this role, understanding the control of ainS/ainR will provide important insight into gene regulation during establishment of the symbiosis. LitR activates the ain system, closing a positive feedback loop2,13, and CRP-cAMP was recently identified as an activator of both ainS/ainR and luxI/luxR 24, although the connections between CRP, cAMP, and carbon source are not entirely clear in V. fischeri 25,26. Given the complex regulation of luxI and luxR 23,24,27,28,29,30,31,32,33, we predict that other regulatory mechanisms of ain regulation await discovery. Indeed, studies have suggested that the presence of ainR in cis somehow affects AinS activity13,18 and that a feedback loop exists between LuxR and the ain system13.
We previously reported that both the lux and ain loci have evolved rapidly and diverged between V. fischeri strains more rapidly than most housekeeping genes34. Moreover, we found that comparison of the luxIR intergenic region between strains provided insight into conserved regulatory sequences34. In this study we similarly used bioinformatic comparisons and targeted experimentation to gain insight into expression of the ain locus.
Results
Comparative analysis of the ainS/ainR locus
Homologs of V. fischeri AinS and AinR include Vibrio harveyi LuxM and LuxN35, which were the first members of this type of PS system described, as well as other pairs of similar ORFs in Vibrio salmonicida, Vibrio parahaemolyticus, Vibrio alginolyticus, Vibrio splendidus, Vibrio sp. MED222, and Photobacterium profundum (Fig. 1). Unlike LuxI and LuxR homologs, signaling systems similar to AinS and AinR have not yet been identified outside the Vibrionaceae. Most of the loci shown in Fig. 1 lack synteny or useful DNA sequence conservation with V. fischeri ainS/ainR. However, we were able to effectively compare this locus in three strains; V. fischeri ES11436,37, which is a dimly luminescent strain characteristic of other isolates from E. scolopes, V. fischeri MJ1138,39, which is a bright isolate from the Japanese pinecone fish Monocentris japonica, and Vibrio salmonicida LFI123840, which was isolated from a diseased cod. ES114 and MJ11 represent different clades of V. fischeri 34,39, whereas V. salmonicda is a closely related fish pathogen. Genome sequences are available for all three strains37,39,40.

Homologs of V. fischeri AinS/AinR and synteny around the ain locus. Aligned sequences are from V. fischeri ES114 and MJ11, V. salmonicidia LF1238, V. parahaemolyticus RMID2210633, V. harveyi ATCC BAA-1116, V. alginolyticus 12G01, V. splendidus 12B01, Vibrio spp. MED222, and Photobacterium profundum SS9. Arrows of the same color share homology, white arrows have no homologs in the figure, and numbers indicate percent identity to AinS or AinR from V. fischeri ES114. The 10-kb region encompassing V. fischeri ES114 ainS is shown, and synteny was assessed using the SEED44 database.
As previously reported34, the ainS and ainR genes have diverged more between ES114 and MJ11 than have most other orthologs in these strains, including the well conserved rluB gene adjacent to ainS (Fig. 2A). This trend was also evident in a comparison of ES114 and V. salmonicida (Fig. 2A). In all three strains there is only an 11-bp gap between the stop codon of ainS and the start codon of ainR, and the DOOR operon-prediction database indicated that ainS and ainR are likely to be co-transcribed41. Consistent with that prediction, RT-PCR indicated that ainS and ainR sequences can be found on the same RNA, as evidenced by an appropriately sized RT-PCR product that was absent in a no-RT control or when mRNA from ∆ainS or ∆ainR mutants was used (see Supplementary Figure S1). Using ARNold42, we further identified a putative Rho-independent transcriptional terminator between ainR (ORF VF_1036) and the adjacent convergent ORF VF_1035 (Fig. 2B). Based on its sequence, this putative terminator appears uni-directional and more likely to terminate the VF_1035 transcript than the ainSR transcript.

Comparison of the ainSR locus in three V. fischeri and V. salmonicida strains. The sequence between the stop codons of VF_1038 and VF_1035 in V. fischeri ES114 was compared to the orthologous regions from V. fischeri MJ11 and V. salmonicida LFI1238. In panel A, arrows show the arrangement of the three complete genes at this locus, which extends to the stop codon of VF_1035 on the right. Bold and italicized letters under the arrows indicate regions for which DNA sequence is shown in the corresponding panels. Homology between ES114 and MJ11 or V. salmonicida is shown for corresponding regions in shaded plots that range from fifty to one hundred percent identity within a 100-bp window as determined by VISTA73 with the LAGAN74 alignment function and default settings. The grey line denoted seventy-five percent identity. Panel B shows a conserved putative Rho-independent transcriptional terminator downstream of VF_1035 that was identified by ARNold42, with arrows indicating inverted repeat stems, with stem mismatches indicated by gaps on the arrows. A bold-lettered run of A’s indicates the canonical string of U’s (on the reverse strand transcript) following a stem loop structure in such terminators. Panel C shows sequences aligned from the start codon of ainR, with arrows indicating inverted repeats and mismatches indicated by gaps on the arrows. Panel D shows an alignment of sequences upstream of and within ainS. Two transcriptional start sites mapped in strain ES114 by 5′ RACE are indicated as TSS1 and TSS2, and putative −10 and −35 promoter elements associated with TSS1 and TSS2 are boxed. Start codons predicted by The SEED44 are indicated with bold and underlined letters. Brackets indicate alternative start codons conserved across all three strains. A CRP binding site24 and possible “lux box” LuxR-binding sequences are highlighted by alignment with the respective consensus binding sequence. In panels B–D, asterisks indicate bases conserved in all three strains.
Despite the relatively low homology between the ainSR loci in ES114 and MJ11, there is a short stretch of high sequence identity spanning the downstream end of ainS and the beginning of ainR (Fig. 2A and C). A striking feature within this portion of ainR is a nearly perfect 32-bp inverted repeat element designated IR1 (Fig. 2C). IR1 is also evident in V. salmonicida, although the sequence has diverged from that of the V. fischeri strains (Fig. 2C). RNA-folding predictions revealed another inverted repeat (designated IR2), although there are more mismatches between repeats and the gap between the repeats in IR2 is over 30 bp (Fig. 2C).
To help identify possible mechanisms of ainSR regulation, we mapped the ainS transcriptional start site(s) in ES114 using 5′-RACE and overlaid the results on an alignment of sequences upstream of ainS. After sequencing ten RACE clones, we found a nearly even distribution of two distinct 5′ transcript ends, which are shown in Fig. 2D labeled as TSS1 (four clones) or TSS2 (six clones). Boxed sequences upstream of TSS1 and TSS2 in Fig. 2D represent possible −10 and −35 promoter elements, based on reasonable matches to these elements and their spacing at Escherichia coli sigma-70 promoters43. Although this analysis must be viewed cautiously, there is enough conservation of key putative-promoter elements between the strains to suggest that TSS1 and TSS2 are not unique to ES114.
The translational start(s) for AinS was difficult to place definitively. Annotation by the SEED44 predicted non-canonical (non-ATG) start codons for ainS in each of the three strains, but the position of the predicted start is different in each strain (Fig. 2D). None of these predicted start sites is conserved between the three strains and none match the translational start suggested by Gilson et al. for AinS in strain MJ145. Furthermore, each of the predicted translational start sites is upstream of TSS2 and would not be present on transcripts that initiate at this position (Fig. 2D). Other recent studies (e.g., Nakahigashi et al.46) have underscored the potential for mistakes with automated annotation of translational starts as well as the prevalence of multiple start sites for particular genes. Accordingly, it seemed worth re-examining possible translational starts for AinS. It seems likely that the canonical E. coli ribosome binding site (RBS) sequence serves the same function in V. fischeri and V. salmonicida, because all three species are identical across the critical 3′ end of the 16S rRNA that forms complementary base pairing with the Shine-Dalgarno sequence on transcripts (data not shown). Among non-ATG start codons, GTG and TTG are the most common and are often the only possibilities considered by automated annotation programs; however, CTG and ATT start codons have been documented as well46,47,48. We identified two ATT codons and one TTG that are conserved across all three strains (Fig. 2D), and a putative ribosome-binding site (RBS) is well conserved across all three strains for the first (furthest upstream) ATT. Moreover, the putative RBS sequences near these potential starts appear to be as good or better matches than those for previously annotated starts. These putative non-ATG start codons also occur downstream of TSS2 but before AinS residues begin to align with similarity to AinS orthologs found in other members of the Vibrionaceae.
Previous examination of the ainS promoter region identified a CRP binding site24 and a putative lux box just upstream of the CRP site45, each of which are reasonably well conserved across the three strains (Fig. 2D). We also identified a second potential lux box overlapping the putative −35 promoter element associated with TSS2 (Fig. 2D). No other potential regulatory sequences were immediately apparent, although implications of our findings for regulation by CRP and LitR are discussed below. We focused experimentally on the putative lux box elements and IR1 for their potential role in ainSR control.
luxR-mediated repression of ainSR
We sought to clarify the role, if any, of LuxR in regulating ainSR. Others had identified one of the putative lux boxes upstream of ainS 45, and we previously reported a LuxR-dependent decrease in P qrr -lacZ reporter activity, which might be due to LuxR repression of the ainSR promoter13. On the other hand, ainS was not identified as part of the LuxR regulon49,50, although as discussed below those studies are arguably not definitive. Testing the effect of LuxR on ainSR is potentially complicated by the role of the Ain system in regulating LuxR, most notably via LitR17, but also potentially via C8-HSL affecting LuxR’s autoregulatory activity32. We eliminated such complicating feedback loops by utilizing a set of engineered strains lacking both AHL synthases and litR while also using a constitutive non-native promoter to drive luxR transcription21. Using these engineered strains, we found that addition of 3OC6-HSL significantly decreased activity of a P ainS -gfp reporter (P < 0.05) only when luxR was present (Fig. 3A). This effect of 3OC6-HSL appeared to be dose-dependent over a physiologically relevant range from 10 to 100 nM, although this effect was more evident with luxR from MJ1 than luxR from ES114 (Fig. 3B). At high concentrations C8-HSL can inhibit activation of the lux operon by 3OC6-HSL-LuxR15,16,21, and we similarly found that C8-HSL could significantly (P < 0.05) alleviate the repressive effect of 3OC6-HSL-LuxR on ainS reporter activity (Fig. 3A). Addition of 500 nM C8-HSL alone had no apparent effect (P > 0.05) on the reporter (Fig. 3A).

LuxR-3OC6-HSL represses a P ainS -gfp reporter. Both panels report fluorescence of cells harboring P ainS -gfp reporter pHK12 grown in SWTO to an OD595 of ~2.5. Strains harboring the reporter lack AHL synthases (∆luxI ∆ainS) and feedback regulation of luxR (litR::ermR Pcon -luxR), and either express luxR ES114 (JHK045), luxR MJ1 (JHK099), or no luxR (JHK046). Panel A: Strains with luxR ES114 (grey bars) or no luxR (white bars) harboring pHK12 and supplemented with 500 nM 3OC6-HSL and/or C8-HSL. Treatments marked with different letters are significantly different (P < 0.05) as determined by one-way ANOVA. Panel B: Strains with luxR ES114 or luxR MJ1, carrying pHK12, in media with varied 3OC6-HSL concentrations. In both panels, bars indicate standard error (n = 3).
As noted above, a previous study45 highlighted a potential lux box upstream of ainS (“lux box 1” - Fig. 2D), and we found a second putative lux box (“lux box 2” - Fig. 2D). The plasmid-based P ainS -gfp reporter described in the experiments above (pHK12) includes both of these lux boxes. We generated a second reporter plasmid (pHK156) containing only lux box 1 and TSS1, without lux box 2 or TSS2 (Fig. 2D), and a comparison of the reporters indicated that repression by 3OC6-HSL-LuxR was lost when only lux box 1 and TSS1 were included (Fig. 4). As discussed below, these data suggest the importance of the downstream lux box and promoter for LuxR-mediated regulation of ainS.

“lux box 2” is required for repression of ainSR promoter-reporter by LuxR-3OC6-HSL. Fluorescence of cells harboring P ainS -gfp reporter pHK12 grown in SWTO to OD595 ~2.5. Strains with luxR ES114 or no luxR harboring P ainS -gfp reporter plasmids pHK156 (with lux box 1) or pHK12 (both lux boxes) were grown with 50 nM 3OC6-HSL (white bars) or no addition (grey bars). Strains harboring these reporters lack AHL synthases (∆luxI ∆ainS) and feedback regulation of luxR (litR::ermR Pcon -luxR). Bars indicate standard error (n = 3), and asterisk indicates a significant difference in reporter activity upon addition of 50 nM 3OC6-HSL (P < 0.05).
We also tested the effect of LuxR-3OC6-HSL on the accumulation of C8-HSL, which is the product of AinS. We used strain JHK091 where luxR is again disconnected from native ain-influenced regulation using a litR::ermR mutation and a constitutive non-native luxR promoter, but in this case ainS was present. When JHK091 was grown with added 3OC6-HSL, it produced significantly less C8-HSL (Fig. 5). Thus, our data show that LuxR can both repress transcription from the ainS promoter (Figs 3 and 4) and diminish output of C8-HSL (Fig. 5).

C8-HSL accumulation by strain JHK091 is affected by 3OC6-HSL (-AHL). The asterisk indicates a significant difference between 0 and 50 nM 3OC6-HSL (P < 0.05). Error bars indicate standard error (n = 2).
Correlation between IR1 and C8-HSL accumulation
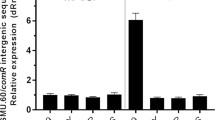
The discovery of IR1 (Fig. 2C) was intriguing given the previous evidence that having ainR present in cis with ainS somehow affected C8-HSL output13. We therefore hypothesized that IR1 in ainR might have a post-transcriptional effect on AinS expression, thus accounting for the decreased C8-HSL production of a ∆ainR mutant. To test the correlation between IR1 and C8-HSL, we constructed ainR variants truncated immediately after IR1 (ainR nat) or in IR1 (ainR trunc) (Fig. 6A). We found that truncation in IR1 resulted in decreased C8-HSL accumulation, as did full deletion of ainR; however, truncation of ainR after IR1 yielded wild-type levels of C8-HSL (Fig. 6B).

Effects of ainR sequence on C8-HSL accumulation. Panel A: Illustration of ainR alleles in this study. The ainS and ainR genes are shown as grey arrows delineated by thin vertical lines. Dashed lines correspond to deletions in ainR. Each repeat in IR1 is shown as a black arrow, and in ainR mut the IR is altered to scramble the inverted repeat without changing the amino acid sequence. In ainR scar, short horizontal lines near the ainR termini indicate 6-bp insertions from restriction enzyme sites. Panel B: C8-HSL accumulation in cultures of strains ES114, JHK003 (∆ainR), JHK055 (ainR trunc), JHK056 (ainR nat), JHK115 (ainR scar), JHK119 (ainR repair), JHK120 (ainR mut) grown with shaking in SWTO medium to an OD595 ~1.5. Letters indicate significant differences (P < 0.05) in ANOVA test. Panel C: Alignment of IR1 region in wild type (ainRWT)and the targeted mutant ainR mut showing conservation of amino acids encoded and increased number of mismatches in the inverted repeat, which are depicted as gaps in arrows. Panel D: C8-HSL accumulation in cultures of E. coli MG1655 carrying the ainR nat and ainR trunc alleles on pHK103 and pHK102, respectively, grown shaking in LB to OD595 ~1.5. Bars indicate standard error (n = 2).
The above experiments with ES114 and the ∆ainR, ainR nat, and ainR trunc mutants showed a correlation between the presence or absence of the full IR1 and higher or lower C8-HSL, respectively; however, three additional experiments suggested a more complex, context-dependent role of IR1. First, we reintroduced full-length ainR into the ∆ainR mutant by cloning ainR into an AvrII restriction site created within the ∆ainR allele. Although this process re-introduced the complete IR1, it also introduced a 6-bp restriction site scar immediately after the ainR start codon, generating a new allele that we designated ainR scar (Fig. 6A). Re-introducing IR1 on the ainR scar allele did not restore wild-type C8-HSL levels (Fig. 6B). To test whether a second-site mutation in the ainR scar mutant was responsible for its unexpected phenotype, we crossed the wild-type ainR sequence into the ainR scar mutant (ainR repair – Fig. 6A), which restored C8-HSL to wild-type levels. We also created an ainR mut allele (Fig. 6A), in which IR1’s DNA symmetry was disrupted while preserving the amino acid sequence of AinR (Fig. 6C). Despite lacking IR1, the strain with ainR mut produced wild-type levels of C8-HSL (Fig. 6B). Taken together, it appears that IR1 in cis can influence C8-HSL output, but this effect is influenced by the context of sequences both in and outside of IR1.
We hypothesized that IR1 preserves ainS mRNA by inhibiting 3′ RNA exonuclease activity from reaching and degrading the ainS coding part of the ainSR transcript, and we hoped to test this by exploiting defined RNase mutants in E. coli. When we placed constructs expressing ainS-ainR trunc and ainS-ainR nat into E. coli MG1655, we observed a similar pattern of C8-HSL accumulation as we saw for these alleles in V. fischeri, with even more dramatic differences (Fig. 6D). However, when we screened C8-HSL output in transgenic ainS-containing E. coli mutants51,52,53 that lack RNases D (rnd), PH (rph), T (rnt), R (rnr), or Z (rnz), we still saw higher C8-HSL output from ainS-ainR nat than from ainS-ainR trunc (see Supplementary Table S1). Furthermore, qRT-PCR revealed indistinguishable levels of ainS transcript copy number in ES114 and all of the ainR mutant variants shown in Fig. 6A (see Supplementary Figure S2). Thus we found no evidence that IR1 influences ainS transcript stability.
Discussion
V. fischeri uses the luxIR and ainSR AHL-based PS systems to control symbiotic phenotypes1,3,54, with the ain system being the first of these to be activated and priming the luxIR system through a signaling cascade conserved amongst members of the Vibrionaceae family. Despite the placement of ainSR atop this signaling cascade, only recently have we begun to understand the regulation of this system2,13,24,55. In this study, we expanded upon those findings by exploiting comparative genomic analyses and mapping the transcriptional start sites for ainSR (Fig. 2), which we confirmed are co-transcribed. Our results shed new light on regulation of the ainSR operon and its connection to luxRI in the PS circuitry of V. fischeri (see Supplementary Figure S3).
Previous studies showed that LitR and cAMP-CRP regulated ainS 2,17,24. A LitR binding site sequence has remained elusive, but binding studies confirmed cAMP-CRP interaction with a near-canonical recognition sequence upstream of ainS 24, at a site that is reasonably well conserved across strains (Fig. 2D). It was previously noted that CRP appeared to elicit both a LitR-dependent activation and LitR-independent repression of the ainS promoter, and the mapping of transcriptional start sites (Fig. 2D) allows further interpretation of these observations. The speculated “class III” CRP-dependent activation in conjunction with LitR24 could only realistically occur at the further downstream transcriptional start site, TSS2 (Fig. 2D). On the other hand, the known CRP binding site appears to overlap the −35 promoter element associated with TSS1 (Fig. 2D), and this might lead to repression. Further study is warranted to make firm conclusions about regulatory mechanisms, and defining LitR-DNA interactions would be especially useful in this regard.
Hierarchical activation of PS systems, such as the jump-starting of LuxIR by AinSR, is a common feature of bacteria possessing multiple PS systems56,57. We have now shown a negative feedback loop also exists between the second system and the first in V. fischeri ES114. LuxR represses the ainS promoter, and this activity responds to 3OC6- and C8-HSL in much the same way that these pheromones activate (3OC6-HSL) or antagonize (C8-HSL in the presence of 3OC6-HSL) LuxR’s stimulation of transcription at the luxI promoter (Fig. 3A). During colonization of the host, LuxR repression of ainS might lead to lower concentrations of C8-HSL, while luxIR expression increases during establishment and progression of the symbiosis.
Negative feedback loops are known in other bacterial cell-cell signaling systems. In Pseudomonas aeruginosa, RsaL serves a negative regulator of the lasIR PS system, binding its activator LasR and preventing it from activating expression of the lasI AHL synthase, thus maintaining signal levels during growth58,59. In a similar but more complicated scheme, Sinorhizobium meliloti ExpR activates expression of the AHL-synthase sinI and represses expression of a second sinI activator, sinR 60, thus, like RsaL, ultimately repressing the expression of its cognate-signal synthase. Perhaps a closer parallel to V. fischeri is found in Burkholderia cenocepacia, which contains CepI/CepR and CciI/CciR AHL-based PS systems. In B. cenocepacia CepR is required for cciIR induction, but CciR represses cepI 61, forming an inter-system negative feedback loop. As more bacteria with multiple PS systems are investigated, such inter-circuit feedback loops may become more apparent.
Identification of this negative feedback regulation by 3OC6-HSL-LuxR on ainSR is somewhat surprising considering that despite such regulation being postulated upon the discovery of these genes45, ainSR was not identified as a target gene in multiple studies of LuxR-dependent regulation49,50,62. In this regard it is worth noting that the effect of LuxR repression is small (Figs 3 and 4) and might be further obscured by feedback loops that we have deliberately removed from our experimental setup. An early proteomic analysis of the LuxR regulon therefore might easily have missed a small change in AinS levels50. In a setup closer to ours, Qin and colleagues saw no effect of LuxR on a P ainS -lacZ transcriptional reporter, but used sequences of V. fischeri MJ1 in transgenic E. coli, which may account for differences between our results in V. fischeri ES11462.
Perhaps the hardest data to reconcile with our own are that of Antunes et al.49, who used a microarray to assess the effect of adding 3OC6-HSL on the ES114 LuxR regulon. That study differed from ours in that it was performed in a background of endogenous 3OC6-HSL and C8-HSL, and perhaps more importantly in that it also included LitR-dependent positive feedback, which as noted above might obscure the negative feedback we observed. One might then question whether or not LuxR-mediated repression of ainSR is relevant in wild type, if its detection requires decoupling from LitR-mediated feedback. While this poses a reasonable question, it seems unlikely that LuxR-mediated repression of ainSR would represent a coincidental artifact, with a LuxR binding site overlapping an ainSR promoter occurring by chance. We speculate that this regulation evolved due to a fitness advantage conferred in some situation(s), for example during host colonization, where conditions undoubtedly differ from any of these experimental setups in batch broth cultures. For example, host-mediated C8-HSL turnover could dramatically affect regulation during the hierarchical activation of the two AHL-based systems. Further studies of this regulatory cascade during a model symbiotic infection will help resolve these issues.
One of the more striking findings of our analysis of the ainSR locus was the presence of a small region of relatively high conservation between ES114 and MJ11, and to a lesser degree conserved in V. salmonicida, including the inverted repeat elements IR1 and IR2 within the 5′ part of ainR (Fig. 2A and C). The sequences of AinS and AinR have diverged more than other protein components of the core Vibrio PS system (i.e. LuxU, LuxO, LitR)34, but IR1 in particular was striking and almost completely conserved.
We suspected that these IR sequences in ainR related to previously unexplained phenotypes of ∆ainR mutants. Ray and Visick reported a luminescence defect in a ∆ainR mutant, which is the opposite of the predicted effect based on our understanding of the regulatory system and its orthologs in V. harveyi 18. We also subsequently showed less C8-HSL accumulation in ∆ainR strains, independent of the positive feedback in the Ain system mediated by LitR13, which again was unexpected and if anything contrary to our understanding of the PS circuitry. Importantly, we found ainR must be present in cis with ainS to alleviate this defect, suggesting that the effects were related to the linkage between ainS and ainR, rather than AinR13. We have now confirmed that ainS and ainR are cotranscribed, and we hypothesized that IR1 in ainR protected the ainS portion of the transcript from degredation, resulting in more AinS, C8-HSL, and luminescence in wild type than in the ∆ainR mutant. However, although truncations in ainR supported this idea, other ainR alleles suggested a more complex regulatory mechanism, as there was not a consistent clear-cut difference between the presence or absence of IR1 (Fig. 6). We also saw no difference in ainS mRNA levels in wild type and the ∆ainR mutant (data not shown). These results indicate that the mechanism by which ainR sequence influences ainS and C8-HSL production is not the simple transcript-stability model that we initially proposed. Further examination of AinS protein levels using these IR1 mutants may reveal clues to how ainR sequence affects C8-HSL production. Whatever the mechanism, post-transcriptional regulation may allow ainSR to be co-transcribed while AinS and AinR are expressed in different stoichiometries.
Sequence comparisons at the luxIR locus revealed conserved regulatory elements34, and here a similar bioinformatic approach helped identify potential regulatory mechanisms in the ainSR PS system. As more genome sequences for different V. fischeri strains become available, this comparative approach may become even more useful in identifying regulatory elements at these and other loci. In this study, new puzzling questions have been revealed and remain unanswered, including; (i) what is the actual translational start(s) of AinS, considering previous annotations are inconsistent with transcripts arising at TSS2, (ii) what is the mechanistic role of IR1 in ainR, and (iii) is the negative feedback loop between LuxR and ainSR relevant to PS during symbiotic infection. Further investigation should also help clarify LitR’s relation to ainSR regulation (e.g. its binding site) and help determine whether any other factors control this locus.
Materials and Methods
Bacteria, growth media, and reagents
Bacterial strains are listed and briefly described in Table 1. V. fischeri ES114 was the wild-type strain used throughout36. Plasmids were transformed into Escherichia coli strain DH5α63 or DH5αλpir 64 in the case of plasmids with the R6K origin of replication. E. coli strain MG165565 and its derivatives were used as recipients for plasmids expressing different ainR alleles as described below. E. coli was grown in LB medium66 or brain heart infusion (BHI) medium, and V. fischeri was grown in LBS67 or SWTO27. Solid media were prepared with 15 g L−1 agar. For selection of E. coli, chloramphenicol (Cam) and kanamycin (Kan) were added to LB at final concentrations of 20 and 100 μg ml−1, respectively, and erythromycin (Erm) was added to BHI at a final concentration of 150 μg ml−1. For selection of V. fischeri on LBS, the concentrations of Cam, Erm, and Kan used were 2, 5, and 100 μg ml−1, respectively. 3OC6-HSL and C8-HSL were obtained from Sigma-Aldrich (St. Louis, MO).
Molecular genetic techniques
Oligonucleotides and plasmids are listed in Table 1 and the latter were constructed using standard techniques, with enzymes and other materials described previously13. To generate the P ainS -gfp reporter pHK156, a 428-bp fragment extending upstream of ainS was PCR amplified with primers pr_HK03 and pr_NL63. The resulting amplicon was digested with SphI and AvrII and ligated into SphI- and XbaI-digested pJLS27.
To generate the ∆ainR deletion allele on allelic exchange vector pHK138, a 1,350-bp fragment upstream of ainR including the start codon was PCR amplified using primers pr_HK146 and pr_NL28.3 and cloned into pCR-Blunt to generate pHK135. The 1,519-bp region downstream of ainR including the stop codon was PCR amplified using primers pr_NL29 and pr_HK126, digested with BamH1 and AvrII, and ligated into BamHI- and AvrII-digested pHK135 to create pHK136. This plasmid contains a unique AvrII restriction site between the ainR start and stop codons along with sequences flanking ainR. To reintroduce ainR into ∆ainR mutants, wild-type ainR was PCR amplified using primers pr_HK136 and pr_HK137 and this fragment was digested with NheI and ligated into AvrII-digested pHK136 to generate pHK137. As a result of these cloning steps, ainR on pHK137 differs from wild type in that it contains two 6-bp insertions, one immediately following the start codon and another preceding the stop codon, and we refer to the corresponding allele as ainR scar. Mobilizable ∆ainR and ainR scar allelic exchange vectors were generated by digesting pHK136 and pHK137 with BamHI and ligating them to BamHI-digested pEVS11864, which contains a conjugative origin of transfer, generating pHK138 and pHK139, respectively.
To generate the ainR trunc allele on an allelic exchange vector, in which ainR is truncated after the first 16-bp of the inverted repeat IR1, 428 bp upstream of ainS through the first half of the ainR inverted repeat (see Results) was PCR amplified using primers pr_HK17.2 and pr_HK28.2. A 1,500-bp region comprising the final 15 bp of ainR and sequence downstream of ainR was PCR-amplified using primers pr_HK40.1 and pr_HK41.4. The two amplicons were digested with BamHI, ligated together, and the combined fragment was blunt-end cloned into pCR-Blunt-II-TOPO to generate pHK34. This plasmid was then digested with KpnI and ligated to KpnI-digested pEVS118 to generate pHK37. To generate the ainR nat allele on allelic exchange vector pHK76, in which ainR is truncated after IR1, 1,500-bp downstream of ainR was again amplified using primer pair pr_HK40.1 and pr_HK41.4. This amplicon was digested with BamHI and SphI and ligated with BamHI and SphI-digested pEVS12264 to generate pHK75. The fragment from 428-bp upstream of ainS through IR1 was PCR amplified with primers pr_HK27.3 and pr_HK28.2. The resulting amplicon was digested with BamHI and KpnI and ligated into similarly digested pHK75 to generate the ainR nat allele on pHK76.
To place ainS and variants of ainR in E. coli on isogenic plasmid constructs, the fragment containing the 428-bp region upstream of ainS through 1,500-bp downstream of the ainR stop codon in strains ES114, JHK055, and JHK056 (described below) were PCR-amplified using primers pr_HK27.3 and pr_HK41.4 and cloned into pCR-Blunt-II-TOPO in the same orientation generating pHK95, pHK93 and pHK94, respectively. To add a selectable marker compatible with kanamycin-resistant E. coli RNase mutants, each plasmid was then digested using KpnI and SpeI and ligated with similarly digested pEVS118, which encodes resistance to Cam, to generate pHK104, pHK102 and pHK103, respectively.
To assess the function of an inverted repeat (IR1) in ainR, a synthetic DNA fragment (Integrated DNA Technologies, Coralville, IA) was designed to preserve the amino acid sequence of AinR while also disrupting the mirror symmetry of IR1 in ainR. This sequence, ainR_IR_conAA, is contained in an 844-bp ClaI to NdeI fragment. The synthetic fragment was cloned into pCR-Blunt to generate pHK129 and then PCR-amplified using primers pr_HK13 and pr_HK14. The resulting amplicon was ClaI- and NdeI-digested and ligated into similarly digested pHK95 to replace the native ainR sequence and generate pHK152. This plasmid was then digested with KpnI and SpeI and ligated with similarly digested pEVS118 to generate the mobilizable allelic exchange vector pHK153.
Mutant alleles were transferred from E. coli into V. fischeri on plasmids by triparental matings using the conjugative helper strain CC118λpir pEVS10468,69. Recombination and marker exchange were identified by screening for antibiotic resistance, and putative mutants were tested by PCR. In this way, the allele on pJLB9513 was introduced into DC43, DJ01, and JHK007 to generate JHK099, JHK045, and JHK046, respectively. To generate strains with different ainR variants, the alleles on pHK36, pHK76, pHK138, or pHK153 were introduced into ES114 to generate strains JHK055, JHK056, JHK114, and JHK120, respectively. JHK114 was subsequently used as the parent strain for the reintroduction of the ainR scar allele on pHK139, thus generating JHK115. The ainR scar allele in JHK115 was then recombinationally repaired using the native ainR locus on pHK104, generating strain JHK119. The Pcon-luxR P luxI -luxCDABEG ∆luxI locus on pDJ01 was introduced into the litR::ermR strain JB1813 to generate strain JHK091.
Luminescence measurements
Overnight V. fischeri cultures were diluted 1:1,000 in 25 ml SWTO in 125-ml flasks and incubated with shaking (200 rpm) at 24 °C. At regular intervals, the optical density at 595 nm (OD595) was measured for 500-μl samples using a BioPhotometer (Brinkman Instruments, Westbury, NY). Relative luminescence was measured with a TD-20/20 luminomenter (Turner Designs, Sunnyvale, CA) immediately following shaking to aerate the sample. Specific luminescence was calculated as the luminescence per OD595.
Transcriptional reporter assays
Strains harboring the P ainS -gfp reporter plasmid pHK1270 or the promoterless parent vector pJLS2726 were grown overnight in LBS and subcultured 1:1,000 into 125-ml flasks containing 25 ml SWTO, with or without 3OC6-HSL or C8-HSL, and incubated with shaking (200 rpm) at 24 °C. At regular intervals, 200-µl samples were aliquoted into clear-bottomed, black-walled, 96-well plates, where green fluorescence and OD595 were measured using a Synergy 2 plate reader (BioTek, Winooski, VT). Fluorescence values are reported from cultures at a similar cell density (OD595), as indicated.
C8-HSL bioassays
C8-HSL accumulation was assessed as previously described24. Briefly, culture supernatants were extracted with acidified ethyl acetate, extracts were dried and resuspended in SWTO, and C8-HSL levels were determined by comparison to standards using the bioassay strain DC22.
Characterization of ain transcript
Overnight cultures were diluted 1:1,000 in SWTO and grown to an OD595 ~0.5, at which point total RNA was extracted using the RNASnap method of Stead et al.71, followed by precipitation with sodium acetate and ethanol. RQ1 DNase (Promega, Madison, WI) was used to remove genomic DNA from samples according to the manufacturer’s protocol. 50 ng of DNA-free RNA was used as template for reverse transcription using the Superscript III First Strand cDNA synthesis kit (Invitrogen, Orange, CA) with random hexamers according to the manufacturer’s protocol. The resulting cDNA was diluted 1/10 in a PCR reaction using the primers pr_HK01 and pr_NL8924, which encompass a 236-bp fragment spanning the junction of ainS and ainR. The resulting amplicon from ES114 cDNA was cloned and sequenced to confirm its identity.
The ainS transcriptional start site was determined by the rapid amplification of cDNA ends (RACE) method of Scotto-Lavino et al.72. DNA-free RNA was prepared from ES114 grown in SWTO medium at 24 °C to an OD595 ~0.5 as described above. One microgram of RNA was used as a template for cDNA generation using the SuperScript III First-Strand Synthesis system (Invitrogen) and the ainS-specific primer pr_NL3523 followed by RNA removal using RNase H (Invitrogen). Poly-A tails were then added to cDNA products using 250 ng cDNA and terminal transferase (New England Biolabs, Ipswich, MA). Tailed cDNAs were then diluted 1:25 and used as template for the first of two nested-PCR reactions using three primers72 QT, QO, and the ainS-specific primer pr_HK02. The PCR products were then cleaned and diluted 1:250 and used as template for a second nested-PCR reaction with primers QI and pr_HK144 followed by cloning into pCR-Blunt and sequencing to determine the origin of the mRNA.
Quantitative RT-PCR analysis of ainS transcript
DNA-free RNA was prepared as it was for RACE analysis. 100 ng RNA was used as template for reverse transcription using either the SuperScript VILO (Invitrogen) or iScript (Bio-Rad, Hercules, CA) kits, according to the manufacturer’s protocol. Ten ng of RNase-treated cDNA was used as a template for qPCR using ainS-specific primers pr_NL108 and pr_NL10924 and the iQ SYBR-green qPCR supermix (Bio-Rad) using the MyiQ real-time PCR detection system. To generate standard curves, 10-fold serial dilutions of pHK95 were included during real-time analysis, and no-template and no-reverse transcriptase controls were included when appropriate.
Data availability statement
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Visick, K. L., Foster, J., Doino, J., McFall-Ngai, M. & Ruby, E. G. Vibrio fischeri lux genes play an important role in colonization and development of the host light organ. J. Bacteriol. 182, 4578–4586 (2000).
Lupp, C. & Ruby, E. G. Vibrio fischeri LuxS and AinS: comparative study of two signal synthases. J. Bacteriol. 186, 3873–3881 (2004).
Lupp, C., Urbanowski, M., Greenberg, E. P. & Ruby, E. G. The Vibrio fischeri quorum-sensing systems ain and lux sequentially induce luminescence gene expression and are important for persistence in the squid host. Mol. Microbiol. 50, 319–331 (2003).
Bose, J. L., Rosenberg, C. S. & Stabb, E. V. Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch. Microbiol. 190, 169–183 (2008).
Koch, E. J., Miyashiro, T. I., McFall-Ngai, M. J. & Ruby, E. G. Features governing symbiont persistence in the squid-vibrio association. Molec. Ecology 23, 1624–1634 (2014).
Stabb, E. V., Schaefer, A., Bose, J. L. & Ruby, E. G. in Chemical Communication Among Bacteria (eds S. C. Winans & B. L. Bassler) 233–250 (ASM Press, 2008).
Stabb, E. V. & Visick, K. L. In The Prokaryotes (eds E. Rosenberg et al.) 497–532 (Springer-Verlag, 2013).
Nealson, K. H., Platt, T. & Hastings, J. W. Cellular control of the synthesis and activity of the bacterial luminescent system. J. Bacteriol. 104, 313–322 (1970).
Kaplan, H. B. & Greenberg, E. P. Diffusion of autoinducer is involved in regulation of the Vibrio fischeri luminescence system. J. Bacteriol. 163, 1210–1214 (1985).
Engebrecht, J., Nealson, K. & Silverman, M. Bacterial bioluminescence: isolation and genetic analysis of functions from Vibrio fischeri. Cell 32, 773–781 (1983).
Eberhard, A. et al. Structural identification of autoinducer of Photobacterium fischeri luciferase. Biochemistry 20, 2444–2449 (1981).
Urbanowski, M. L., Lostroh, C. P. & Greenberg, E. P. Reversible acyl-homoserine lactone binding to purified Vibrio fischeri LuxR protein. J. Bacteriol. 186, 631–637 (2004).
Kimbrough, J. H. & Stabb, E. V. Substrate specificity and function of the pheromone receptor AinR in Vibrio fischeri ES114. J. Bacteriol. 195, 5223–5232 (2013).
Kuo, A., Blough, N. V. & Dunlap, P. V. Multiple N-acyl-L-homoserine lactone autoinducers of luminescence in the marine symbiotic bacterium Vibrio fischeri. J. Bacteriol. 176, 7558–7565 (1994).
Schaefer, A. L., Hanzelka, B. L., Eberhard, A. & Greenberg, E. P. Quorum sensing in Vibrio fischeri: probing autoinducer-LuxR interactions with autoinducer analogs. J. Bacteriol. 178, 2897–2901 (1996).
Eberhard, A., Widrig, C. A., McBath, P. & Schineller, J. B. Analogs of the autoinducer of bioluminescence in Vibrio fischeri. Arch. Microbiol. 146, 35–40 (1986).
Fidopiastis, P. M., Miyamoto, C. M., Jobling, M. G., Meighen, E. A. & Ruby, E. G. LitR, a new transcriptional activator in Vibrio fischeri, regulates luminescence and symbiotic light organ colonization. Mol. Microbiol. 45, 131–143 (2002).
Ray, V. A. & Visick, K. L. LuxU connects quorum sensing to biofilm formation in Vibrio fischeri. Mol. Microbiol. 86, 954–970 (2012).
Miyashiro, T., Wollenberg, M. S., Cao, X., Oehlert, D. & Ruby, E. G. A single qrr gene is necessary and sufficient for LuxO-mediated regulation in Vibrio fischeri. Mol. Microbiol. 77, 1556–1567 (2010).
Miyamoto, C. M., Lin, Y. H. & Meighen, E. A. Control of bioluminescence in Vibrio fischeri by the LuxO signal response regulator. Mol. Microbiol. 36, 594–607 (2000).
Colton, D. M., Stabb, E. V. & Hagen, S. J. Modeling analysis of signal sensitivity and specificity by Vibrio fischeri LuxR variants. PLoS One 10, e0126474 (2015).
Kuo, A., Callahan, S. M. & Dunlap, P. V. Modulation of luminescence operon expression by N-octanoyl-L-homoserine lactone in ainS mutants of Vibrio fischeri. J. Bacteriol. 178, 971–976 (1996).
Lyell, N. L., Dunn, A. K., Bose, J. L. & Stabb, E. V. Bright mutants of Vibrio fischeri ES114 reveal conditions and regulators that control bioluminescence and expression of the lux operon. J. Bacteriol. 192, 5103–5114 (2010).
Lyell, N. L. et al. Cyclic AMP receptor protein regulates pheromone-mediated bioluminescence at multiple levels in Vibrio fischeri ES114. J. Bacteriol. 195, 5051–5063 (2013).
Colton, D. M. & Stabb, E. V. Rethinking the roles of CRP, cAMP, and sugar-mediated global regulation in the Vibrionaceae. Curr Genet (2015).
Colton, D. M., Stoudenmire, J. L. & Stabb, E. V. Growth on glucose decreases cAMP-CRP activity while paradoxically increasing intracellular cAMP in the light-organ symbiont Vibrio fischeri. Mol. Microbiol. 97, 1114–1127 (2015).
Bose, J. L. et al. Bioluminescence in Vibrio fischeri is controlled by the redox-responsive regulator ArcA. Mol. Microbiol. 65, 538–553 (2007).
Septer, A. N. et al. Bright luminescence of Vibrio fischeri aconitase mutants reveals a connection between citrate and the Gac/Csr regulatory system. Mol. Microbiol. 95, 283–296 (2015).
Dolan, K. M. & Greenberg, E. P. Evidence that GroEL, not sigma 32, is involved in transcriptional regulation of the Vibrio fischeri luminescence genes in Escherichia coli. J. Bacteriol. 174, 5132–5135 (1992).
Dunlap, P. V. & Greenberg, E. P. Control of Vibrio fischeri luminescence gene expression in Escherichia coli by cyclic AMP and cyclic AMP receptor protein. J. Bacteriol. 164, 45–50 (1985).
Dunlap, P. V. & Greenberg, E. P. Control of Vibrio fischeri lux gene transcription by a cyclic AMP receptor protein-LuxR protein regulatory circuit. J. Bacteriol. 170, 4040–4046 (1988).
Shadel, G. S. & Baldwin, T. O. Positive autoregulation of the Vibrio fischeri luxR gene. LuxR and autoinducer activate cAMP-catabolite gene activator protein complex-independent and -dependent luxR transcription. J. Biol. Chem. 267, 7696–7702 (1992).
Ulitzur, S. H-NS controls the transcription of three promoters of Vibrio fischeri lux cloned in Escherichia coli. J Biolumin Chemilumin 13, 185–188 (1998).
Bose, J. L. et al. Contribution of rapid evolution of the luxR-luxI intergenic region to the diverse bioluminescence outputs of Vibrio fischeri strains isolated from different environments. Appl. Environ. Microbiol. 77, 2445–2457 (2011).
Bassler, B. L., Wright, M., Showalter, R. E. & Silverman, M. R. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol. Microbiol. 9, 773–786 (1993).
Boettcher, K. J. & Ruby, E. G. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J. Bacteriol. 172, 3701–3706 (1990).
Ruby, E. G. et al. Complete genome sequence of Vibrio fischeri: A symbiotic bacterium with pathogenic congeners. Proc. Natl. Acad. Sci. USA 102, 3004–3009 (2005).
Mandel, M. J., Stabb, E. V. & Ruby, E. G. Comparative genomics-based investigation of resequencing targets in Vibrio fischeri: focus on point miscalls and artefactual expansions. BMC Genomics 9, 138 (2008).
Mandel, M. J., Wollenberg, M. S., Stabb, E. V., Visick, K. L. & Ruby, E. G. A single regulatory gene is sufficient to alter bacterial host range. Nature 458, 215–218 (2009).
Hjerde, E. et al. The genome sequence of the fish pathogen Aliivibrio salmonicida strain LFI1238 shows extensive evidence of gene decay. BMC Genomics 9, 616 (2008).
Mao, F., Dam, P., Chou, J., Olman, V. & Xu, Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 37, D459–463 (2009).
Naville, M., Ghuillot-Gaudeffroy, A., Marchais, A. & Gautheret, D. ARNold: a web tool for the prediction of Rho-independent transcription terminators. RNA Biol 8, (11–13 (2011).
Shultzaberger, R. K., Chen, Z., Lewis, K. A. & Schneider, T. D. Anatomy of Escherichia coli σ 70 promoters. Nucleic Acids Res. 35, 771–788 (2007).
Overbeek, R. et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702 (2005).
Gilson, L., Kuo, A. & Dunlap, P. V. AinS and a new family of autoinducer synthesis proteins. J. Bacteriol. 177, 6946–6951 (1995).
Nakahigashi, K. et al. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res 23, 193–201 (2016).
Binns, N. & Masters, M. Expression of the Escherichia coli pcnB gene is translationally limited using an inefficient start codon: a second chromosomal example of translation initiated at AUU. Mol. Microbiol. 44, 1287–1298 (2002).
Sacerdot, C. et al. Sequence of a 1.26-kb DNA fragment containing the structural gene for E.coli initiation factor IF3: presence of an AUU initiator codon. EMBO J 1, 311–315 (1982).
Antunes, L. C. et al. Transcriptome analysis of the Vibrio fischeri LuxR-LuxI regulon. J. Bacteriol. 189, 8387–8391 (2007).
Callahan, S. M. & Dunlap, P. V. LuxR- and acyl-homoserine-lactone-controlled non-lux genes define a quorum sensing regulon in Vibrio fischeri. J. Bacteriol. 182, 2811–2822 (2000).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2, 2006–0008 (2006).
Deana, A., Celesnik, H. & Belasco, J. G. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451, 355–358 (2008).
Perwez, T. et al. Intragenic suppressors of temperature-sensitive rne mutations lead to the dissociation of RNase E activity on mRNA and tRNA substrates in Escherichia coli. Nucleic Acids Res. 36, 5306–5318 (2008).
Lupp, C. & Ruby, E. G. Vibrio fischeri uses two quorum-sensing systems for the regulation of early and late colonization factors. J. Bacteriol. 187, 3620–3629 (2005).
Septer, A. N., Lyell, N. L. & Stabb, E. V. The iron-dependent regulator Fur controls pheromone signaling systems and luminescence in the squid symbiont Vibrio fischeri ES114. Appl. Environ. Microbiol. 79, 1826–1834 (2013).
Latifi, A., Foglino, M., Tanaka, K., Williams, P. & Lazdunski, A. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol. Microbiol. 21, 1137–1146 (1996).
Lithgow, J. K. et al. The regulatory locus cinRI in Rhizobium leguminosarum controls a network of quorum-sensing loci. Mol. Microbiol. 37, 81–97 (2000).
de Kievit, T., Seed, P. C., Nezezon, J., Passador, L. & Iglewski, B. H. RsaL, a novel repressor of virulence gene expression in Pseudomonas aeruginosa. J. Bacteriol. 181, 2175–2184 (1999).
Rampioni, G. et al. RsaL provides quorum sensing homeostasis and functions as a global regulator of gene expression in Pseudomonas aeruginosa. Mol. Microbiol. 66, 1557–1565 (2007).
McIntosh, M., Meyer, S. & Becker, A. Novel Sinorhizobium meliloti quorum sensing positive and negative regulatory feedback mechanisms respond to phosphate availability. Mol. Microbiol. 74, 1238–1256 (2009).
Malott, R. J., Baldwin, A., Mahenthiralingam, E. & Sokol, P. A. Characterization of the cciIR quorum-sensing system in Burkholderia cenocepacia. Infect. Immun. 73, 4982–4992 (2005).
Qin, N., Callahan, S. M., Dunlap, P. V. & Stevens, A. M. Analysis of LuxR regulon gene expression during quorum sensing in Vibrio fischeri. J. Bacteriol. 189, 4127–4134 (2007).
Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580 (1983).
Dunn, A. K., Martin, M. O. & Stabb, E. V. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54, 114–134 (2005).
Blattner, F. R. et al. The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462 (1997).
Miller, J. H. A short course in bacterial genetics. (Cold Spring Harbor Laboratory Press, 1992).
Stabb, E. V., Reich, K. A. & Ruby, E. G. Vibrio fischeri genes hvnA and hvnB encode secreted NAD+-glycohydrolases. J. Bacteriol. 183, 309–317 (2001).
Herrero, M., De Lorenzo, V. & Timmis, K. N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in Gram-negative bacteria. J. Bacteriol. 172, 6557–6567 (1990).
Stabb, E. V. & Ruby, E. G. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Meth. Enzymol. 358, 413–426 (2002).
Kimbrough, J. H. & Stabb, E. V. Antisocial luxO Mutants Provide a Stationary-Phase Survival Advantage in Vibrio fischeri ES114. J. Bacteriol. 198, 673–687 (2015).
Stead, M. B. et al. RNAsnap™: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 40, e156 (2012).
Scotto-Lavino, E., Du, G. & Frohman, M. A. 5′ end cDNA amplification using classic RACE. Nat. Protoc. 1, 2555–2562 (2007).
Mayor, C. et al. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, (1046–1047 (2000).
Brudno, M. et al. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13, 721–731 (2003).
Acknowledgements
We thank Nicholas Wiese and Sidney Kushner for providing E. coli strains and Christy Hartman for technical assistance. The National Science Foundation supported this research under grants IOS-1121106, IOS-1557964, and MCB-1716232. J.H.K. was partially supported with funds awarded by the University of Georgia Presidential Graduate Fellows Program.
Author information
Authors and Affiliations
Contributions
Both authors conducted bioinformatic analyses, prepared figures and manuscript text, and reviewed the final manuscript. J.H.K. conducted experiments.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kimbrough, J.H., Stabb, E.V. Comparative analysis reveals regulatory motifs at the ainS/ainR pheromone-signaling locus of Vibrio fischeri . Sci Rep 7, 11734 (2017). https://doi.org/10.1038/s41598-017-11967-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11967-7
- Springer Nature Limited
This article is cited by
-
Dimension-reduction simplifies the analysis of signal crosstalk in a bacterial quorum sensing pathway
Scientific Reports (2021)
-
A lasting symbiosis: how Vibrio fischeri finds a squid partner and persists within its natural host
Nature Reviews Microbiology (2021)