
Abstract
Exendin-4 is a long acting glucagon-like peptide 1 (GLP-1) analogue that is an agonist for the GLP-1 receptor, a G-protein coupled receptor (GPCR). Exendin-4 is used to clinically improve glucose tolerance in diabetic patients due to its ability to enhance insulin secretion. In rodents, and possibly in humans, exendin-4 can stimulate β-cell proliferation. The exact mechanism of action to induce β-cell proliferation is not well understood. Here, using a β-cell specific epidermal growth factor receptor (EGFR) null mouse, we show that exendin-4 induced an increase in proliferation and β-cell mass through EGFR. Thus, our study sheds light on the role of EGFR signaling in the effects of exendin-4 on the control of blood glucose metabolism and β-cell mass.
Similar content being viewed by others


Introduction
Inducing β-cell proliferation is an attractive treatment modality in the diabetic patient1. Exogenous insulin aids in glucose control, but is inherently different than endogenous insulin, which prevents the microvascular and macrovascular complications of diabetes. Recent studies have begun to elucidate the signaling pathways involved in β-cell proliferation, particularly in the mature β-cell2.
The epidermal growth factor receptor 1 (EGFR) is one of four erbB transmembrane tyrosine kinase receptors. Binding of ligand leads to dimerization and autophosphorylation of a tyrosine kinase, which in turn leads to activation of the epidermal growth factor (EGF) intracellular signaling pathway3. Within the pancreas, EGFR is predominantly expressed in islets, exerting its effects on β-cell development, function, and proliferation4. EGFR null mutant mice have delayed β-cell differentiation and impaired β-cell proliferation during organogenesis5. Additional evidence of this impairment was displayed with inhibition of EGFR postnatally6, in pregnancy7, and with high fat diet7. Recently, we found that EGFR was upregulated in islets after partial pancreatectomy. In the absence of β-cell EGFR, β-cell proliferation was reduced at baseline and after partial pancreatectomy8.
Glucagon-like peptide 1 (GLP-1) is an incretin hormone that is a potent β-cell mitogen, and also stimulates insulin synthesis and secretion. Exendin-4, a long-acting GLP-1 analogue, is resistant to DPP-IV cleavage and therefore more useful clinically9. It is not clear how GLP-1 signaling leads to the PI3-kinase activity seen in β-cell proliferation. Exendin-4 appears to activate the β-cell EGFR to promote β-cell proliferation4, 7, 10, 11. In vitro, it appears that GLP-1 or exendin-4 treatment leads to release of betacellulin and EGF ligands from the peri-islet extracellular matrix, with subsequent stimulation of β-cell proliferation12.
Here, we show that EGFR is necessary in vivo for these exendin-4-mediated effects. We employ a pancreatic β-cell specific EGFR null mouse (β-EGFRfx/fx) and exogenous exendin-4 treatment to show a role for β-cell EGFR in β-cell mass, islet size, β-cell proliferation, and insulin content.
Materials and Methods
Mouse Manipulations
The Animal Research and Care Committee at the Children’s Hospital of Pittsburgh and the University of Pittsburgh IACUC approved all mouse experiments. All experiments were performed in accordance with relevant guidelines and regulations. C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Transgenic mice expressing EGFRfx/fx have been previously described13. EGFRfx/fx mice were crossed with Pdx1-CreERT mice, which also have been previously described, to get Pdx1-CreERT; EGFRfx/fx mice8, 14. The CreERT-negative littermates of same age were used as controls, which showed no phenotypic difference from wild-type C57/BL6 mice. All of these mice have a C57/BL6 background. The terminology used henceforth to distinguish amongst groups is as follows: Pdx1-CreERT; EGFRfx/fx without exendin treatment (Cre+Ex−), Pdx1-CreERT; EGFRfx/fx with exendin treatment (Cre+Ex+), EGFRfx/fx without exendin treatment (Cre−Ex−), and EGFRfx/fx with exendin treatment (Cre−Ex+).
Measurement of Glucose Tolerance, β-cell Mass, and Insulin Content
Glucose tolerance and β-cell mass were measured as previously described1, 15. Pancreatic samples were cut on the longitudinal axis allowing for calculation of β-cell mass as well as insulin content. Insulin content was determined from homogenate pancreatic tissue samples normalized to weight with the Mouse Insulin ELISA Kit (MyBioSource Inc, San Diego, CA, USA).
Tamoxifen and Exendin-4 Injection
Tamoxifen (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 10 mg/ml corn oil (Sigma-Aldrich, St. Louis, MO, USA) and 1 mg/20 g body weight per day of intraperitoneal tamoxifen was given to 6-week-old mice for 5 consecutive days in order to induce Cre recombination. Control mice also received tamoxifen at the same dose and frequency. Intraperitoneal injection of exendin-4 (Sigma-Aldrich, St. Louis, MO, USA) was performed 2 weeks after tamoxifen injection. Exendin-4 (0.1 μg/mg of body weight) was administered twice daily, while saline was administered to control mice.
BrdU Incorporation, Quantification of Proliferating β-Cells
Mice were given BrdU drinking water for 1 week after they received 2 days of exendin-4 injection or saline injection, as described before15, 16. BrdU water was replaced every 3 days. Quantification of β-cell proliferation was based on the percentage of BrdU+ β-cells. At least 2,000 β-cells were counted for each mouse. If the percentage of BrdU+ or β-cells was low, more cells were counted until at least 50 BrdU+ β cells or 20 Ki-67+ β-cells were counted15.
Pancreatic Digestion and Islet Isolation
Pancreas digestion and islet isolation procedures were performed as described before17,18,19. The pancreas was infused with 0.25 mg/ml collagenase (Sigma-Aldrich) for 40 min to obtain a single cell population. Islets were handpicked at least three times to avoid contamination from non-islet fractions. Total RNA was extracted from isolated islets and analyzed by rt-PCR to confirm the purity of the islets by the absence of amylase and CK19 at the transcriptional level.
Western Blot Analysis
Western blotting analysis was performed as previously described (23, 38). Primary antibodies for Western blotting analysis were rabbit polyclonal anti-GAPDH (Cell Signaling, San Jose, CA) and rabbit anti-EGFR (Santa Cruz Biotechnology). Secondary antibody is HRP-conjugated anti-rabbit (Dako, Carpinteria, CA).
Isolation of RNA and RT-qPCR
Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA), and then quantified with the Nanodrop 1000 (Thermo Fisher Scientific, Inc., Waltham, MA), followed by cDNA synthesis (Qiagen) and RT-qPCR. RT-qPCR was done as described before17,18,19. The primers used were EGFR (QT00101584, Quiagen, Hilden, Germany), GLP1R (Fisher Scientific, Waltham, MA), and GAPDH (Fisher Scientific, Waltham, MA). Results were first normalized against the housekeeping gene GAPDH, which is stable across all samples, and then against the experimental controls.
Glucose Stimulated Insulin Secretion (GSIS)
For in vitro static GSIS, digestion and islet isolation was performed as previously described1, 15, 16 for each condition. Mouse islets were cultured in Ham’s F10 medium (Life Technologies, St. Louis, MO, USA) supplemented with 0.5% BSA (Sigma-Aldrich, St. Louis, MO, USA), 2 mmol/L glutamine, 2 mmol/L calcium, and 5 mmol/L glucose at 37 °C, 95% air/5% CO2. After overnight culture, 200 islets per condition were transferred to new plates and treated with low glucose (2.8 mmol/L) and high glucose (16.7 mmol/L) conditions. Islets were pelleted by centrifugation and lysed in acid ethanol for assessment of insulin in media and islets by radioimmunoassay (Linco Research Inc., St. Charles, Missouri, USA). Results were reported as insulin secreted (ng/mL) per hour normalized to number of islets.
Immunohistochemistry
Tissues were fixed in 4% paraformaldehyde overnight at 4 °C, followed by cryoprotection in 30% sucrose overnight, and finally snap-freezing. All samples were sectioned at 6 μm. For antigen retrieval, BrdU immunostaining slides were incubated in 2 mmol/liter HCl for 40 min. All slides were incubated with primary antibodies at 4 °C overnight, then incubated with secondary antibodies for 2 h at room temperature. Primary antibodies used in this study were: guinea pig polyclonal insulin (Dako), rabbit Ki-67 (Abcam), and rat BrdU (Abcam, Cambridge, MA). The secondary antibodies were: Cy2, Cy3, or Cy5-conjugated donkey streptavidin, anti-rabbit, anti-guinea pig, and anti-rat. Nuclear staining was performed with Hoechst staining (Becton-Dickinson Biosciences, San Jose, CA). Cryosection imaging was performed with the AxioImager Z.1 microscope (Zeiss).
Data Analysis
GraphPad Prism 6.0 (GraphPad Software, Inc. La Jolla, CA) was used for statistical analyses. All values are depicted as mean ± S.E. Five repeats were analyzed in each condition. All data were statistically analyzed using one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test to compare two groups. Significance was considered when p < 0.05.
Results
Impaired glucose tolerance in a β-cell specific EGFR null mouse
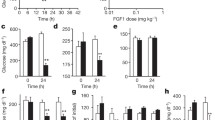
Male pdx1-creERT;EGFRfx/fx mice (Cre+) and control Cre−;EGFRfx/fx mice received exendin-4 (Cre+Ex+ and Cre−Ex+) or saline injection (Cre+Ex− and Cre−Ex−) for 5 days starting at 8 weeks of age, 2 weeks after tamoxifen treatment. There is loss of EGFR mRNA and protein expression after two weeks of tamoxifen treatment (Supplementary Fig. 1A,B). After 5 days of exendin-4 (Cre+Ex+ and Cre−Ex+) or saline (Cre+Ex− and Cre−Ex−) intraperitoneal injection, glucose tolerance tests were performed to assess the ability of the mice to respond to a glucose load. Fasting glucose values were significantly higher in Cre+Ex− mice compared to Cre−Ex− controls at baseline (142 ± 11.4 mg/dL vs. 90 ± 3.3 mg/dL)(Fig. 1A). The fasting glucose levels when the Cre+ mice were treated with exendin-4 (Cre+Ex+) remained elevated (137.3 ± 3.3 mg/dL). However treatment of Cre− mice with exendin-4 (Cre−Ex+) led to significantly lowered blood glucose values (Fig. 1B). The Cre+Ex− mice displayed a decreased ability to respond to a glucose load, producing a diabetic phenotype (Fig. 1B). This diabetogenic effect was not rescued by the addition of exendin-4 (Cre+Ex+), suggesting that exendin-4 exerts its hypoglycemic effect through EGFR. These differences were significant at 15, 30, and 60 min (p < 0.05). These data suggest that EGFR is necessary for a normal insulin response, and that exendin-4 amplifies the insulin response through β-cell EGFR.

(A) Fasting glucose levels were increased in Cre+Ex−mice. This increase was not reversed by exendin-4 treatment (Cre+Ex+). (B) Glucose tolerance tests showed an enhanced ability to respond to a glucose load in control Cre− mice with exendin-4 treatment (Cre−Ex+). A defect in glucose tolerance was seen in Cre+Ex− mice that could not be salvaged with exendin-4 (Cre+Ex−). (C) β-cell mass was significantly increased in control mice as compared with Cre+ mice. (D) Insulin content mirrors β-cell mass. (E) Islet size was significantly increased in control mice after exendin-4 treatment (Cre−Ex+), as compared with Cre+Ex+ and Cre+Ex− mice. n = 5. All results were mean ± S.E. * #, p < 0.05, NS, no significance). (F) In vitro GSIS shows significantly increased insulin release in control mice with exendin-4 treatment (Cre−Ex+) at low and high glucose concentrations. At high glucose concentrations, there is significantly less insulin secretion in Cre+ mice, regardless of treatment with exendin-4, as compared to control Cre− mice.
Effect of EGFR on glucagon like peptide 1 receptor (GLP1-R)
As discussed above, there is loss of EGFR mRNA and protein with Cre recombination in the β-cell in this model. The recombination event has no effect on glucagon like peptide 1 receptor (GLP-1R) mRNA (Supplementary Fig. 1C). Therefore, there should be no differential effect on β-cell mass, islet size, β-cell proliferation, and insulin content with exendin-4 treatment due to GLP1-R expression.
Effect of exendin-4 on β-cell mass
In accordance with previously published data20, β-cell mass was roughly doubled with exendin-4 treatment (Cre−Ex+, 3.12 ± 0.28 mg vs 1.63 ± 0.21 mg, p < 0.05)(Fig. 1C). Very large islets were seen throughout the pancreas after 5 days of exendin-4 injection (see below). However, the Cre+Ex− mice showed no difference in β-cell mass from Cre−Ex− controls at baseline (1.65 ± 0.18 mg). Interestingly, in Cre+Ex+mice, exendin-4 had no enhancing effect on β-cell mass (1.63 ± 0.14 mg, p < 0.05), further suggesting that EGFR is required for GLP-1 signaling to induce β-cell proliferation.
Effect of exendin-4 on insulin content
The pancreas of Cre− mice treated with exendin-4 (Cre−Ex+) had significantly higher insulin content than saline-treated Cre−Ex− controls (68.2 ± 6.3 μg/mg of pancreas vs 37.2 ± 3.1 μg/mg of pancreas, p < 0.05) (Fig. 1D). This ratio corresponds well with the almost 2-fold increase in β-cell mass in Cre−Ex+ mice discussed above. This effect of exendin-4 is lost in the Cre+ mice (37.0 ± 2.8 μg/mg of pancreas, p < 0.05) supporting that EGFR is necessary for the insulinotropic effects of exendin-4.
Effect of exendin-4 on islet size
The pancreas of Cre− mice treated with exendin-4 (Cre−Ex+) had significantly larger-sized islets than controls (134.1 ± 12.2 μm vs 62.9 ± 5.6 μm, p < 0.05) (Fig. 1E). This 2-fold ratio fits well with the increases in β-cell mass and insulin content. There is no effect of exendin-4 in Cre+Ex+ mice (63.1 ± 5.5 μm, p < 0.05) furthering the hypothesis that EGFR mediates the effects of exendin-4.
Effect of exendin-4 on glucose-stimulated insulin secretion (GSIS)
At low glucose concentrations (2.8 mmol/L), exendin-4 treated control islets in vitro (Cre−Ex+) have a significantly increased insulin secretion compared to control (Cre−Ex−) islets, and to Cre+ islets with or without exendin-4 treatment. At high glucose concentrations (16.7 mmol/L), exendin-4 treated control islets (Cre−Ex+) again showed increased insulin secretion. Interestingly, insulin secretion by Cre+ islets is reduced compared to Cre−Ex− controls, and is further 2-fold less than controls treated with exendin-4 (Cre−Ex+). This result suggests a baseline dependence on EGFR to respond to a glucose load. The lack of a rescue effect with exendin-4 treatment implies that the GLP-1 functions through EGFR in this effect.
Exendin-4-induced β-cell proliferation
Mice received BrdU in the drinking water for 1 week beginning on day 3 of exendin-4 or saline injection to aid in the analysis of β-cell replication. Ki-67 expression in β-cells from these mice was also analyzed at harvest. BrdU should label all of the proliferating cells during the 7-day exposure to BrdU, while Ki-67 labels cells within the G1 to M phase of the cell cycle at the time the pancreas is harvested16. In saline-injected mice, the ratio of BrdU+/Insulin+ cells to all Insulin+ cells at baseline was 1.2 ± 0.1% in control Cre−Ex− mice, and 0.8 ± 0.1% in Cre+Ex−mice (Fig. 2A,C). In exendin-4 treated control mice (Cre−Ex+), the ratio was strikingly elevated, at 38 ± 3.4%, but remained only 0.9 ± 0.1% in Cre+Ex+mice. Ki-67 data showed similar results (Fig. 2B).

BrdU and Ki67 positive cells within each treatment group. (A) There is a 20-fold increase in %BrdU+/insulin+ cells in control Cre−Ex+ mice when treated with exendin-4 as compared to untreated Cre−Ex− controls. There is no effect of exendin-4 in Cre+Ex+ mice. (B) There is a 20-fold increase in %Ki67+/insulin+ cell numbers in control mice treated with exendin-4 (Cre−Ex+) as compared to saline control (Cre−Ex−). Again, there is no effect of exendin-4 in Cre+Ex+ mice. (C) Representative sections show the large islets in exendin-4 treated control Cre-Ex + mice, with many BrdU+ proliferating cells. n = 5. All results were mean ± S.E. *p < 0.05, NS, no significance).
Discussion
Reagents that promote β-cell proliferation are potentially very important as treatment modalities for diabetes mellitus. GLP-1 is one hormone signaling pathway useful for therapeutic intervention21, although the exact mechanism of action has yet to be completely elucidated. We show that a long acting GLP-1 analogue improves glucose tolerance, increases insulin content, and increases proliferation. Further, by employing a β-cell specific EGFR null mouse, we show that β-cell EGFR is necessary for these effects of GLP-1/exendin-4.
Previous studies have shown that GLP-1 signaling stimulates insulin gene expression and biosynthesis22, enhances glucose-stimulated insulin secretion23, restores glucose competence in glucose-resistant β-cells24, and stimulates β-cell mass expansion20. In addition, GLP-1 receptor null mice develop glucose intolerance with dysmorphic islets25. GLP-1 clearly acts as a β-cell growth factor in animal models, leading to increased insulin synthesis and β-cell mass. When administered to type 2 diabetics, GLP-1 increases insulin secretion and decreases blood glucose and glucagon9, 23.
Here, we confirm a robust improvement in glucose tolerance and insulin content (in direct proportion to β-cell mass) as a result of exendin-4 treatment. The increase in β-cell mass can result from either hypertrophy of existing β-cells or from replication and neogenesis of β-cells. The high percentage of BrdU-incorporated β-cells in exendin-4-treated mice suggests that the increased β-cell mass mainly results from augmentation of β-cell proliferation. The relatively high percentage of Ki67 positive cells that represent cells in an active cell cycle, as compared to BrdU positive cells that represent any proliferation over the 7 days, implies that maximal replication likely occurs at 7 days (time of analysis). This increased β-cell mass correlates with increased insulin content and enhanced glucose tolerance. These effects are limited to the β-cells, as increases in proliferation were not seen in non- β-cell islet cells, nor in acinar nor ductal cells. The absence of proliferation within the acinar or ductal compartments also relates to the current controversy about the potential risk for neoplastic proliferation in patients treated with GLP-1 analogues. Without significant increases in BrdU or Ki67 staining, neoplastic changes in the exocrine compartment would seem unlikely, although endocrine neoplasia would still seem to be a possibility.
GLP-1 is thought to exert its effects by interacting with its high affinity receptor (GLP-1R), which is a member of the G-protein coupled receptor (GPCR) family. The G-protein activation results in the second messenger cascade of cAMP and protein kinase A activation26. There is also some evidence that cytosolic calcium may be increased via the cAMP second messenger27. The end result is activation of PI3-kinase and its downstream effectors that lead to β-cell proliferation28. The precise mechanism of activating the proliferative pathway via the PI3-kinase pathway is not clear. Recent studies suggest that GLP-1R lacks kinase activity, and may rely on EGFR to promote proliferation29. Other GPCR agonists have shown reliance upon the EGFR to activate PI3-kinase, including lysophosphatidic acid29, thrombin30, and angiotensin II29.
In this study, exendin-4 seems to fit with the other GPCR agonists in a dependence on EGFR. After 5 consecutive days of tamoxifen injection followed by a 2-week waiting period to allow for degradation of pre-existing EGFR, the majority of the EGFR in the β-cell specific EGFR knockout mice is gone as shown previously8. Tamoxifen injection in adult mice to activate the Cre system avoids the effects of EGFR deletion on embryological development that occur with the insulin promoter driving cre, without an ERT, and specifically β-cell development and maturation. Importantly, our previous work also demonstrated that the Pdx1-CreERT line employed here does not contain the human growth hormone mini-gene that could confound the study8 nor does the system have leakage into the hypothalamus8, as reported in a recent study31. In β-cell specific null mutants, the increase in β-cell mass with exendin-4 treatment is lost along with the increases in β-cell proliferation and insulin content. A state of relative glucose intolerance develops when EGFR is deleted from β-cells, suggesting a direct link to insulin secretion and a diminished level of baseline β cell proliferation, consistent with previous work8. This result is independent of the proliferative effects of EGFR on β-cell function, which suggests an integral role for EGFR in the coordination of insulin secretion. Although pronounced improvements in β-cell mass and glucose homeostasis are seen with exendin-4 administration in control mice, none of those improvements are seen in the β-cell EGFR null mutants. These data taken together implicate EGFR as a critical pathway from GLP-1 to PI3-kinase and β-cell proliferation. Activation of the EGFR pathway alone may not be sufficient to produce the effects of GLP-1 and exendin-4, but these data suggest that it is necessary. Continued study of the crosstalk among signaling pathways is vital to fully understand β-cell proliferation.
References
Xiao, X. et al. No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest 123, 2207–2217, doi:10.1172/JCI66323 (2013).
Saunders, D. & Powers, A. C. Replicative capacity of beta-cells and type 1 diabetes. J Autoimmun 71, 59–68, doi:10.1016/j.jaut.2016.03.014 (2016).
Yarden, Y. & Sliwkowski, M. X. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2, 127–137, doi:10.1038/35052073 (2001).
Buteau, J., Foisy, S., Joly, E. & Prentki, M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 52, 124–132 (2003).
Huotari, M. A. et al. ErbB signaling regulates lineage determination of developing pancreatic islet cells in embryonic organ culture. Endocrinology 143, 4437–4446, doi:10.1210/en.2002-220382 (2002).
Miettinen, P. J. et al. Downregulation of EGF receptor signaling in pancreatic islets causes diabetes due to impaired postnatal beta-cell growth. Diabetes 55, 3299–3308, doi:10.2337/db06-0413 (2006).
Hakonen, E. et al. Epidermal growth factor (EGF)-receptor signalling is needed for murine beta cell mass expansion in response to high-fat diet and pregnancy but not after pancreatic duct ligation. Diabetologia 54, 1735–1743, doi:10.1007/s00125-011-2153-1 (2011).
Fusco, J. et al. Epidermal Growth Factor Receptor Signaling Regulates beta Cell Proliferation in Adult Mice. J Biol Chem 291, 22630–22637, doi:10.1074/jbc.M116.747840 (2016).
Holst, J. J. & Orskov, C. The incretin approach for diabetes treatment: modulation of islet hormone release by GLP-1 agonism. Diabetes 53(Suppl 3), S197–204 (2004).
MacDonald, P. E. et al. Antagonism of rat beta-cell voltage-dependent K+ currents by exendin 4 requires dual activation of the cAMP/protein kinase A and phosphatidylinositol 3-kinase signaling pathways. J Biol Chem 278, 52446–52453, doi:10.1074/jbc.M307612200 (2003).
Miettinen, P., Ormio, P., Hakonen, E., Banerjee, M. & Otonkoski, T. EGF receptor in pancreatic beta-cell mass regulation. Biochem Soc Trans 36, 280–285, doi:10.1042/BST0360280 (2008).
Miyagawa, J. et al. Immunohistochemical localization of betacellulin, a new member of the EGF family, in normal human pancreas and islet tumor cells. Endocr J 46, 755–764 (1999).
Lee, T. C. & Threadgill, D. W. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 47, 85–92, doi:10.1002/dvg.20464 (2009).
Wicksteed, B. et al. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59, 3090–3098, doi:10.2337/db10-0624 (2010).
Xiao, X. et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci USA 111, E1211–1220, doi:10.1073/pnas.1321347111 (2014).
Xiao, X. et al. TGFbeta receptor signaling is essential for inflammation-induced but not beta-cell workload-induced beta-cell proliferation. Diabetes 62, 1217–1226, doi:10.2337/db12-1428 (2013).
Li, D. S., Yuan, Y. H., Tu, H. J., Liang, Q. L. & Dai, L. J. A protocol for islet isolation from mouse pancreas. Nat Protoc 4, 1649–1652, doi:10.1038/nprot.2009.150 (2009).
Xiao, X. et al. Neurogenin3 activation is not sufficient to direct duct-to-beta cell transdifferentiation in the adult pancreas. J Biol Chem 288, 25297–25308, doi:10.1074/jbc.M113.484022 (2013).
Xiao, X. et al. Pancreatic duct cells as a source of VEGF in mice. Diabetologia 57, 991–1000, doi:10.1007/s00125-014-3179-y (2014).
Stoffers, D. A. et al. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes 49, 741–748 (2000).
Holst, J. J. Gut hormones as pharmaceuticals. From enteroglucagon to GLP-1 and GLP-2. Regul Pept 93, 45–51 (2000).
Fehmann, H. C. & Habener, J. F. Insulinotropic hormone glucagon-like peptide-I(7-37) stimulation of proinsulin gene expression and proinsulin biosynthesis in insulinoma beta TC-1 cells. Endocrinology 130, 159–166, doi:10.1210/endo.130.1.1309325 (1992).
Gutniak, M., Orskov, C., Holst, J. J., Ahren, B. & Efendic, S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med 326, 1316–1322, doi:10.1056/NEJM199205143262003 (1992).
Holz, G. Gt., Kuhtreiber, W. M. & Habener, J. F. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37). Nature 361, 362–365, doi:10.1038/361362a0 (1993).
Scrocchi, L. A. et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med 2, 1254–1258 (1996).
Widmann, C., Burki, E., Dolci, W. & Thorens, B. Signal transduction by the cloned glucagon-like peptide-1 receptor: comparison with signaling by the endogenous receptors of beta cell lines. Mol Pharmacol 45, 1029–1035 (1994).
Dillon, J. S. et al. Cloning and functional expression of the human glucagon-like peptide-1 (GLP-1) receptor. Endocrinology 133, 1907–1910, doi:10.1210/endo.133.4.8404634 (1993).
Buteau, J. et al. Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia 47, 806–815, doi:10.1007/s00125-004-1379-6 (2004).
Voisin, L. et al. EGF receptor transactivation is obligatory for protein synthesis stimulation by G protein-coupled receptors. Am J Physiol Cell Physiol 283, C446–455, doi:10.1152/ajpcell.00261.2001 (2002).
Daub, H., Weiss, F. U., Wallasch, C. & Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557–560, doi:10.1038/379557a0 (1996).
Brouwers, B. et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab 20, 979–990, doi:10.1016/j.cmet.2014.11.004 (2014).
Author information
Authors and Affiliations
Contributions
The study was conceived and designed by G.K.G. Acquisition of data was by J.F., X.X., K.P., Q.S., C.C., and Y.C.M. Analysis of data was carried out by J.F., X.X., K.P., Q.S., C.C., Y.C.M. and G.K.G. J.F. and G.K.G. interpreted the data. J.F. drafted the manuscript, and all authors revised the article and approved the final version to be published. G.K.G.is the guarantor of this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fusco, J., Xiao, X., Prasadan, K. et al. GLP-1/Exendin-4 induces β-cell proliferation via the epidermal growth factor receptor. Sci Rep 7, 9100 (2017). https://doi.org/10.1038/s41598-017-09898-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09898-4
- Springer Nature Limited
This article is cited by
-
Exendin-4 Prevents Memory Loss and Neuronal Death in Rats with Sporadic Alzheimer-Like Disease
Molecular Neurobiology (2024)
-
The role of insulin and incretin-based drugs in biliary tract cancer: epidemiological and experimental evidence
Discover Oncology (2022)
-
The Biological Role of Optimized Recombinant Oral Long-Acting Glucagon Like Peptide-1 and Its Impact on the Expression of Genes Associated with Glucose Metabolism of Diabetes
International Journal of Peptide Research and Therapeutics (2021)