
Abstract
Intracerebral hemorrhage (ICH) is one of the most devastating and disabling forms of stroke, yet effective treatments are still lacking. Prostaglandins and their receptors have been implicated in playing vital roles in ICH outcomes. Recently, laropiprant, a DP1 receptor antagonist, has been used in combination with niacin to abolish the prostaglandin D2-(PGD2)-induced flushing. Here, we test the hypothesis that laropiprant limits bleeding and rescues the brain from ICH. Wildtype (WT) and DP1−/− mice were subjected ICH and neurologic deficits and hemorrhagic lesion outcomes were evaluated at 72 hours after the ICH. To test the therapeutic potential of laropiprant, WT mice subjected to ICH were treated with laropiprant at 1 hour after the ICH. The putative effect of laropiprant on limiting hematoma expansion was tested by an in vivo tail bleeding cessation method and an ex vivo coagulation method. Finally, the roles of laropiprant on gliosis and iron accumulation were also investigated. A significant decrease in the injury volume was observed in DP1−/− as well as laropiprant-treated WT mice. The tail bleeding time was significantly lower in laropiprant group as compared with the vehicle group. Significantly lower Iba-1 and Perls’ iron staining in DP1−/− and laropiprant-treated WT groups were observed. Altogether, the data suggest that laropiprant treatment post-ICH attenuates brain damage by targeting primary as well as secondary injuries.
Similar content being viewed by others


Introduction
Intracerebral hemorrhage (ICH) is one of the most fatal stroke subtype and account for about 10–30% of all stroke patients1, 2. Only about 20% of people who survive ICH achieve functional independence at 6 months post-ICH3,4,5. The fatality rate at 1 month post-ICH is about 40% and the incidence is increasing every year due to the increasing number of aging population1, 5. ICH can lead to primary or secondary brain injury which is irrespective of the cause of the injury6,7,8,9,10. Primary injury is caused by the hemorrhage and hematoma expansion resulting in the increased intracranial pressure and mechanical injury to the tissue11. Secondary brain injury is the result of the physiological and pathological response to the hematoma7. Thus, therapeutic strategies have been developed to target primary and secondary brain injury following ICH6, 11,12,13,14. Therapies against primary injury target limiting the intracranial bleeding, hematoma expansion, and intracranial pressure11; secondary injuries are countered by targeting excitotoxicity, oxidative stress, and neuroinflammation7, 11, 15, 16.
An important feature of the pathophysiological response to ICH is the inflammatory cascade triggered by the degeneration and rupture of the blood vessels and hematoma formation. This inflammatory cascade involves the prostaglandin (PG), a family of lipid mediators, as well as other pathways16,17,18. Prostaglandins execute their effects through their respective receptors. The role of the prostaglandins and their specific receptors in acute neurologic conditions is actively being investigated. Interestingly, data so far suggests a paradoxical role of these prostaglandins and their receptors based on the nature, type, and location of the injury18,19,20,21,22,23,24,25.
PGD2 is the most abundant prostaglandin in the brain and exerts most of its biological effects by binding to the DP1 receptor. This receptor couples with Gs protein and activates adenylyl cyclase, resulting in an increase in cAMP which is reported to be involved with increases blood flow and platelet aggregation inhibition26. We have previously shown that DP1 receptor activation augments cerebral blood flow, which formed the basis for our hypothesis that absence of the DP1 receptor or its blockade by laropiprant, a selective DP1 receptor antagonist, would minimize brain injury and neurologic deficits following intracerebral hemorrhage. Interestingly, laropiprant, also known as MK-0524, has been used in pre-clinical and clinical settings alone or in combination with niacin27, 28 essentially to attenuate the PGD2-induced vasodilation that results in hot flashes on skin (a common undesirable side effect of niacin treatment). In humans, laropiprant has been found to be well tolerated, and has very decent bioavailability and a long half-life26, 29.
In the current study, a single dose of collagenase was injected into the striatum of WT and DP1−/− mice to induce ICH. Neurologic deficits and lesion volumes were assessed at 72 hours. To test whether the beneficial effects of DP1 receptor inhibition could be the result of a reduction in hematoma expansion, non-ICH WT and DP1−/− mice and laropiprant-treated WT mice were tested for tail bleeding time and ex vivo coagulation. Finally, the role of laropiprant treatment and DP1 receptor deletion was also assessed in gliosis through the analysis of Iba-1 and GFAP immunoreactivity, and iron content through Perls’ staining.
Materials and Methods
Mice
All procedures were performed in accordance with the guidelines of the National Institutes of Health (Bethesda, MD) and were approved by the Institutional Animal Care and Use Committee at the University of Florida. Adult male WT and DP1−/− C57BL/6 mice of 11–13 weeks old (24–28 g) were bred and maintained in our animal facility. The DP1−/− mice grow normally and have no gross abnormalities in behavior and brain macroscopic vasculature anatomy indices when compared to WT mice30, 31. Mice were given food and water ad libitum and housed under controlled reversed light cycle conditions (23 ± 2 °C; 12-hour light/dark cycle).
Randomization, Exclusion, and Blinding
Randomization was performed by a person who was not involved in the study and who had no knowledge of experimental groups. The preset exclusion criteria included any mouse with apparent abnormal conditions (e.g. dermatitis, red eye or discharge, abdominal or whisker abnormalities, signs of infection, etc.), ear puncture during placement in the stereotactic setup, bleeding upon drilling or on needle insertion, or an animal reaching a moribund condition requiring euthanasia. The number of mice used for the WT, Vehicle, DP1−/−, and laropiprant groups were 12, 10, 12, and 10 respectively. The number of animals used for tail bleeding assay were 10, 8, 10, 10, and 5 for the WT, Vehicle, DP1−/−, WT with laropiprant treatment, and DP1−/− with laropiprant treatment groups respectively. For the ex vivo coagulation study 5 mice/group were used. For in vivo thrombosis, five mice per group were used. The surgeon and investigators performing neurobehavioral testing were blinded to the experimental groups. Additionally, all histologic outcomes were quantified in a blinded manner.
Collagenase-induced ICH Model and Laropiprant Treatment
Mice were anesthetized with isoflurane (3% for induction, 1 to 1.5% for maintenance) and their heads were immobilized on a stereotaxic frame. A small incision was made at the overlying skin over the skull and a single unilateral, intrastriatal injection of collagenase VII-S (0.04 units in 0.2 μL saline, Sigma) was given at the following stereotactic coordinates relative to bregma: 0.5 mm anterior, 2.4 mm lateral, 3.2 mm from dura19. Rectal temperature was monitored and maintained at 37.0 °C ± 0.5 °C throughout the surgery. After the surgery, mice were moved to a temperature and humidity regulated recovery chamber for 2 hour then moved to their home cages and were allowed to survive up to 72 hours after ICH.
In a separate cohort of similarly ICH-induced WT mice, a single dose of 0.4 mg/kg laropiprant was given at 1 hour after the collagenase injection. Mice were allowed to survive up to 72 hours after ICH. The dose chosen is based on reports showing the effect of this compound in attenuating PGD2 or niacin-induced cutaneous vasodilation in mice29. Laropiprant is reported to have a half-life up to approximately 18 hours as well as good bioavailability26.
Neurological Deficit Scores (NDS)
The neurologic deficits were assessed at 72 hours post-ICH using the 24-point scale19, 32. The tests included body symmetry, gait, climbing, circling behavior, front limb symmetry, and compulsory circling. Each test was graded from 0 to 4, establishing a maximum deficit score of 24.
Histology and Immunohistochemistry
At 72 hours after ICH, mice were deeply anesthetized and transcardially perfused with phosphate-buffered saline (PBS, pH 7.4) followed by fixation with 4% paraformaldehyde. The brains were quickly harvested and processed for histology and immunohistochemistry using Leica CM 1850 cryostat. The mounted sections were stained with Cresyl violet to estimate the lesion volume following the detail protocol described by us recently19, 33, 34. To identify microgliosis and astrogliosis sections were processed for immunohistochemistry as described by us previously33 by using the following primary antibodies: ionized calcium-binding adaptor protein 1 (Iba1; 1:1,000; Wako, Richmond, VA) and glial fibrillary acidic protein (GFAP; 1:1,000; Dako, Carpinteria, CA) respectively. For the ferric iron content, Perls’ iron staining was performed by incubating the sections in a mixture containing equal ratio of 2% hydrochloric acid and 2% potassium ferrocyanide for 20 min, followed by counterstaining with nuclear fast red as previously reported by us34. All stained sections were imaged by using ScanScope CS and analyzed with ImageScope software (Aperio Technologies, Inc., Vista, CA).
Tail Bleeding Monitoring
To determine whether DP1 inhibition plays a role on platelet adhesion at the site of injury, a tail bleeding test was performed following the protocol we described earlier35, 36. This techniques is widely used to determine the ability of platelets to form a hemostatic plug37, 38. Briefly, the mice were either non-treated or given an i.p. injection of the vehicle (1% DMSO) or 0.4-mg/kg laropiprant. At 30 minutes from the injection, the mice were anesthetized and 2 mm of the tail tip was excised. The tail was immediately placed in warm PBS (37.0 ± 0.5 °C) and the time until bleeding ceased was recorded.
Ex Vivo Thrombus Formation
A modified ex vivo clot formation model, as we35 as well as Prasad et al.39 have described in the past, was used to test whether laropiprant can prevent hematoma expansion. Whole blood from non-ICH WT mice was harvested by cardiac puncture and 200 µL of the blood was mixed and vortexed with 600 µL of the vehicle or 600 µL of 30.6 µM laropiprant. The mixture was kept at room temperature for 2 minutes, followed by centrifugation for 30 seconds at 4000 g. The tubes were gently removed and the amount of uncoagulated supernatant was quantified.
Statistics
Statistical analyses were performed using Prism 5 (GraphPad, San Diego, CA). Neurologic deficit scores were analyzed by the non-parametric Kruskal–Wallis test followed by Dunn’s multiple comparison test and are presented as medians with interquartile ranges (25th and 75th percentiles). The remaining data sets were checked for differences in variances between groups and normality, and a one-way ANOVA test was performed, followed by Newman-Keuls multiple comparison test or an unpaired two-tailed Student’s t test with Welch’s correction. Data are expressed as Mean ± SD with P < 0.05 considered statistically significant.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Results
Absence or Blockade of PGD2 DP1 Receptor Limits Hemorrhagic Brain Damage
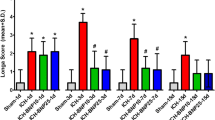
As expected, the collagenase injection in WT and DP1−/− mice resulted in significant brain damage. The lesion volume calculated in DP1−/− was 32.74 ± 16.31% lower than the lesion volume in WT mice. Similarly, the NDS were also significantly lower in DP1−/− as compared to the WT mice (Fig. 1).

Laropiprant treatment or deletion of DP1 receptor attenuates ICH outcomes. WT and DP1−/− mice were subjected to ICH by giving a single intrastriatal injection of collagenase. Another cohort of similarly ICH-induced WT mice were given vehicle or laropiprant at 1 hour after the ICH. Neurological deficits were scored at 72 hour post-ICH; thereafter, brains were harvested by perfusion and fixation. Cryosections were stained with Cresyl violet for lesion volume quantification. (A) Representative macrographs of CV-stained sections obtained from WT (n = 9), DP1−/− (n = 9), and laropiprant (n = 8) groups. Black dotted line encompasses lesion area. (B) ICH-induced neurological deficits were significantly lower in DP1−/− and laropiprant-treated WT groups. (C) DP1−/− mice and the WT mice treated with laropiprant also resulted in a significant decrease in the lesion volume as compared with the WT or vehicle (n = 8) treatment groups. *P < 0.05, ***P < 0.001 compared with the WT group; † P < 0.05 compared with the vehicle group. Scale bar = 2 mm.
Then, to determine if pharmacological inhibition of the receptor will lead to similar outcomes, ICH-induced mice were given a single dose of 0.4 mg/kg laropiprant 1 hour after ICH. Interestingly, laropiprant was able to attenuate the NDS by 40.32 ± 18.78% and lesion volume by 26.59 ± 16.80% as compared with the vehicle treated group (Fig. 1).
Laropiprant Treatment or Deletion of DP1 Receptor Attenuates Bleeding
To determine whether the beneficial effects of laropiprant are due to its role in limiting the bleeding, we monitored tail bleeding time. We found that bleeding caused by tail clipping ceased significantly faster in groups that were treated with laropiprant or lacked the DP1 receptor. The bleeding time was 36.45 ± 21.79% and 26.51 ± 13.31% lower in laropiprant treatment and DP1−/− groups, respectively (Fig. 2A). To further evaluate whether the effects of laropiprant are mediated through the DP1 receptor, the tail bleeding assay was also performed in the DP1−/− mice treated with laropiprant. Interestingly, there was no significant difference between the bleeding time in DP1−/− and DP1−/− treated with laropiprant (247.4 ± 43.2 minute vs 224.6 ± 82.7 minute; P = 0.48). Then, considering that laropiprant use could putatively lead to microhemorrhage or microthrombosis in vivo, non-ICH mice were given intraperitoneal injection of 0.4 mg/kg/day laropiprant for 7 days, and brains were then harvested and their hemoglobin content was quantified. Interestingly, there was no significant difference (P = 0.68) in the hemoglobin content in vehicle or laropiprant-treated groups (Fig. 2D).

Laropiprant treatment affects hemostasis under hemorrhagic condition. (A) To determine blood coagulation in vivo, the tail tip of anesthetized mice was excised, and bleeding time was recorded. To determine the effect of laropiprant on bleeding, vehicle or laropiprant was given intraperitoneal and a tail bleeding test was performed 30 min after the injection. DP1−/− and laropiprant-treated WT groups exhibited a significantly faster cessation of bleeding (n = 8–10/group). Interestingly, the DP1−/− mice treated with laropiprant (n = 5) exhibited no difference as compared with either the DP1−/− mice or the laropiprant treated WT mice. (B) To determine blood coagulation in vivo, blood was harvested by cardiac puncture from WT mice and mixed with vehicle or laropiprant. After incubation and centrifugation, the uncoagulated content was found to be significantly lower in the laropiprant-treated group (n = 5/group). (C) Chronic use of laropiprant in vivo did not induce microhemorrhage or microthrombus formation, suggesting that its effect on coagulation is only under hemorrhagic condition (n = 4/group, P = 0.68). **P < 0.01 compared with the WT group; †† P < 0.01, ††† P < 0.001 when compared with the vehicle group; ns = non-significant.
Laropiprant Treatment Facilitates Ex Vivo Coagulation
To determine whether the beneficial effects of laropiprant are due to its influence on coagulation, which would thus limit hematoma expansion, whole blood was incubated with laropiprant. Treatment of the whole blood with laropiprant led to larger clot formation ex vivo. The uncoagulated content after the clot formation was quantified and found to be 26.77 ± 11.02% lower in the laropiprant-treated group (Fig. 2B).
DP1 Receptor Deletion Augments Microgliosis
Harvested brain sections were immunostained for Iba-1 and GFAP immunoreactivity. The Iba-1 immunopositive cells were significantly higher in the ipsilateral side with predominant amoeboid or activated morphology in the preihematoma region. We found that DP1−/− or WT treated with laropiprant has significantly lower Iba-1 immunoreactivity as compared with the WT mice (Fig. 3A and B). The immunoreactivity for Iba-1 decreased by 48.63 ± 24.40 and 47.94 ± 24.40 in DP1−/− and laropiprant groups respectively (Fig. 3C). Two distinct morphological characteristics of Iba-1 immunopositive cells were detected representing classically activated microglial form and amoeboid shaped microglia/macrophages. Interestingly, there was strong GFAP immunoreactivity on the ipsilateral hemisphere along the perihematoma in both groups; however, there was no significant difference in immunoreactivity between the between the WT and DP1−/− mice (Fig. 4A–C).

Laropiprant attenuates microgliosis. The brain sections were immunostained for Iba-1 immunoreactivity to monitor microgliosis. (A) Representative macrographs at 1x showing Iba-1 immunoreactivity in WT, DP1−/−, and WT mice treated with laropiprant. Scale bar = 2 mm. (B) The regions in black boxes in panel A were magnified at 10x. The black dotted line demarcates the lesion core from the perihematoma area. Scale bar = 200 µm. The perihematoma and the intact area in WT mice exhibit two distinct morphologies of microglia. The microglia closer to the perihematoma exhibited amoeboid morphology whereas, those in the intact area show activated morphology. (C) Quantification of the Iba-1 immunoreactivity shows significant reduction in Iba-1 immunoreactivity in DP1−/− and WT treated with laropiprant. **P < 0.01 compared with the WT group. n = 6–7/group.

Astrogliosis remain unaffected by DP1 receptor deletion. The brain sections were immunostained for GFAP immunoreactivity to determine astrogliosis. (A) Representative macrographs at 1x showing GFAP immunoreactivity in WT and DP1−/−, mice. Scale bar = 2 mm. (B) The region in black boxes in panel A were magnified at 10x. The black dotted line demarcates the lesion core from the perihematoma area. No immunoreactivity toward astrocytes was detected in lesion core, whereas the perihematoma and intact area shows the astrocytes’ immunoreactivity. Scale bar = 200 µm. (C) Quantification of the GFAP immunoreactivity show no difference in WT and DP1−/−. ns = non-significant, P = 0.11. n = 8/group.
Laropiprant Treatment or Deletion of DP1 Receptor Attenuates Iron Content
One of the major causes of secondary injury following ICH is hemolysis, which results in iron accumulation and augments neuroinflammation and oxidative stress. With Perls’ staining, the results suggest that DP1 receptor deletion or WT treatment with laropiprant resulted in a significant decrease in iron content (Fig. 5A and B). Such staining in DP1−/− and laropiprant treatment groups decreased by 80.13 ± 26.03% and 76.25 ± 16.27%, respectively (Fig. 5C).

Laropiprant attenuates iron content in perihematoma. The brain sections were stained with Perls’ stain for iron content quantification. (A) Macrographs showing the sections obtained from WT, DP1−/−, and WT mice treated with laropiprant with Perls’ stain. Scale bar = 2 mm. (B) The regions in the black boxes in panel A were magnified at 10x. Blue dots represent iron staining, which was along the perihematoma region. Scale bar = 200 µm. Black dotted line demarcates lesion core from perihematoma area. (C) Quantification of Perls’ staining shows a significant decrease in iron content in DP1−/− and laropiprant-treated groups. *P < 0.05, **P < 0.01 compared with the WT group. n = 6–7/group.
Discussion
Intracerebral hemorrhage is a devastating acute neurological condition with high mortality and morbidity and limited treatment options. In this study, we used the selective PGD2 DP1 receptor antagonist laropiprant to test the hypothesis whether DP1 inhibition or absence will prevent hemorrhagic brain damage. We found that deletion of the DP1 receptor significantly attenuated the induced-ICH lesion volume, and similar effects were also observed with the laropiprant treatment. We also found that laropiprant augmented the ex vivo coagulation; whereas, deletion of the DP1 receptor or laropiprant treatment also attenuated the tail bleeding time, microgliosis, and iron content. These data support the usefulness of laropiprant as a therapeutic tool to resolve primary as well as secondary brain damage following ICH. We believe that this is the first study to show the beneficial effects of clinically tested laropiprant in ICH treatment.
Primary damage caused by ICH occurs within the first few hours and is mainly caused by bleeding and hematoma expansion, resulting in poor prognosis and high mortality. Immediate interventions to remove the hematoma or stop the bleeding are important therapeutic considerations. Therefore, one focus of this study was to test the feasibility of laropiprant in limiting bleeding during the initial phase of ICH. Interestingly, laropiprant has been tested in clinics for its role in niacin-induced flushing and nasal airway obstruction in humans29, 40,41,42; however, it has not been tested in neurologic conditions. Laropiprant is reported to abolish the PGD2-induced platelet inhibition43, thereby suggesting its potential in minimizing hemorrhage. Similarly, our data shows that treatment of ICH one hour after the onset can limit intracranial bleeding and lesion volume and improve neurologic deficits.
The first attempt to minimize ICH is to reduce the intracranial bleeding by regulating hemostasis, which is maintained through a network of processes including the platelet system, coagulation, and anticoagulant and fibrinolytic pathways44,45,46. The interaction of the downstream cascades of these pathways facilitates the adhesion of platelets with the sub-endothelial collagen in damaged vessels, forming a hemostatic vascular plug47. These plugs prevent further leakage of the blood into the brain parenchyma and limits hematoma expansion, consequently limiting secondary brain damage. Because PGD2 through DP1 receptor activation is reported to inhibit at least one of these processes43, we next tested whether DP1 receptor inhibition can limit bleeding and found that DP1 receptor inhibition or its deletion can indeed attenuate bleeding and limit ICH lesion volume. However, a few clinical trials indicate that attenuation of hematoma expansion or removal of a blood clot may not necessarily improve functional outcomes. For example, in clinical settings, the Surgical Trial in Intracerebral Hemorrhage (STICH) did not provide convincing evidence supporting the efficacy of early surgical removal of hematomas48. Similarly, use of recombinant activated factor VII did not improve survival or functional outcomes in ICH patients, although it did successfully reduce hematoma enlargement49. Nevertheless, the observations in our current study indicate that laropiprant treatment may have broader therapeutic effects. Such observation requires further investigation by testing the effect of laropiprant on long-term outcomes. Moreover, the observed Perls’ staining data also suggests that there is lower iron content in DP1−/− mice and WT mice treated with laropiprant. It is yet to be confirmed whether the lower iron levels in DP1−/− or laropiprant treatment groups are due to the restricted bleeding or if they are a result of the better microglial phagocytosis in DP1−/− and laropiprant treatment groups than in WT mice, as the role of microglial phagocytosis in hematoma clearance has been reported.
The current study supports a clear role of laropiprant in attenuating ICH-induced brain injury by limiting hematoma expansion; however, its efficacy for human use has been potentially challenged by a recent clinical trial. The HPS2-THRIVE trial was conducted to investigate the favorable effects of niacin/laropiprant combination to supplement the beneficial effects of statin in minimizing cardiovascular events. However, the trial concluded that this combination did not have a useful beneficial effect, but rather produced some side effects including hemorrhage in some cases28, 50. Careful review of the published data suggests that the side effects observed in the HPS2-THRIVE trial could be due to the fact that both niacin and statin are vasodilators and could have additive or synergistic effects leading to exacerbated hemorrhagic conditions40, 51,52,53. Moreover, in an attempt to achieve the pharmacologic dose that can minimize the niacin-induced flushing, the trial may have used higher doses (20 or 40 mg) of laropiprant rather than testing its lower effective dose26. It was reported that a dose of 6 mg or higher effectively suppressed PGD2-induced platelets aggregation. It is noteworthy that at higher concentrations, laropiprant could potentially also act as a TxA2 TP receptor antagonist54, though no clinically significant effect on bleeding times was observed with multiple doses of laropiprant alone adding up to 200 mg26. However, it is likely that the combination of high dose of laropiprant with niacin may have had an additive effect with statin in the HPS2-THRIVE trial, leading to hemorrhagic conditions. Interestingly, in our models, we found that 0.4 mg/kg, which is equivalent to 1.94 mg for a 60-kg human based on the body surface area dose conversion method55, had a protective effect against ICH. Furthermore, we also tested whether sub-chronic treatment of laropiprant could lead to microthrombosis or microhemorrhage in non-ICH naïve WT mice. Estimation of brain hemoglobin content on day 7 of the laropiprant treatment reveals no difference in the optical density of the brain samples obtained from vehicle or laropiprant-treated groups suggesting that laropiprant has no detrimental effect on hemostasis under non-pathologic (i.e. non-hemorrhagic) conditions.
The beneficial effects of laropiprant observed here could also be through regulating secondary injuries such as neuroinflammation. Microglial activation in response to injury could have beneficial or detrimental effects. Data from various labs, including ours, suggest that microglial activation in ICH results in phagocytosis and hematoma clearance11, 19, 56. However, generally chronic microglial activation is associated with increased inflammation and augmented brain damage25, 57, 58. Under neurologic conditions activated microglia produce proinflammatory cytokines such as TNFα and IL-1β, reactive oxygen/nitrogen species, and chemokines thereby augments brain damage16, 59, 60. Interestingly, our current data shows that DP1 receptor inhibition decreases microglial activation (Fig. 3). This decrease could be a result of lower lesion volume or lower iron content which then resulted in lower neuroinflammation. Interestingly, there was strong GFAP immunoreactivity in the ipsilateral side; however there was no difference between the WT and DP1−/− mice groups.
Limitations
This study shows for the first time proof-of-concept that DP1 antagonism, by clinically administered drug, could limit hematoma expansion and ICH brain damage. However, there are certain limitations that warrant further investigation. For example, (1) the clinical dose with and without niacin combination also needs to be investigated in ICH condition. (2) Because often older populations or populations with comorbidities are at greater risk of ICH, similar studies in older animals and animals with comorbidities such as diabetes or hypertension will provide more information regarding the therapeutic benefits of laropiprant. (3) To test the proof of the concept and to avoid any confounding effects due to sex differences, only male mice were used in this study. Therefore, additional studies are needed to test the therapeutic efficacy of laropiprant in young and old females. (4) Although we found that laropiprant affects bleeding and regulates hematoma volume, a detailed study on hemostasis would provide mechanisms associated with laropiprant treatment. (5) We also reported attenuated microgliosis in laropiprant treatment groups; however, a detailed study on acute and chronic effects of laropiprant on macrophage and microglial polarization will provide additional associated mechanisms.
Conclusions
Our data reveal for the first time that DP1 receptor inhibition or deletion is beneficial against ICH. Although in the recent trials, use of laropiprant with niacin along with statin therapy did not provide the desired beneficial effects, the efficacy of laropiprant alone in minimizing intracranial bleeding can still be considered a vital therapeutic strategy. To draw more reliable conclusions, further studies are needed to investigate the mechanism(s) associated with laropiprant treatment and its role in regulating primary and secondary brain injury following ICH.
References
Qureshi, A. I., Mendelow, A. D. & Hanley, D. F. Intracerebral haemorrhage. Lancet 373, 1632–1644, doi:10.1016/S0140-6736(09)60371-8 (2009).
Sacco, S., Marini, C., Toni, D., Olivieri, L. & Carolei, A. Incidence and 10-Year Survival of Intracerebral Hemorrhage in a Population-Based Registry. Stroke 40, 394–399, doi:10.1161/strokeaha.108.523209 (2009).
Razinia, T. et al. Body mass index and hospital discharge outcomes after ischemic stroke. Archives of Neurology 64, 388–391, doi:10.1001/archneur.64.3.388 (2007).
Benjamin, E. J. et al. Heart Disease and Stroke Statistics—2017 Update: A Report From the American Heart Association. Circulation, doi:10.1161/cir.0000000000000485 (2017).
Feigin, V. L., Lawes, C. M., Bennett, D. A., Barker-Collo, S. L. & Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurology 8, 355–369 (2009).
Aronowski, J. & Hall, C. E. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurological Research 27, 268–279 (2005).
Aronowski, J. & Zhao, X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42, 1781–1786, doi:10.1161/STROKEAHA.110.596718 (2011).
Wu, J. et al. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Research 953, 45–52 (2002).
Wang, X., Mori, T., Sumii, T. & Lo, E. H. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke 33, 1882–1888 (2002).
Wagner, K. R., Sharp, F. R., Ardizzone, T. D., Lu, A. & Clark, J. F. Heme and iron metabolism: role in cerebral hemorrhage. Journal of Cerebral Blood Flow & Metabolism 23, 629–652 (2003).
Keep, R. F., Hua, Y. & Xi, G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurology 11, 720–731, doi:10.1016/s1474-4422(12)70104-7 (2012).
Keep, R. F., Xi, G., Hua, Y. & Hoff, J. T. The deleterious or beneficial effects of different agents in intracerebral hemorrhage: think big, think small, or is hematoma size important? Stroke 36, 1594–1596 (2005).
Matsushita, K. et al. Evidence for apoptosis after intercerebral hemorrhage in rat striatum. Journal of Cerebral Blood Flow & Metabolism 20, 396–404 (2000).
Stocchetti, N. et al. Neuroprotection in acute brain injury: an up-to-date review. Critical Care 19, 186, doi:10.1186/s13054-015-0887-8 (2015).
Katsu, M. et al. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. Journal of Cerebral Blood Flow & Metabolism 30, 1939–1950, doi:10.1038/jcbfm.2010.45 (2010).
Wang, J. & Doré, S. Inflammation after intracerebral hemorrhage. Journal of Cerebral Blood Flow & Metabolism 27, 894–908 (2007).
Zhao, X. et al. Toxic role of prostaglandin E receptor EP1 after intracerebral hemorrhage in mice. Brain, Behavior, and Immunity 46, 293–310, doi:10.1016/j.bbi.2015.02.011 (2015).
Mohan, S., Ahmad, A. S., Glushakov, A., Chambers, C. & Dore, S. Putative role of prostaglandin receptor in intracerebral hemorrhage. Frontiers in Neurology 3, 1–17, doi:10.3389/fneur.2012.00145 (2012).
Singh, N. et al. Role of PGE(2) EP1 receptor in intracerebral hemorrhage-induced brain injury. Neurotoxicity Research 24, 549–559, doi:10.1007/s12640-013-9410-7 (2013).
Ahmad, A. S., Kim, Y. T., Ahmad, M., Maruyama, T. & Doré, S. Selective blockade of PGE2 EP1 receptor protects brain against experimental ischemia and excitotoxicity, and hippocampal slice cultures against oxygen-glucose deprivation. Neurotoxicity Research 14, 343–351 (2008).
Glushakov, A. V., Fazal, J. A., Narumiya, S. & Dore, S. Role of the prostaglandin E2 EP1 receptor in traumatic brain injury. PLoS One 9, e113689, doi:10.1371/journal.pone.0113689 (2014).
Kawano, T. et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nature Medicine 12, 225–229, doi:10.1038/nm1362 (2006).
McCullough, L. et al. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. The Journal of Neuroscience 24, 257–268, doi:10.1523/JNEUROSCI.4485-03.2004 (2004).
Liang, X., Wu, L., Hand, T. & Andreasson, K. Prostaglandin D2 mediates neuronal protection via the DP1 receptor. Journal of Neurochemistry 92, 477–486, doi:10.1111/j.1471-4159.2004.02870.x (2005).
Carlson, N. G. et al. Microglial inhibition of neuroprotection by antagonists of the EP1 prostaglandin E2 receptor. Journal of Neuroinflammation 6, 5, doi:10.1186/1742-2094-6-5 (2009).
Lai, E. et al. Pharmacokinetics, Pharmacodynamics, and Safety of a Prostaglandin D2 Receptor Antagonist. Clinical Pharmacology & Therapeutics 83, 840–847, doi:10.1038/sj.clpt.6100345 (2008).
Maccubbin, D. L. et al. Effectiveness and Safety of Laropiprant on Niacin-Induced Flushing. The American Journal of Cardiology 110, 817–822, doi:10.1016/j.amjcard.2012.05.009 (2012).
Haynes, R. et al. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. European Heart Journal 34, 1279–1291, doi:10.1093/eurheartj/eht055 (2013).
Cheng, K. et al. Antagonism of the prostaglandin D2 receptor 1 suppresses nicotinic acid-induced vasodilation in mice and humans. Proceedings of the National Academy of Sciences 103, 6682–6687, doi:10.1073/pnas.0601574103 (2006).
Saleem, S. et al. PGD2 DP1 receptor protects brain from ischemia-reperfusion injury. European Journal of Neuroscience 26, 73–78, doi:10.1111/j.1460-9568.2007.05627.x (2007).
Ahmad, A. S., Ahmad, M., Maruyama, T., Narumiya, S. & Doré, S. Prostaglandin D2 DP1 receptor is beneficial in ischemic stroke and in acute exicitotoxicity in young and old mice. Age 32, 271–282, doi:10.1007/s11357-010-9135-y (2010).
Clark, W., Gunion-Rinker, L., Lessov, N. & Hazel, K. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke 29, 2136–2140 (1998).
Leclerc, J. L., Lampert, A. S., Diller, M. A. & Doré, S. Genetic deletion of the prostaglandin E2 E prostanoid receptor subtype 3 improves anatomical and functional outcomes after intracerebral hemorrhage. European Journal of Neuroscience 41, 1381–1391, doi:10.1111/ejn.12909 (2015).
Leclerc, J. L. et al. Increased brain hemopexin levels improve outcomes after intracerebral hemorrhage. Journal of Cerebral Blood Flow & Metabolism, 0271678X16679170, doi:10.1177/0271678x16679170 (2016).
Ahmad, A. S. PGD2 DP1 receptor stimulation following stroke ameliorates cerebral blood flow and outcomes. Neuroscience 279, 260–268, doi:10.1016/j.neuroscience.2014.08.050 (2014).
Faraday, N. et al. Cathepsin G-Dependent Modulation of Platelet Thrombus Formation In Vivo by Blood Neutrophils. PLoS One 8, e71447, doi:10.1371/journal.pone.0071447 (2013).
Quick, A. J. The Bleeding Time as a Test of Hemostatic Function. American Journal of Clinical Pathology 64, 87–94, doi:10.1093/ajcp/64.1.87 (1975).
Lauer, A. et al. Antiplatelet pretreatment does not increase hematoma volume in experimental intracerebral hemorrhage. Journal of Cerebral Blood Flow & Metabolism 31, 1736-1742, doi:http://www.nature.com/jcbfm/journal/v31/n8/suppinfo/jcbfm201122s1.html (2011).
Prasad, S. et al. Development of an in vitro model to study clot lysis activity of thrombolytic drugs. Thrombosis Journal 4, 1–4, doi:10.1186/1477-9560-4-14 (2006).
Lai, E. et al. Suppression of Niacin-induced Vasodilation with an Antagonist to Prostaglandin D(2) Receptor Subtype 1. Clinical Pharmacology & Therapeutics (2007).
Lauring, B. et al. Laropiprant in combination with extended-release niacin does not alter urine 11-dehydrothromboxane B2, a marker of in vivo platelet function, in healthy, hypercholesterolemic, and diabetic subjects. Journal of Clinical Pharmacology 49, 1426–1435, doi:10.1177/0091270009339593 (2009).
Van Hecken, A. et al. The effect of MK-0524, a prostaglandin D2 receptor antagonist, on prostaglandin D2-induced nasal airway obstruction in healthy volunteers. European Journal of Clinical Pharmacology 63, 135–141 (2007).
Schuligoi, R. et al. PGD2 metabolism in plasma: Kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochemical Pharmacology 74, 107–117, doi:10.1016/j.bcp.2007.03.023 (2007).
Davì, G. & Patrono, C. Platelet Activation and Atherothrombosis. New England Journal of Medicine 357, 2482–2494, doi:10.1056/NEJMra071014 (2007).
Rosenberg, R. D. & Aird, W. C. Vascular-Bed–Specific Hemostasis and Hypercoagulable States. New England Journal of Medicine 340, 1555–1564, doi:10.1056/NEJM199905203402007 (1999).
Borissoff, J. I., Spronk, H. M. H. & ten Cate, H. The Hemostatic System as a Modulator of Atherosclerosis. New England Journal of Medicine 364, 1746–1760, doi:10.1056/NEJMra1011670 (2011).
Furie, B. & Furie, B. C. Mechanisms of Thrombus Formation. New England Journal of Medicine 359, 938–949, doi:10.1056/NEJMra0801082 (2008).
Mendelow, A. D. et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 365, 387–397, doi:10.1016/S0140-6736(05)17826-X (2005).
Mayer, S. A. et al. Efficacy and Safety of Recombinant Activated Factor VII for Acute Intracerebral Hemorrhage. New England Journal of Medicine 358, 2127–2137, doi:10.1056/NEJMoa0707534 (2008).
Landray, M. J. et al. Effects of Extended-Release Niacin with Laropiprant in High-Risk Patients. New England Journal of Medicine 371, 203–212, doi:10.1056/NEJMoa1300955 (2014).
Warnholtz, A. et al. Effects of oral niacin on endothelial dysfunction in patients with coronary artery disease: Results of the randomized, double-blind, placebo-controlled INEF study. Atherosclerosis 204, 216–221, doi:10.1016/j.atherosclerosis.2008.08.003 (2009).
Ling, M. C. et al. Early effects of statin therapy on endothelial function and microvascular reactivity in patients with coronary artery disease. American Heart Journal 149, 1137, doi:10.1016/j.ahj.2005.02.033 (2005).
Lorkowska, B. & Chlopicki, S. Statins as coronary vasodilators in isolated bovine coronary arteries—involvement of PGI2 and NO. Prostaglandins, Leukotrienes and Essential Fatty Acids 72, 133–138, doi:10.1016/j.plefa.2004.10.012 (2005).
Philipose, S. et al. Laropiprant Attenuates EP3 and TP Prostanoid Receptor-Mediated Thrombus Formation. PLoS One 7, e40222, doi:10.1371/journal.pone.0040222 (2012).
Reagan-Shaw, S., Nihal, M. & Ahmad, N. Dose translation from animal to human studies revisited. The FASEB Journal 22, 659–661, doi:10.1096/fj.07-9574LSF (2008).
Zhao, X., Grotta, J., Gonzales, N. & Aronowski, J. Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke 40, S92–94, doi:10.1161/STROKEAHA.108.533158 (2009).
Ransohoff, R. M. Microgliosis: the questions shape the answers. Nature Neuroscience 10, 1507–1509, doi:10.1038/nn1207-1507 (2007).
del Zoppo, G. J. et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke 38, 646–651, doi:10.1161/01.STR.0000254477.34231.cb (2007).
Matsushita, H. et al. Suppression of CXCL2 upregulation underlies the therapeutic effect of the retinoid Am80 on intracerebral hemorrhage in mice. Journal of Neuroscience Research 92, 1024–1034, doi:10.1002/jnr.23379 (2014).
Yang, Z. et al. Sinomenine inhibits microglia activation and attenuates brain injury in intracerebral hemorrhage. Molecular Immunology 60, 109–114, doi:10.1016/j.molimm.2014.03.005 (2014).
Acknowledgements
This work was supported by the National Institutes of Health R01NS046400 (S.D.), the McKnight Brain Research Foundation, Brain and Spinal Cord Injury Research Trust Fund (S.D.), and the Department of Anesthesiology, University of Florida College of Medicine. The authors would like to express their gratitude to the members of Dore Lab for providing their feedback.
Author information
Authors and Affiliations
Contributions
A.S.A. performed surgeries and experimental procedures. M.M. and D.H. performed functional assessments, tissue sectioning, staining and quantification, and bleeding and coagulation studies. A.S.A., M.M., and D.H. analyzed the data, prepare the figures, and wrote the paper. A.S.A. and S.D. designed research, planned all the experiments and wrote the paper. A.S.A. and S.D. conceived and led the project. All the authors read and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmad, A.S., Mendes, M., Hernandez, D. et al. Efficacy of Laropiprant in Minimizing Brain Injury Following Experimental Intracerebral Hemorrhage. Sci Rep 7, 9489 (2017). https://doi.org/10.1038/s41598-017-09994-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09994-5
- Springer Nature Limited